
Abstract
Genomic studies provide deeper insights into secondary metabolites produced by diverse bacterial communities, residing in various environmental niches. This study aims to understand the potential of a biosurfactant producing Bacillus sp. AM13, isolated from soil. An integrated approach of genomic and chemical analysis was employed to characterize the antibacterial lipopeptide produced by the strain AM13. Genome analysis revealed that strain AM13 harbors a nonribosomal peptide synthetase (NRPS) cluster; highly similar with known biosynthetic gene clusters from surfactin family: lichenysin (85 %) and surfactin (78 %). These findings were substantiated with supplementary experiments of oil displacement assay and surface tension measurements, confirming the biosurfactant production. Further investigation using LCMS approach exhibited similarity of the biomolecule with biosurfactants of the surfactin family. Our consolidated effort of functional genomics provided chemical as well as genetic leads for understanding the biochemical characteristics of the bioactive compound.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bacterial natural products have gained wide attention in recent years due to their extensive applications in food and pharmaceutical industries. These industrially and biologically important biomolecules encompass variety of active compounds like terpenoids, bacteriocins, lipopeptides, etc (Nerurkar 2010; Raaijmakers et al. 2010). Biosurfactants, specifically the ‘Lipopeptides’, are amphiphilic cyclic peptides and usually linked with a fatty acid chain (Nerurkar 2010). Surfactants have been widely studied for years, both with structural and functional aspects. Apart from environmental and industrial uses, these biomolecules have wide biological applications (Rodrigues et al. 2006; Ongena and Jacques 2008). Biosurfactants are known to have antibacterial, hemolytic, and antiviral activity, possessing a specific mechanism that affects the membrane permeability and eventually leading to cell disruption (Heerklotz and Seelig 2007).
These structurally complex biomolecules require equally complex machinery for their biosynthesis. Nonribosomal peptide synthetases (NRPS) are one of the complex set of proteins amongst the several known machineries required to produce bacterial natural products (Ansari et al. 2004; Wang et al. 2014). Various studies have been carried out to understand the biosynthetic gene clusters harbored by bacteria (mainly Bacillus genera) for actively synthesizing such compounds (Yakimov et al. 1995; Mukherjee et al. 2009; Al-Bahry et al. 2013; Donio et al. 2013). Although, gene clusters responsible for the production of NRPS molecules share very similar modules, most of these have a potential to produce isoforms of the same biomolecule (Naruse et al. 1990; Konz et al. 1999; Domingos et al. 2015). Considering the diversity in their structures and functions, it is necessary to carry out extensive screening and characterization using unified methodology of chemical genomics and thus to create a platform for breakthrough understanding of such biomolecules (Payne et al. 2007; Zerikly and Challis 2009; Lu et al. 2014; Aleti et al. 2015). Furthermore, such efforts could eventually lead to the development of better drugs, considering both the production cost and improvement in their effectiveness.
The present study involves the use of an integrated approach to understand the functional genomics of a biosurfactant producing bacterial isolate, Bacillus sp. AM13. Moreover, this study aims to understand the genetic basis of biosynthetic machinery involved in the production of the bioactive molecule, along with comprehending its chemical nature.
Material and methods
Sample collection and bacterial isolation
Soil samples near a pond situated at Ambala, Haryana, India (30° 21’ 23.64” N, 76° 51’ 28.26” E) were collected in triplicate. Samples were transported to the laboratory at 4 °C and were processed immediately. For bacterial isolation, 1 g soil sample was serially diluted in sterile saline solution. Each dilution was spread onto NA and R2A medium (pH 7.2 ± 0.2) and incubated at 30 °C for 2–7 days.
Primary screening of antibacterial activity
Bacterial isolates were screened for antibacterial activity against Escherichia coli ATCC 25922 and Bacillus subtilis ATCC 10876 (data not shown). Isolates demonstrating desired zones were further screened by the same protocol against two multi-drug resistant bacteria (MDR), Enterobacter sp., resistant to sulfamethoxazole, ampicillin, azithromycin, and tetracycline, and Serratia sp. GMX1, resistant to sulfamethoxazole, ampicillin, azithromycin, tetracycline, and netilmicin as described in the previous study (Kapley et al. 2016). Isolates with antibacterial activity against both the MDR strains were selected for further characterization. However, this study focuses only on strain AM13. This strain was deposited at Microbial Culture Collection, Pune, India, and catalogued with the accession number MCC 2971.
Genome sequencing
Genomic DNA extraction was carried out using the method described in the previous study (Prakash et al. 2012). The genomic DNA was fragmented using Ion Shear Plus Reagents Kit (Life technologies, USA) and purified using Agencourt Ampure XP DNA reagent (Beckman Coulter, USA). The fragmented DNA was ligated with barcode adapters. Adaptor-DNA constructs were purified and size selected and amplified via PCR as per manufacturer’s instructions. Quantification and size distribution analysis was carried out on high sensitivity DNA chip kit on Agilent Bioanalyzer 2100 (Agilent technologies, Santa Clara, USA). Emulsion PCR and sequencing of the DNA libraries were carried out using Ion PI chip Ion Proton system (Life technologies, USA).
Genomic data preprocessing, assembly, and annotation
All Ion-proton quality-approved, trimmed, and filtered data were exported as BAM files for further bioinformatics analysis. The De Novo assembly method was employed to carry out the assembly of quality-filtered reads using MIRA assembler version 4.9.3 (Chevreux et al. 1999). Genome annotation was carried out using Rapid Annotation using Subsystem Technology (RAST) (Aziz et al. 2008) and Prokaryotic Genome Automatic Annotation Pipeline (PGAAP) (Angiuoli et al. 2008). tRNAScan (Lowe and Eddy 1997) and RNAmmer 1.2 (Lagesen et al. 2007) were used to predict transfer RNA (tRNA) and ribosomal RNA (rRNA) genes, respectively.
Nucleotide sequence submission
This Whole Genome Shotgun project has been deposited at GenBank under the accession LKCP00000000.1. The version described in this paper is version LKCP01000000.
Phylogenetic analysis and comparative genomics
16S ribosomal sequence retrieved from the AM13 genome was used to carry out phylogenetic analysis. Sequence similarity search was carried out using EzTaxon database with available type strains (Chun et al. 2007). Additionally, Multi Locus Sequence Typing (MLST) approach (using seven housekeeping genes) was used to determine the taxonomic identity of the isolate. For MLST analysis, sequences for nearest neighbors were retrieved from the available genomes. Average nucleotide identity (ANI) and tetra-nucleotide correlation index (Richter et al. 2015) were calculated with the closest species using an online tool, JSpeciesWS (http://jspecies.ribohost.com/jspeciesws). CGview, a comparative genomics tool (Grant and Stothard 2008), was used to compare and visualize the genomes of closely related species with Bacillus sp. AM13.
Prediction and analysis of genomic features of Bacillus sp. AM13
Specialized pipelines like antibiotics and secondary metabolite analysis SHell (antiSMASH) version 3.0.4 (Weber et al. 2015) and Natural Product Domain Seeker (NapDos) (Ziemert et al. 2012) were used to predict and annotate genes involved in production of secondary metabolites. Additionally, genomic data was screened for the presence of Insertion sequence (IS) elements and Phages using, IS Finder (Siguier et al. 2006), and PHAge Search Tool (PHAST) (Zhou et al. 2011), respectively. Screening of Genomic islands (GI) was carried out using the integrated method available in IslandViewer tool (Dhillon et al. 2015), which uses all three methods namely IslandPick, SIGI-HMM, and IslandPath-DIMOB for prediction of Genomic Islands (GI).
Extraction of biosurfactant
A cell-free supernatant from the culture broth was obtained by centrifugation at 10,000 rpm for 20 min at 4 °C. pH of the supernatant was adjusted to 2, using 6 M HCl and was subjected to acid precipitation by placing it at 4 °C overnight. The off-white precipitate was separated by centrifugation at 10,000 rpm for 30 min at 4 °C as described by Domingos et al. (2015). The precipitate was extracted thrice with methanol. Solvent was evaporated using a rotary evaporator at 40 °C, leaving behind relatively pure biosurfactant, which was dissolved in methanol for further use.
Hemolytic activity and oil displacement assay
As biosurfactants are known to lyse erythrocytes, blood agar method is often used for preliminary screening of such microorganisms. A fresh single colony of the culture was taken and spotted onto sheep blood agar plate (HiMedia, India). The plates were incubated for 24–48 h at 37 °C. Hemolytic activity was detected as the occurrence of a defined clear zone around a colony, as described in previous studies (Carrillo et al. 1996). Additionally, bacterial extract was tested for its ability to displace oil from a watery surface (Morikawa and Hirata 2000). Petri plates (diameter 90 mm) were filled with 30 ml of distilled water. Twenty microliters of sterile (filtered, 0.45 μm) crude oil was carefully layered on top of the water phase with a micropipette. Ten microliters of bacterial extract was added in the center of the oil surface. Clearing zone that appeared around the application site was observed. The diameter of this clearing zone on the oil surface correlates to surfactant activity.
Surface tension measurement
Biosurfactant production of strain AM13 was determined by its ability to reduce the surface tension of the culture medium. The reduction in surface tension was measured by Platinum Iridium Wilhelmy plate method using a DCAT11 digital surface tensiometer (Data Physics, USA). The strain AM13 was grown in Tryticase Soy Broth (TSB) at three different time intervals (24, 48, and 72 h) to observe the pattern of biosurfactant production. A cell free broth was obtained by centrifuging culture broth (50 ml) at 10,000 rpm for 20 min. The surface tension measurements were done by using cell free broth of all three-time points. The instrument was calibrated beforehand using Milli-Q water. All measurements were done at room temperature (25 °C). To increase the accuracy of the surface tension measurements, readings were taken in triplicates for each time interval.
Thin layer chromatography
Preliminary characterization of the biosurfactant was done by TLC method. Surfactin standard (Sigma Aldrich, St. Louis, MO, USA) of 1 mg ml−1 was used as control. Three microliters of methanolic extract was spotted on TLC plates Merck silica gel 60; mixture of chloroform and methanol was used as mobile phase in the ratio 7.5:2.5 v/v. Iodine was used to develop the plate. Simultaneously, preparative TLC was carried out as described in the previous study (Lavermicocca et al. 2000) to partially purify the extract. The band with R f value corresponding to the surfactin standard was collected by scrapping off the band and eluted with methanol. This methanolic extract was later checked for its antibacterial activity and also used for LC-MS analysis.
LC-MS analysis
Methanolic extract was diluted as 1:10 methanol/water (v/v) before injecting into LC-MS apparatus. An Agilent 1260 binary LC system (Agilent technologies, Waldbronn, Germany) consisting of a binary pump, thermostatted autosampler, and column compartment was used. The separation was performed at 40 °C on an RRHT Zorbax SB column, 100 × 2.1 mm, 1.8 μm (Agilent technologies, Santa Clara, CA, USA). Mobile phase A consisted of 0.1 % Formic acid), whereas mobile phase B was acetonitrile. The flow rate was 0.3 ml/min with a linear gradient from 5 to 95 % in 13 min. The total time of analysis was 30 min. Mass spectrometry was carried out using 6540 Ultra- High definition accurate mass QTOF LC/MS system (Agilent technologies, Santa Clara, USA), equipped with a dual AJS-ESI. The LC-MS/MS was operated in full scan mode (from 400 to 1700 m/z ratio) to determine molecular mass of the biosurfactant molecule. Surfactin (Sigma Aldrich, St. Louis, MO, USA) of concentration 1 mg ml−1 was used as standard. The extracted ion chromatograms with mass spectra were used for further analysis.
Results
Antibacterial activity
Strain AM13 exhibited antibacterial activity against all test organisms including Multi-Drug Resistant (MDR) bacterial strains (Fig. S1). Moreover, the fraction obtained from preparative thin-layer chromatography (TLC) showed antibacterial activity against test bacterial strains.
Genome sequence and general features
Genome sequencing of strain AM13 using Ion Proton platform yielded 4,654,704 reads with a mean read length of 168 bp. Quality filtered reads (>Q30) used for assembly resulted into 104 contigs. However, only 99 good quality contigs, with an average coverage of 163.24 X and genome size of 3,734,657 bp (Table 1), were used for further analysis. PGAAP revealed that the genome harbored 3791 genes (3481 CDS), while 4030 genomic features were annotated by RAST server (RAST Genome ID: 1386.173) as described in Table 1. Based on the genomic data, GC content was found to be 41.6 %, which is similar to most of the closely related species (Villanueva et al. 2014).
Phylogenetic analysis and comparative genomics
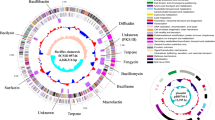
A full-length 16S rRNA gene sequence of 1537 bp (retrieved using RNAmmer 1.2) was selected for phylogenetic analysis. It was found that the strain AM13 shared high similarity with Bacillus safensis FO-36bT (99.93 % similarity), followed by Bacillus pumilus ATCC 7061T (99.87 %) and Bacillus altitudinis 41KF2bT (99.54 %). Phylogenetic analysis indicated that strain AM13 formed a separate clade with B. safensis FO-36bT, within the B. pumilus group (Fig. 1a). Phylogenetic analysis using concatenated dataset of seven MLST genes highlighted its relatedness with B. safensis (Fig. 1b, Table S1). Furthermore, the genome comparison approach employed using JSpeciesWS (results shown in Table S2), also substantiated its high similarity with B. safensis. Further confirmation of the taxonomic identity based on genome comparison using CGview tool supported these findings. Figure 2 represents a circular map of the genome illustrating various coding regions along with the location of tRNA, rRNA, and other genes. The comparison revealed high similarity between genomes in the regions harboring tRNA and rRNA genes along with few other regions as seen in Fig. 2. The height of the BLAST hits (which is drawn proportional to the percent identity of the hits) shows that the strain AM13 (BLAST 1) and B. safensis (BLAST 3) share high genome similarity as compared to the genome of B. pumilus (BLAST 2).

Phylogentic analysis. a Phylogenetic relationship of isolate AM13 with closely related taxa based on 16S rRNA gene sequences (16S rRNA gene sequence of Aeribacillus pallidus was used as outgroup). b Phylogenetic relationship of isolate AM13 with closely related taxa within Bacillus pumilus group, based on MLST approach. The phylogenetic trees were constructed using Neighbor-Joining method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches. The evolutionary distances were computed using the Kimura 2-parameter method and are in the units of the number of base substitutions per site. The rate variation among sites was modeled with a gamma distribution (shape parameter = 1)

Circular genome plot. The figure represents comparison of genomes (BLAST) between isolate AM13 (BLAST1), Bacillus pumilus SAFR032 (BLAST2) and Bacillus safensis FO-36b (BLAST3). The arrows in the outer rings indicate the features (CDS) within the genome of strain AM13, while the height of the BLAST hits is proportional to percent identity of the hit
Genome profile: screening of GI, IS elements, and pro-phage
The presence of two pro-phages, one intact pro-phage and an incomplete pro-phage, was observed in the genome, as revealed by PHAST (Fig. 3a). Screening of horizontal gene transfer (HGT) events (mobile genetic elements), predicted the presence of nine regions of genomic islands (see Fig. 3b). Functional features of the genes in these predicted GI regions were determined using annotation files obtained from RAST and PGAAP. Our analysis suggested that these regions harbored genes mainly encoding few ribosomal, hypothetical, and viral proteins. However, some GI regions also included transport-related genes and Plantazolicin synthase D subunit. The list of predicted genes in the probable GI’s is described in Table S3. Furthermore, very few significant hits for IS (Insertion Sequence) elements related to Prophage origin were found using the IS Finder tool.

Prophage and Genomic islands analysis. Genome contigs of the isolate AM13 were screened for any horizontal gene transfer profiles using PHAST and Islandviewer. a The figure illustrates the circular map of the genome and the predicted prohages along with their positions in the genome. b Circular plot of the genome contigs along with the predicted genomic islands using IslandViewer tool
Secondary metabolite prediction and NRPS cluster analysis
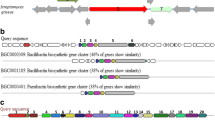
Secondary metabolite prediction using antiSMASH revealed the presence of a biosynthetic gene cluster, with a similarity of 85 % (gene similarity) with known lichenysin cluster and 78 % with the known surfactin cluster (Fig. 4). This gene cluster was found to be highly similar to the genome of B. pumilus W8 (Fig. 4). Domains and their substrate specificity predicted using antiSMASH and NaPDoS suggested that the predicted gene cluster belonged to NRPS family, specifically to the biosurfactant group.

NRPS cluster analysis. The figure depicts the presence of bio-surfactant (NRPS Non-ribosomal peptide synthetase) synthesizing gene cluster in the genome of isolate AM13. NRPS gene cluster predicted and annotated by antiSMASH along with their closest genome (A1) and biosynthetic gene cluster homologs (A2) are represented in the above figure. The section A3 in the figure is an illustration of the predicted arrangement of the domains along with the annotated modules
Biosurfactant activity
It is evident from the study carried out by Morikawa and Hirata (2000) that the extent of oil displacement is directly proportional to the amount of biosurfactant produced. Methanolic extract of strain AM13 displaced the oil layer completely to the circumference of the petri dish, indicating its capacity to produce biosurfactant. Additionally, strain AM13 demonstrated β-hemolytic activity, producing a clear zone of 1.7 cm around the colony on blood agar plate, which reasserts biosurfactant production of strain AM13. Biosurfactant activity of the strain AM13 was also confirmed by its ability to reduce the surface tension of the culture medium to 31.233, 29.185, and 29.055 mN m−1 in 24, 48, and 72 h, respectively.
Chemical characterization of biosurfactant
The TLC technique used showed the presence of a biomolecule (R f = 0.9) similar to the standard surfactin (R f = 0.9). Chemical characterization of the fraction (with antibacterial activity) obtained from preparative TLC was further deciphered by mass spectrometry. Although, the data obtained from ESI (+)-MS showed few differences in ratio, four major ions of m/z 1036, 1044, 1058, and 1072 were observed to be similar and predominant, confirming its relatedness with members of the surfactin family (Fig. 5).

LCMS analysis. Mass spectrometry profile for the biosurfactant produced by isolate AM13. X-axis represents the m/z ratio; Y-axis represents the abundance (count) of each fragment. a Mass spectrum of the extract of isolate AM13. b Mass spectrum of the standard surfactin used
Discussion
The advent in sequencing technologies has revolutionized our understanding about several microbial mechanisms. Along with revealing several hidden genomic features, it has dramatically improved various approaches like deciphering novel metabolic pathways, genome-based phylogeny, and to carry out comparative genomics to understand the genome wide variations in closely related organisms (Land et al. 2015). Recent developments in genomic studies have helped researchers to reveal complexity of metabolic pathways involved in the production of wide variety of secondary metabolites produced by bacteria (Van Lanen and Shen 2006; Lam 2007; Doroghazi et al. 2014). Although, various efforts have been made to decipher the functionality of these bioactive molecules, genomic information has increased the magnitude of our understanding about varied types of biomolecules that could be synthesized by identical genetic machinery (Bergmann et al. 2007; Harvey et al. 2015). Previous studies suggest that genome sequencing efforts have enabled to understand the intricacies involved in metabolic pathways of the bacterium (Pal et al. 2015; Kapley et al. 2016). The present study uses an integrative approach of functional genomics, not only to characterize the bioactivity but also to understand the genomic features of this complex gene clusters. Our study focuses on one such potential bacterial isolate with antibacterial activity.
Primarily, various phylogenetic studies have revealed the complexity in taxonomic classification of bacterial isolates belonging to genus Bacillus (Liu et al. 2013; Branquinho et al. 2014). Several groups in this genus, especially the Bacillus cereus and B. pumilus group, are known to be the most complex groups for taxonomic classification, based on 16S rRNA gene sequencing (Liu et al. 2013). Although, MLST approach may improve the taxonomic resolution of these groups, genomic data is known to overcome all the drawbacks associated with the former method (Rey et al. 2004). Our integrative approach, which includes use of 16S rRNA gene, seven MLST genes, ANI, and Tetra-nucleotide Correlation index method, helped us to confirm the taxonomic classification of strain AM13 as B. safensis (Fig. 1 and Table S2). The genome wide comparison carried out emphasized the level of similarity and complexity in genomes of these closely related species within B. pumilus group.
Furthermore, our efforts to carry out genome sequencing of the strain AM13 helped us to decipher the gene repertoire involved in synthesis of bioactive molecules. The secondary metabolite prediction and annotation revealed the presence of a NRPS gene cluster, similar to known lichenysin biosynthetic gene cluster (85 % of the genes showed similarity), followed by surfactin biosynthetic gene cluster (78 % similarity). Additionally, the identity of each individual domain of the gene cluster was confirmed using NapDos and BLAST tool from NCBI database, which also revealed its similarity with gene clusters responsible for synthesizing bacterial lipopeptides (specifically of surfactin family). The arrangement of domains was observed as illustrated in Fig. 4, with predictions of the substrate of each adenylation domain (AMP binding site). It was noted that predicted gene cluster consists of six domains (Fig. 4). Domain 1, 2, 3, and 6 are typical NRPS domains, which include modules for condensation (C), AMP binding sites, peptide carrier proteins (PCP), epimerization (E), and thioesterase (TE). Interestingly, ketoreductase (KR) module was found in domain 4 and 5 along with these typical NRPS modules. Previous studies suggest that domain 4 and 5 are found in species of B. pumilus group producing similar biosurfactants (Konz et al. 1999; Koumoutsi et al. 2004; Domingos et al. 2015). From substrate prediction analysis, amino acid sequence of the lipopeptide was found to be Glu/Gln-Leu-Leu-Val/Leu/Ile-Asp-Leu-Ile (domain 1–3, see Fig. 4).
Although few potential GI were evident in the genome, region harboring the biosynthetic gene cluster was devoid of any mobile elements (Fig. 3). This finding further confirmed the inheritance of biosynthetic gene cluster, ruling out any possibility of HGT in strain AM13. Other experiments carried out based on the genomic insights obtained to understand and characterize the bioactive compound provided further leads about the chemical nature of the compound. Strain AM13 exhibited β-hemolytic and oil displacement activity, which are amongst the key tests to determine the production of biosurfactants, as suggested in previous studies (Carrillo et al. 1996; Satpute et al. 2010). Additionally, mass spectrometry (MS) analysis revealed its similarity with the standard surfactin used. Specifically, the predominant peaks with m/z ratio 1022, 1036, 1044, 1058, and 1072 were present in common and thus could be attributed to the surfactin family (Fig. 5). Previous studies carried out on characterization of biosurfactants have also reported the presence of similar mass profiles for lipopetides primarily belonging to the surfactin family (Yakimov et al. 1995; Ben Ayed et al. 2014; Domingos et al. 2015). Phylogenetic analysis carried out along with the ANI approach successfully revealed the taxonomic classification of the isolate to be B. safensis. The NRPS cluster prediction and characterization of the antimicrobial biomolecule confirmed its chemical nature to be a biosurfactant belonging to surfactin family.
In summary, this study reports an integrative approach to characterize the antimicrobial compound along with insights into genomic features of strain AM13. Such methodological approaches will help researchers to explore the variety of biosurfactants and their potential usage in human health, industry, and environmental protection.
References
Al-Bahry SN, Al-Wahaibi YM, Elshafie AE, Al-Bemani AS, Joshi SJ, Al-Makhmari HS, Al-Sulaimani HS (2013) Biosurfactant production by Bacillus subtilis B20 using date molasses and its possible application in enhanced oil recovery. Int Biodeterior Biodegrad 81:141–146. doi:10.1016/j.ibiod.2012.01.006
Aleti G, Sessitsch A, Brader G (2015) Genome mining: prediction of lipopeptides and polyketides from bacillus and related firmicutes. Comput Struct Biotechnol J 13:192–203. doi:10.1016/j.csbj.2015.03.003
Angiuoli SV, Gussman A, Klimke W, Cochrane G, Field D, Garrity G, Kodira CD, Kyrpides N, Madupu R, Markowitz V, Tatusova T, Thomson N, White O (2008) Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. Omi A J Integr Biol 12:137–141. doi:10.1089/omi.2008.0017
Ansari MZ, Yadav G, Gokhale RS, Mohanty D (2004) NRPS-PKS: a knowledge-based resource for analysis of NRPS-PKS megasynthases. Nucleic Acids Res 32:405–413. doi:10.1093/nar/gkh359
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F et al (2008) The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi:10.1186/1471-2164-9-75
Ben Ayed H, Jridi M, Maalej H, Nasri M, Hmidet N (2014) Characterization and stability of biosurfactant produced by bacillus mojavensis A21 and its application in enhancing solubility of hydrocarbon. J Chem Technol Biotechnol 89:1007–1014. doi:10.1002/jctb.4192
Bergmann S, Schümann J, Scherlach K, Lange C, Brakhage AA, Hertweck C (2007) Genomics-driven discovery of PKS-NRPS hybrid metabolites from Aspergillus nidulans. Nat Chem Biol 3:213–217. doi:10.1038/nchembio869
Branquinho R, Sousa C, Lopes J, Pintado ME, Peixe LV, Osorio (2014) Differentiation of Bacillus pumilus and Bacillus safensis using MALDI-TOF-MS. PLoS One 9:e116426. doi:10.1371/journal.pone.0110127
Carrillo PG, Mardaraz C, Pitta-Alvarez SI, Giulietti AM (1996) Isolation and selection of biosurfactant-producing bacteria. World J Microbiol Biotechnol 12:82–4. doi:10.1007/BF00327807
Chevreux B, Wetter T, Suhai S (1999) Genome sequence assembly using trace signals and additional sequence information. Computer Science and Biology: Proceedings of the German Conference on Bioinformatics (GCB) 99:45–56
Chun J, Lee JH, Jung Y, Kim M, Kim S, Kim BK, Lim YW (2007) EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol 57:2259–2261. doi:10.1099/ijs.0.64915-0
Dhillon BK, Laird MR, Shay JA, Winsor GL, Lo R, Nizam F, Pereira SK, Waglechner N, McArthur AG, Langille MG, Brinkman FS (2015) IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res 43:W104–8. doi:10.1093/nar/gkv401
Domingos DF, de Faria AF, de Souza GR, Eberlin MN, Greenfield P, Zucchi TD, Melo IS, Tran-Dinh N, Midgley D, de Oliveira VM (2015) Genomic and chemical insights into biosurfactant production by the mangrove-derived strain Bacillus safensis CCMA-560. Appl Microbiol Biotechnol 99:3155–3167. doi:10.1007/s00253-015-6377-8
Donio MBS, Ronica FA, Viji VT, Velmurugan S, Jenifer JS, Michaelbabu M, Dhar P, Citarasu T (2013) Halomonas sp. BS4, a biosurfactant producing halophilic bacterium isolated from solar salt works in India and their biomedical importance. Springerplus 2:149. doi:10.1186/2193-1801-2-149
Doroghazi JR, Albright JC, Goering AW, Ju KS, Hainess RR, Tchalukov KA, Labeda DP, Kelleher NL, Metcalf WW (2014) A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol 10:963–8. doi:10.1038/nchembio.1659
Grant JR, Stothard P (2008) The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res 36:181–184. doi:10.1093/nar/gkn179
Harvey AL, Edrada-Ebel R, Quinn RJ (2015) The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov 14:111–129. doi:10.1038/nrd4510
Heerklotz H, Seelig J (2007) Leakage and lysis of lipid membranes induced by the lipopeptide surfactin. Eur Biophys J 36:305–314. doi:10.1007/s00249-006-0091-5
Kapley A, Tanksale H, Sagarkar S, Prasad AR, Kumar RA, Sharma N, Qureshi A, Purohit HJ (2016) Antimicrobial activity of Alcaligenes sp. HPC 1271 against multidrug resistant bacteria. Funct Integr Genomics 16:57–65. doi:10.1007/s10142-015-0466-8
Konz D, Doekel S, Marahiel M a (1999) Molecular and biochemical characterization of the protein template controlling biosynthesis of the lipopeptide lichenysin molecular and biochemical characterization of the protein template controlling biosynthesis of the lipopeptide lichenysin. J Bacteriol 181:133–140, doi: 0021-9193/99/$04.0010
Koumoutsi A, Chen X, Henne A, Lieseqang H, Hitzeroth G, Franke P, Vater J, Borriss R (2004) Structural and functional characterization of gene clusters directing nonribosomal synthesis of bioactive cyclic lipopeptides in Bacillus amyloliquefaciens strain FZB42. J Bacteriol 186:1084–1096. doi:10.1128/JB.186.4.1084
Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. doi:10.1093/nar/gkm160
Lam KS (2007) New aspects of natural products in drug discovery. Trends Microbiol 15:279–289. doi:10.1016/j.tim.2007.04.001
Land M, Hauser L, Jun SR, Nookaew I, Leuze MR, Ahn TH, Karpinets T, Lund O, Kora G, Wassenaar T, Poudel S, Ussery DW (2015) Insights from 20 years of bacterial genome sequencing. Funct Integr Genomics 15:141–61. doi:10.1007/s10142-015-0433-4
Lavermicocca P, Valerio F, Evidente A, Lazzaroni S, Corsetti A, Gobbetti M (2000) Purification and characterization of novel antifungal compounds from the sourdough lactobacillus plantarum strain 21B. Appl Environ Microbiol 66:4084–4090. doi:10.1128/AEM.66.9.4084-4090.2000
Liu Y, Lai Q, Dong C, Sun F, Wang L, Li G, Shao Z (2013) Phylogenetic diversity of the Bacillus pumilus group and the marine ecotype revealed by multilocus sequence analysis. PLoS One 8:1–11. doi:10.1371/journal.pone.0080097
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. doi:10.1093/nar/25.5.955
Lu L, Wang J, Xu Y, Wang K, Hu Y, Tian R, Yang B, Lai Q, Li Y, Zhang W, Shao Z, Lam H, Qian PY (2014) A high-resolution LC-MS-based secondary metabolite fingerprint database of marine bacteria. Sci Rep 4:6537. doi:10.1038/srep06537
Morikawa M, Hirata Y (2000) A study on the structure-function relationship of lipopeptide biosurfactants. 1488:211–218. doi:10.1016/S1388-1981(00)00124-4
Mukherjee S, Das P, Sivapathasekaran C, Sen R (2009) Antimicrobial biosurfactants from marine bacillus circulans: extracellular synthesis and purification. Lett Appl Microbiol 48:281–288. doi:10.1111/j.1472-765X.2008.02485.x
Naruse N, Tenmyo O, Kobaru S, Kamei H, Miyaki T, Konishi M, Oki T (1990) Pumilacidin, a complex of new antiviral antibiotics. Production, isolation, chemical properties, structure and biological activity. J Antibiot (Tokyo) 43:267–280. doi:10.7164/antibiotics.43.267
Nerurkar AS (2010) Structural and Molecular Characteristics of Lichenysin and Its Relationship with Surface Activity. In: Sen R (ed) Biosurfactants, vol 672. New York, Springer, pp 304–315. doi:10.1007/978-1-4419-5979-9_23
Ongena M, Jacques P (2008) Bacillus lipopeptides: versatile weapons for plant disease biocontrol. Trends Microbiol 16:115–125. doi:10.1016/j.tim.2007.12.009
Pal RR, Khardenavis AA, Purohit HJ (2015) Identification and monitoring of nitrification and denitrification genes in Klebsiella pneumoniae EGD-HP19-C for its ability to perform heterotrophic nitrification and aerobic denitrification. Funct Integr Genomics 15:63–76. doi:10.1007/s10142-014-0406-z
Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL (2007) Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6:29–40. doi:10.1038/nrd2201
Prakash O, Green SJ, Jasrotia P, Ovreholt WA, Canion A, Watson DB, Brooks SC, Kostka JE (2012) Rhodanobacter denitrificans sp. nov., isolated from nitrate-rich zones of a contaminated aquifer. Int J Syst Evol Microbiol 62:2457–2462. doi:10.1099/ijs.0.035840-0
Raaijmakers JM, de Bruijn I, Nybroe O, Ongena M (2010) Natural functions of lipopeptides from bacillus and pseudomonas: More than surfactants and antibiotics. FEMS Microbiol Rev 34:1037–1062. doi:10.1111/j.1574-6976.2010.00221.x
Rey MW, Ramaiya P, Nelson BA, Brody-Karpin SD, Zaretsky EJ, Tang M, Lopez de Leon A, Xiang H, Gusti V, Clausen IG et al (2004) Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillus species. Genome Biol 5:R77. doi:10.1186/gb-2004-5-10-r77
Richter M, Rosselló-Móra R, Glöckner FO, Peplies J (2015) JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32:929–31. doi:10.1093/bioinformatics/btv681
Rodrigues L, Banat IM, Teixeira J, Oliveira R (2006) Biosurfactants: potential applications in medicine. J Antimicrob Chemother 57:609–618. doi:10.1093/jac/dkl024
Satpute SK, Banpurkar AG, Dhakephalkar PK, Banat IM, Chopade BA (2010) Methods for investigating biosurfactants and bioemulsifiers: a review. Crit Rev Biotechnol 30:127–144. doi:10.3109/07388550903427280
Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M (2006) ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34:D32–6. doi:10.1093/nar/gkj014
Van Lanen SG, Shen B (2006) Microbial genomics for the improvement of natural product discovery. Curr Opin Microbiol 9:252–260. doi:10.1016/j.mib.2006.04.002
Villanueva J, Switala J, Ivancich A, Loewen PC (2014) Genome sequence of Bacillus pumilus MTCC B6033. Genome Announc 2:e00327–14. doi:10.1128/genomeA.00327-14
Wang H, Fewer DP, Holm L, Rouhiainen L, Sivonen K (2014) Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc Natl Acad Sci U S A 111:9259–9264. doi:10.1073/pnas.1401734111
Weber T, Blin K, Duddela S, Krug D, Hu K, Bruccoleri R, Lee SY, Fischback MA, Muller R, Wohlleben W, Breitling R, Takano E, Medema MH (2015) antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res 43:1–7. doi:10.1093/nar/gkv437
Yakimov MM, Timmis KN, Wray V, Fredrickson HL (1995) Characterization of a new lipopeptide surfactant produced by thermotolerant and halotolerant subsurface Bacillus licheniformis BAS50. Appl Environ Microbiol 61:1706–1713, doi: 0099-2240/95/$04.0010
Zerikly M, Challis GL (2009) Strategies for the discovery of new natural products by genome mining. ChemBioChem 10:625–633. doi:10.1002/cbic.200800389
Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS (2011) PHAST: a fast phage search tool. Nucleic Acids Res 39:1–6. doi:10.1093/nar/gkr485
Ziemert N, Podell S, Penn K, Badger JH, Allen E, Jensen PR (2012) The natural product domain seeker NaPDoS: a phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS One 7:1–9. doi:10.1371/journal.pone.0034064
Acknowledgments
Authors are thankful to Dr. Kiran Mahale (National Centre for Cell Science, Pune) and Dr. Sneha Sagarkar (Department of Biotechnology, Savitribai Phule University of Pune) for their valuable technical inputs. Authors are grateful to Mrs. Ashwini Rane (Department of Microbiology, Savitribai Phule University of Pune) for her assistance in conducting biosurfactant assay, and Datar Genetics Limited, Nashik, India for their support in Genome sequencing. We would also like to thank Dr. Ranadhir Chakraborty (North Bengal University) for providing the MDR isolates used in the study. This study was funded by Department of Biotechnology, Government of India, under the project of Microbial Culture Collection (Grant number: BT/PR10054/NDB/52/94/2007).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s10142-016-0528-6.
Rights and permissions
About this article

Cite this article
Shaligram, S., Kumbhare, S.V., Dhotre, D.P. et al. Genomic and functional features of the biosurfactant producing Bacillus sp. AM13. Funct Integr Genomics 16, 557–566 (2016). https://doi.org/10.1007/s10142-016-0506-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-016-0506-z