
Abstract
The selection of homozygous lines is a crucial step in the characterization of newly generated transgenic plants. This is particularly time- and labor-consuming when transgenic stacking is required. Here, we report a fast and accurate method based on quantitative real-time PCR with a rice gene RBE4 as a reference gene for selection of homozygous lines when using multiple transgenic stacking in rice. Use of this method allowed can be used to determine the stacking of up to three transgenes within four generations. Selection accuracy reached 100 % for a single locus and 92.3 % for two loci. This method confers distinct advantages over current transgenic research methodologies, as it is more accurate, rapid, and reliable. Therefore, this protocol could be used to efficiently select homozygous plants and to expedite time- and labor-consuming processes normally required for multiple transgene stacking. This protocol was standardized for determination of multiple gene stacking in molecular breeding via marker-assisted selection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genetic transformation is widely used in basic and applied research to generate genetically modified plants. It has led to the development of commercial crops with improved agronomic characteristics [1]. Rapidly obtaining homozygous lines shortens the breeding duration for generating transgenes, particularly when multiple genes or transgenes are stacked once the viability of primary transformants has been obtained. Screening of homozygotes and estimation of transgene copy number are vital for the selection and cultivation of transgenic plants, making them indispensable research techniques. Fast and accurate identification of transgenic homozygotes in subsequent generations is both highly desirable and beneficial, especially in crops requiring large amounts of planting space to produce their next generation. Therefore, a robust and reliable method of identifying homozygotes, especially for transgenic pyramiding or stacking, and for ascertaining transgene copy number in early generations is important for molecular marker-assisted selection (MAS) in transgenic breeding programs. Traditionally, target gene PCR and Southern blot are used to analyze transgenic homozygotes and copy number. Unfortunately, these methods are both laborious and time-consuming, requiring considerable amounts of DNA from fresh or frozen samples, and may involve hazardous radioisotopes [2].
To overcome these limitations, quantitative real-time PCR (qPCR) is often applied to analyze transgene integration. qPCR collects data throughout the PCR process, thus combining amplification and detection in a single step. Detection has been achieved using a variety of fluorescent molecules, in which PCR product concentration is correlated with fluorescence intensity [3]. The quantitative endpoint for qPCR is the threshold cycle (Ct) [4], and this is defined as the PCR cycle during which the fluorescent signal of the reporter dye crosses an arbitrarily determined threshold. Presenting data as a Ct ensures that quantification occurs during the exponential phase of amplification. The value of the Ct is inversely related to the amount of amplicons in the reaction [5]. The greater the quantity of target DNA in the starting material, the faster a significant increase in fluorescent signal will appear, thus yielding a lower Ct [6].
Although qPCR has been used to screen homozygous transgenic plants [7–9], accuracy and reliability for transgenic pyramiding or stacking have not been well resolved and practiced in breeding programs. In this study, we optimized the rice endogenous gene RBE4 as a reference gene for determination of transgenically stacked homozygous lines. Accuracy reached 100 % for a single locus and 92.3 % for two loci. The accuracy of insertion locus/loci and copy number determination reached 100 %. The reliability of the homozygous lines was verified by PCR, and the accuracy of the insertion locus/loci was confirmed by Southern blotting and genetic analysis in the next generation. This methodology was successfully applied to three transgenic stackings and had the distinct advantages of higher accuracy, speed, and reliability and less time- and labor-consuming over Southern blotting process and other current transgenic research methodologies. This protocol was standardized for multiple gene stacking in molecular breeding programs via MAS.
Materials and Methods
Plasmid Construction and Generation of Transgenic Rice Plants
α-1,6-Fucosyltransferase gene (FUT8; GenBank accession No. D89289.1) and β-1,4-galactosyltransferase gene (GalT; GenBank accession No. M22921.1) were synthesized by GenScript Corporation (NJ, USA) using rice-preferred genetic codons. A mediated construct with endosperm-specific expression cassette with a globulin (Glb) promoter designated as pOsPMP512 was used in this study. The synthesized genes were obtained by sequential digestion with SchI and XhoI and then cloned into pOsPMP512 that was digested by NaeI and XhoI, resulting in pOsPMP513 (FUT8) and pOsPMP514 (GalT). The plasmids of pOsPMP513 and pOsPMP514 were digested with HindIII and EcoRI and cloned into a binary vector JH2600 that was digested by the same restriction enzymes. The resulting binary vectors were designated as pOsPMP515 and pOsPMP516 (Supplementary Fig. S1). The plasmids were transformed into Agrobacterium tumefaciens strain EHA 105. A pOsPMP122 containing a hygromycin phosphotransferase (HPT) gene under control of a CP promoter was used as selective marker (Supplementary Fig. S1). The target gene plasmid and the selective marker pOsPMP122 were co-transformed into calli derived from a rice variety TP 309 through Agrobacterium-mediated transformation [10].
The hygromycin resistance-positive transformants were identified by PCR using the gene-specific primer pair HPT-F/HPT-R (Supplementary Table 1). A transgenic line (132-17) expressing a human α1-antitrypsin (AAT) was used in this study [11]. Co-transformants containing HPT gene and either FUT8 or GalT gene were identified by PCR using target gene-specific primers FUT8-F2/FUT8-R2 or GalT-F2/GalT-R2 (Supplementary Table 1). A homozygous transgenic line 515 crossed with a homozygous line 516 first, and then, the F1 of 515 × 516 crossed with line 132-17. An F2 population derived from the cross (515 × 516) × 132-17 was used for identifying and confirming homozygous and heterozygous plants through application of the method developed.
DNA Extraction
Rice genomic DNA used for qPCR analysis was extracted from fresh leaves using the cetyltriemethylammonium bromide (CTAB) method [12]. The concentration of genomic DNA was measured with UV absorption at 260 nm, and DNA quality was evaluated using UV absorption ratio at 260/280 nm. DNA samples were analyzed using 1 % agarose gel electrophoresis.
PCR Amplification and Primers
Oligonucleotide primers were designed using Primer 5.0 software (Supplementary Table 1). For enhanced amplification efficiency, amplicons for the SYBR Green qPCR primers were designed to be smaller than 200 bp. An RBE4-F/RBE4-R primer pair for the endogenous reference gene RBE4, a FUT8-F1/FUT8-R1 primer pair for the target gene FUT8, a GalT-F1/GalT-R1 primer pair for the target gene GalT, and an AAT-F1/AAT-R1 primer pair for the target gene AAT were used for the SYBR Green qPCR assay, yielding amplified fragments of 106, 118, 96, and 91 bp, respectively. A FUT8-F2/FUT8-R2 primer pair for the target gene FUT8, a GalT-F2/GalT-R2 primer pair for the target gene GalT, and an AAT-F2/AAT-R2 primer pair for the target gene AAT were used for PCR, yielding amplified fragments of 965, 645, and 644 bp, respectively. The qPCR efficiency for each gene was determined using serial dilutions to obtain appropriate standard curves.
SYBR Green qPCR
Amplification reactions were performed on a qPCR platform (Applied Biosystems, Foster City, CA, USA). qPCR was carried out in 10-μl reaction mixtures containing 5-μl PCR buffer mix (Invitrogen, Grand Island, NY, USA), 1-μl DNA template (30 ng), and 500-nM gene-specific primers. The PCR program comprised 1 cycle of 10 min at 95 °C, followed by 40 cycles of 10 s at 95 °C and 20 s at 60 °C. The Ct value of the gene was determined systematically. Amplification data were analyzed using StepOne software (Applied Biosystems, Foster City, CA, USA). Each sample was quantified using three replicates and in triplicate for each replicate.
Determination of Transgene Zygosity
Relative quantification by the comparative Ct (2−ΔΔCt) method was used for identification of the homozygous plants. Because a homozygous plant contains twice as many transgenes as a heterozygous one, the comparison of relative copy ratios of FUT8 and RBE4 between the transgenic plants is sufficient to quantitatively calculate the PCR products; the 2−ΔΔCt value of a homozygote should be twice that of a heterozygote. The comparative Ct of FUT8 and RBE4 could be calculated using the following formula:
To calibrate qPCR efficiencies of the target and internal reference genes, the amplification efficiency of the internal reference gene was used for normalization of the amplification efficiency of the target gene. Each reaction had three technological replicates, repeated in triplicate for each replicate.
PCR for Homozygote Identification
Reactions were performed in 20-μl reaction mixtures (Fisher Scientific, Pittsburgh PA, USA) containing 1-μl DNA template (30 ng), 2 μl 10× PCR buffer, 0.8 μl 1 mmol l−1 dNTP, 1.2 μl 25 mmol l−1 Mg2+, 1 unit Taq DNA polymerase, and 500-nM primers. The PCR program comprised 1 cycle of 10 min at 95 °C, followed by 32 cycles of 30 s at 95 °C, 30 s at 56 °C, and 30 s at 72 °C, and a final extension of 10 min at 72 °C. The sample was stored at 25 °C. Each reaction was repeated in triplicate.
For verification of homozygous plants identified in the previous generation, zygosity of the lines was monitored in the T2 generation. T1 plants were considered homozygous if no segregation was observed in the T2 generation and were considered heterozygous if segregation was observed in the T2 generation.
Determination of Transgene Copy Number
The absolute quantification calculated with the standard curves was used for determination of transgene copy number [4, 13]. UV absorption at 260 nm was used to calculate the copy number of the genes using the formula:
Two standard curves for the two genes were obtained by plotting Ct values against log-transformed concentrations of serial tenfold dilutions (105, 104, 103, 102, and 10 copies μl−1) from the genomic and plasmid DNA solutions. The absolute copy number of the target gene in each transgenic plant was calculated using Ct values based on their standard curves. RBE4 is a single-copy gene, and the relative copy number of the target gene was calculated using the following formula:
where A is the initial absolute copy number of the target gene and B is the initial copy number of the internal reference gene, RBE4.
RBE4 is unique in the rice genome. In the formulas, when a target gene is heterozygous, the copy number was multiplied by two. Each reaction had three replicates and was repeated three times.
Southern Blot Analysis
Genomic DNA (10 μg) was obtained from fresh young leaves using the CTAB method [12]. Genomic DNA was digested with HindIII, EcoRI, or a HindIII/EcoRI and then separated using 0.8 % agarose gel electrophoresis alongside relevant DNA size markers (λ DNA digested with HindIII). Separated DNA was transferred to a nitrocellulose membrane according to the manufacturer’s instructions (Gene Company, Hong Kong, China). A 965 bp fragment derived from FUT8 coding region was used as a probe, which was prepared by PCR using primers FUT8-F2/FUT8-R2. The DNA probe was labeled using a random primer labeling kit according to the manufacturer’s instructions (Roche, Basel, Switzerland). Membrane baking, pre-hybridization, and hybridization were performed according to the manufacturer’s instructions (DIG High Prime DNA Labeling and Detection Starter Kit I, Roche, Basel, Switzerland).
Results
Optimization of the Internal Reference for the Identification of Homozygotes
To reliably identify homozygous plants using qPCR, it is critical that an appropriate endogenous reference gene is used to normalize data. This eliminates sample-to-sample variations and allows calibration of the Ct value. It is important that the amplification of the internal reference does not change, even under conditions when target gene amplification may alter dramatically [14]. Ideally, an internal reference gene should be species-specific, having single or low copy number per haploid genome, and exhibiting low heterogeneity across genotypes within a species [1, 15]. To screen genes appropriate for use as an internal reference, the following eight genes, present as one or two copies in the rice genome, were chosen: starch branching enzyme (RBE4) [16], sucrose phosphate synthase (SPS), rice root-specific gene (gos9), eukaryotic elongation factor 1-alpha (eEF1α), rice actin gene (RAc1), 1-deoxy-d-xylulose-5-phosphate reductoisomerase (dxr), and trs-like genes (Os3bet3 and Os4trs20). To achieve optimal amplification efficiency, 21 primer pairs were tested. Serial dilutions of the DNA template were used in the qPCR assay. The amplification plot, standard curve (including the slope of the line, average correlation coefficients (R 2), and PCR amplification efficiency), and melting curve of the eight genes were compared. The standard amplicons curve of RBE4 had a high correlation coefficient (R 2 = 0.994; Fig. 1b). The PCR amplification efficiency was nearly 100 %. The other genes tested did not completely satisfy all criteria as an internal reference gene or were not as efficient as RBE4 (Table 1). Therefore, the single-copy gene, RBE4, was considered the best for use as an internal reference gene and chosen for further analysis (Fig. 1).

Amplification plot, standard curve, and melting curve of RBE4. a Amplification plot. b Standard curve. c Melting curve. Ct represents threshold cycle, and ΔRn represents normalized reporter − baseline
Establishment of Ct (2−ΔΔCt) for Identification of Homozygous and Heterozygous Plants
To test whether the internal reference gene RBE4 could be applied in the identification of homozygous and heterozygous plants, we examined its accuracy in T1 transgenic lines. Relative quantification was performed using the comparative Ct (2−ΔΔCt) method. Firstly, standard curves were validated and qPCR amplification efficiencies confirmed for both internal reference and target genes. The R 2 of the standard curves for RBE4 and for the FUT8 target gene was 0.998 and 0.994, respectively. PCR amplification efficiencies for both genes reached 99 and 101 %, respectively, indicating that both the internal reference gene and the target gene were effective for homozygous plant identification. To determine the feasibility of the identification of homozygous plants, 100 individual plants each from five transgenic lines derived from the T1 generation were used. It was assumed that the FUT8/RBE4 gene ratio could be quantitatively determined by the ΔCt value [17]. Thus, the ratio of the absolute copy numbers of FUT8/RBE4 between homozygotes and heterozygotes was reflected in their ΔCt. It was assumed that the value of 2−ΔΔCt for a homozygote was twice that of a heterozygote. The 2−ΔΔCt value for each sample was approximately 1 or 2 (Table 2), exactly reflecting the differences between the homozygotes and the heterozygotes.
Accuracy of Homozygous Plant Identification Validated in the T2 Generation
To verify the reliability of the homozygous or the heterozygous plants identified by qPCR, we performed accuracy verification by monitoring genetic segregation in the T2 generation that derived from the T1 plants. Offspring from self-pollinated homozygous, heterozygous, or negative plant lines were used for verification by PCR using gene-specific primers (Table 3). The results indicated that 100 % accuracy for homozygous and negative line determination and 93.33 % for heterozygous line determination were reached when the transgenic lines carrying a single locus. The 92.31 % accuracy for homozygous lines and 86.67 % for heterozygous lines determination were confirmed for the transgenic line carrying two loci. These results demonstrated that this protocol was accurate for identifying homozygous lines in an early generation.
Determination of Homozygous Lines for Transgene Stacking
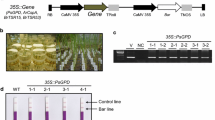
To determine the feasibility of this protocol for gene stacking, we tested its reliability for homozygous plant identification when stacking three genes of FUT8, GalT, and AAT (Fig. 2a). As shown in Table 4, a step-down approach was used for screening homozygous plants. Five hundred individual plants from an F2 population derived from (515 × 516) × 132-17 were tested. Firstly, the plants containing FUT8 from the F2 population of the cross (515 × 516) × 132-17 were screened. A total of 133 FUT8 homozygous plants were identified. These plants were used for a second round of screening to identify GalT homozygous plants. Thirty-six homozygous plants containing both FUT8 and GalT were obtained. Finally, these plants were screened using AAT gene-specific primers, and eight homozygous plants stacked with FUT8, GalT, and AAT were obtained (Table 4).

Transgene stacking and PCR amplification scheme. a Scheme of determining transgene stacking. b Diagrams of four plasmids and the examples of PCR using gene-specific primers for verification of the homozygous individuals that were determined by this protocol in F2 generation. The size of PCR products and entire expression cassette between HindIII and EcoRI were shown. A represents FUT8 gene, B represents GalT gene, and C represents AAT gene. Upper and lower letters represent dominant locus and recessive locus, respectively
To verify the reliability of the homozygous plants identified by this protocol, a homozygous line stacked with the FUT8, GalT, and AAT genes was randomly chosen and used to produce a F2/F3 population. We checked genetic segregation by PCR using the target gene-specific primers. No segregation was found in the 100 plants tested in the F2/F3 population (Fig. 2b). The results demonstrated that the protocol was effective and reliable in the identification of homozygous lines for multiple gene stacking.
Estimation and Validation of Reliability for Insertion Locus/Loci Determination
Because the internal reference gene RBE4 is single-copied in the rice genome, the copy number of a target gene can be determined by comparison with RBE4. To verify the feasibility of this method to determine copy number, we chose five independent FUT8 transgenic lines with different loci, which were previously determined to be heterozygous by this protocol and late confirmed by genetic analysis. Thirty T2 individual plants each line derived from the heterozygous line out of the T1 generation were analyzed. We detected the genetic segregation ratio of the positive and the negative plants for locus/loci determination of the target genes. The copy number of FUT8 was determined by calculating the absolute initial quantity of each gene using its average Ct value from the standard curve with the internal reference gene RBE4. The results indicated that three lines possessed a single locus, and two lines presented two loci (Table 5). These results were consistent with the results from the genetic analysis.
To further validate the reliability of the method, Southern blotting was performed for a single-locus line (515-1) and a two-locus line (515-2). As shown in Fig. 3, there are two different size hybridization bands in line 515-2 when the genomic DNA was digested by HindIII and EcoRI, respectively. There is one hybridization bands with similar size in line 515-1 when the genomic DNA was digested by HindIII and EcoRI, respectively. In addition, both lines showed the single band when digested by HindIII/EcoRI, indicating that these loci have an entire expression cassette. The Southern analysis confirmed that 515-2 carried two loci and 515-1 carried one locus, which was consistent with the qPCR and genetic analysis (Table 5). These results again demonstrated that this qPCR protocol was an effective and accurate way to determine integration locus/loci of the transgene.

Southern blot analyses of the transgenic lines 515-2 and 515-1. Genomic DNA was digested with HindIII, EcoRI, or HindIII/EcoRI and probed with the FUT8 gene. A binary plasmid pOsPMP515 digested by HindIII, EcoRI, and HindIII/EcoRI, respectively, was used as positive control. M represents λ DNA digested with HindIII, lanes 2–4 and 8–10 are the plasmid DNAs digested by HindIII (H3), EcoRI (R1), and HindIII/EcoRI (H3/R1), respectively, lanes 5–7 are the genomic DNAs of the transgenic line 515-2 digested by HindIII, EcoRI, and HindIII/EcoRI, respectively, and lanes 11–13 are the genomic DNAs of the transgenic line 515-1 digested by HindIII, EcoRI, and HindIII/EcoRI, respectively
Discussion
A transgenic homozygote can be obtained from a T1 generation; however, zygosity needs to be confirmed in the T2 generation. Owing to difficulties in distinguishing plants with two identical copies from those with only one copy, neither PCR nor genomic Southern blot hybridization is typically used to identify transgenic homozygotes in a T1 population [9]. Since there are no obvious visual differences between a homozygote and a heterozygote, there is a practical need to identify zygosity as early as possible. It usually takes an additional generation to confirm homozygosity through genetic analysis of transgene segregation in the T2 population. In this study, we optimized a method that could effectively identify homozygous, heterozygous, and negative lines in the early segregation generation; this method could also determine transgene insertion locus/loci. Our results indicated that this protocol was accurate, effective, and reliable.
Although qPCR has previously been used to identify homozygous plants in early generations [7–9, 18], none of the previously reported qPCR methods can simultaneously achieve high accuracy, reliability, and simplicity for determining transgene homozygosis and transgene stacking (Supplementary Table 2). For example, only 15–46 % of qPCR for copy number determination has been confirmed by Southern blotting analysis in a study involving qPCR screening [18]. In another qPCR-based method, the accuracy only reached 83 % [8]. Although different internal reference genes, qPCR parameters, and the formula of analysis were used in those methods, the accuracy has not reached a satisfactory level. In the present study, the accuracy of homozygote and negative plant identification reached 100 % when the line had a single insertion locus, and this was confirmed by PCR. Accuracy of homozygous line identification reached 92.3 % when the line had two insertion loci. qPCR for copy number determination reached 100 %, consistent with the genetic segregation ratio and Southern blotting analysis. We successfully demonstrated that this protocol is a practical and reliable protocol for three-gene stacking.
In addition, none of the qPCR-based method has been utilized for gene stacking to show the accuracy highly consistent among qPCR, genetic segregation, and Southern Blotting analysis [7–9, 19]. Our results indicated that our protocol not only was more effective and accurate than previously published qPCR methods, but also offered the distinct advantages of relative simplicity, rapid screening, and comparable accuracy over traditional methods such as Southern Blotting and previously published qPCR methods. The higher accuracy obtained in the present method is largely attributed to the selection of the appropriate internal reference gene, optimizing the qPCR parameters and primer design. This protocol could be also used in other crops with diploid genomes via minor modifications. For crops with polyploid genomes, this method would be expected to work well if an appropriate internal reference gene is selected and qPCR parameters and primer design are optimized. Furthermore, this method could be used to determine the zygosity of the endogenous gene of interest, induced mutation by TILLING, insertional mutation, or site-directed mutation introduced through a genome editing.
Furthermore, cost is an important issue when using qPCR methods at a large-scale screening homozygous plants in breeding programs. Most current qPCR methods have used TaqMan probes [8, 9]. We choose SYBR Green rather than TaqMan probe in this protocol because it is much cheaper, easier to use than TaqMan probe. In addition, TaqMan probe is used only once, requiring new synthesis and luminophores each time, while SYBR Green is nonspecific as it binds to any double-stranded DNA.
It is essential to develop a fast, accurate, and precise method for identifying homozygous lines for transgenic and molecular breeding. This would accelerate breeding programs and shorten breeding time. Furthermore, these requirements are especially important for breeding programs that use gene stacking or pyramiding. The proposed protocol, using SYBR Green qPCR, provides a simple and feasible approach applicable to molecular breeding and transgenic research. When three-gene stacking is used, breeding programs could be possibly shortened by between 6 months and 2 years, along with savings in labor and costs associated with field trials. For example, in a conventional three-gene stacking program, the selection of a homozygous line requires an F2 generation for the first crossing. And then, a homozygous line from the F3 generation is crossed with a line containing the third gene. This follows the same time course as the first round of crossing and selection. Therefore, at least 7 years/generations are required. However, three-gene stacking or pyramiding, selection of homozygous plants, and crossing of the line with the third gene can simultaneously be accomplished in the F2 generation using our procedure (Fig. 2a). Therefore, only 4 years/generations are required when using this protocol. Additionally, fewer F2 and F3 populations and field trials are required in comparison to conventional breeding programs. Conventionally, to obtain homozygous lines from a segregation population containing three genes, a minimum of ten lines is required from the F2 generation and at least 500 individual plants from each line are required in the F3 population for each cross. This requires 5,000 PCR reactions for a single gene, and the workload is also similarly tremendous for second- and third-round identifications of homozygous plants. Therefore, this protocol has numerous advantages over conventional breeding programs and traditional molecular biology approaches.
References
Baeumler, S., Wulff, D., Tagliani, L., & Song, P. (2006). Journal of Agricultural and Food Chemistry, 54, 6527–6534.
De Preter, K., Speleman, F., Combaret, V., Lunec, J., Laureys, G., Eussen, B. H., Francotte, N., Board, J., Pearson, A. D., De Paepe, A., Van Roy, N., & Vandesompele, J. (2002). Modern Pathology, 15, 159–166.
Higuchi, R., Fockler, C., Dollinger, G., & Watson, R. (1993). Biotechnology (NY), 11, 1026–1030.
Ahmed, F. E. (2002). Trends in Biotechnology, 20, 215–223.
Schmittgen, T. D., & Livak, K. J. (2008). Nature Protocols, 3, 1101–1108.
Heid, C. A., Stevens, J., Livak, K. J., & Williams, P. M. (1996). Genome Research, 6, 986–994.
Bubner, B., & Baldwin, I. T. (2004). Plant Cell Reports, 23, 263–271.
Yang, L., Ding, J., Zhang, C., Jia, J., Weng, H., Liu, W., & Zhang, D. (2005). Plant Cell Reports, 23, 759–763.
Yi, C. X., Zhang, J., Chan, K. M., Liu, X. K., & Hong, Y. (2008). Analytical Biochemistry, 375, 150–152.
Chen, S., Li, X., Liu, X., Xu, H., Meng, K., Xiao, G., Wei, X., Wang, F., & Zhu, Z. (2005). Plant Cell Reports, 23, 625–631.
Zhang, L., Shi, J., Jiang, D., Stupak, J., Ou, J., Qiu, Q., An, N., Li, J., & Yang, D. (2013). Journal of Biotechnology, 164, 300–308.
Sambrook, J., Russell, D.W. (2001). CSHL press.
Livak, K. J., & Schmittgen, T. D. (2001). Methods, 25, 402–408.
Hein, J., Schellenberg, U., Bein, G., & Hackstein, H. (2001). Scandinavian Journal of Immunology, 54, 285–291.
Zhang, Z., & Hu, J. (2007). Toxicological Sciences, 95, 356–368.
Mizuno, K., Kobayashi, E., Tachibana, M., Kawasaki, T., Fujimura, T., Funane, K., Kobayashi, M., & Baba, T. (2001). Plant and Cell Physiology, 42, 349–357.
Ji, W., Cai, L., Wright, M. B., Walker, G., Salgam, P., Vater, A., & Lindpaintner, K. (2000). Journal of Structural and Functional Genomics, 1, 1–7.
Bubner, B., Gase, K., & Baldwin, I. T. (2004). BMC Biotechnology, 4, 14.
Mieog, J. C., Howitt, C. A., & Ral, J. P. (2013). BMC Plant Biology, 13, 71.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
(DOC 48 kb)
Supplementary Table 2
(DOCX 55 kb)
Supplementary Fig. S1
(GIF 112 kb)
Rights and permissions
About this article

Cite this article
Wang, X., Jiang, D. & Yang, D. Fast-Tracking Determination of Homozygous Transgenic Lines and Transgene Stacking Using a Reliable Quantitative Real-Time PCR Assay. Appl Biochem Biotechnol 175, 996–1006 (2015). https://doi.org/10.1007/s12010-014-1322-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-014-1322-3