
Abstract
We generated 383 independent transgenic lines that contained the PsGPD (Glyceraldehyde-3-Phosphate Dehydrogenase), ArCspA (Cold Shock Protein), BrTSR15 (Triple Stress Resistance 15) and BrTSR53 (Triple Stress Resistance 53) genes under the control of a constitutive (CaMV 35S) promoter to generate genetically modified (GM) rice. TaqMan copy number assay was performed to determine the copy numbers of inserted T-DNA. Flanking sequence tags (FSTs) were isolated from 203 single copy T-DNA lines of transgenic plants, and their sequences were mapped to the rice chromosomes. Of the 157 flanking sequence tags that were isolated from single copy lines, transgenes were found to be integrated into genic regions in 58 lines (36 %), whereas 97 lines (62 %) contained transgene insertions in intergenic regions. Approximately 27 putative homozygous lines were obtained through multi-generations of planting, resistance screening and TaqMan copy number assays. To investigate the transgene expression patterns, quantitative real-time PCR analysis was performed using total RNA from leaf tissue of homozygous T1 plants with a single copy and an intergenic insertion of T-DNA. The mRNA expression levels of the examined transgenic rice were significantly increased in all transgenic plants. In addition, myc-tagged 35S:BrTSR15 and 35S:BrTSR53 transgenic plants displayed higher levels of transgene protein. Using numerical data for the mass production of transgenic plants can reduce the time required to obtain a genetically modified plant. Moreover, the duration, cost, and efforts required for transformation can be deliberately predicted. These results may be useful for the large-scale production of transgenic plants or T-DNA inserted rice mutants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genetically modified (GM) research and development of GM crops could contribute to global food production systems and sustainable development. The global area of genetically modified (GM) crop cultivation has increased considerably from 1.7 million hectares in 1996 to 170.3 million hectares in 2012 (James 2012). Because the utilization of GM crops provides substantial economic benefits, the production of genetically modified (GM) plants is being improved, and transgenic crops are increasingly cultivated (Halpin 2005; Rommens 2007; Travella et al. 2005). However, little is known about effective GM plant generation strategies.
Rice (Oryza sativa L.) is one of the major staple food crops and is a wholesome and nutritious cereal grain that is grown worldwide. Moreover, the rice genome has been sequenced (International Rice Genome Sequencing Project, 2005), and rice can be easily transformed (Toki et al. 2006; Hiei and Komari 2008). With the advancements in plant biotechnology over the past decade, it is possible to make step-wise increases in crop yield to obtain high-yield and high-quality rice. GM rice strains KMD1, TT51-1, and KF6 are currently three of the most famous examples (Wu et al. 2002; Tu et al. 1998, 2000; Li et al. 2004). For the efficient production of GM rice, transformation technologies with rapid and reliable assays for putative transformants are needed. The enforcement of threshold values has created a demand for the development of reliable GMO analysis methods that are both rapid and inexpensive. In transgenic plants, one or two copy insertion events generally yield a high level of expression of the exogenous gene (Flavell 1994; Vaucheret et al. 1998). Thus, determining the transgene copy number is vital to the selection and cultivation of GM plants. Although Southern blot analysis has been traditionally used to determine the copy numbers of transgenes, this method is laborious and time-consuming, requires a considerable amount of DNA and often involves the use of hazardous radioisotopes (De Preter et al. 2002). To overcome this obstacle, TaqMan quantitative real-time PCR has been developed as a fast, high-throughput, and quantitatively accurate method for the characterization of transgene insertions (Ingham et al. 2001; Mason et al. 2002; Song et al. 2002). To determine the copy number and insertion site of the transgenes, T-DNA insertion patterns in the rice genome are examined by flanking sequence tag (FST) analysis. The insertion sites and flanking sequence tag (FST) information have been deposited into public domains such as the Rice Genomic Annotation Project (RiceGE, http://rice.plantbiology.msu.edu/) and the Rice Annotation Project Database (RAP-DB, http://rapdb.dna.affrc.go.jp/).
In the present study, we generated 383 independent transgenic rice plants that over-express PsGPD, ArCspA, BrTSR15 and BrTSR53 by using Agrobacterium-mediated transformation and examined their molecular characteristics. Our results suggest that the T-DNA tagged rice population and the database of rice genome-sequences flanking T-DNA inserts will be useful for GM event research and development.
Materials and methods
Plant materials and growth conditions
Transgenic and wild-type japonica rice (O. sativa L. cv Dongjin) were used in these experiments. Sterilized seeds of wild-type and transgenic rice plants were sown on soil and grown in a greenhouse at 30 °C during the day and 20 °C at night in a light/dark cycle of 16 h/8 h or in an experimental field of the National Academy of Agricultural Science (NAAS) under natural environmental conditions.
Construction of plasmid vectors
Full-length cDNAs for abiotic stress resistance genes, glyceraldehyde-3-phosphate dehydrogenase (GPD, AF087676) from Pleurotus sajor-caju and cold shock protein (Csp, WP_056346630) from Arthrobacter sp. A2-5, were isolated by PCR using gene-specific primers and cloned into the pGEM-T easy vector (Cho et al. 2014). To generate transformants over-expressing the abiotic stresses related genes from Brassica rapa, full-length cDNAs of BrTSR15 (BRAS0001S030022015) and BrTSR53 (BRAS0001S00024153) genes were amplified by RT-PCR from the total RNA isolated from leaves of 3-week-old B. rapa plants and subcloned into the pGEM-T Easy vector (Lee et al. 2008). To construct over-expression vectors of PsGPD, ArCspA, BrTSR15 and BrTSR53, each cDNA fragment containing the entire open reading frame was cloned between the CaMV35S promoter and PinII terminator of the pPZP vector that carries Bar as a plant selectable marker (Fuse et al. 2001). The primers used to construct the over-expression vectors for 35S::PsGPD, 35S:: ArCspA, 35S::BrTSR15 and 35S::BrTSR53 were as follows: PsGPD, GPD-F 5′-TCTAGAATGGTCAACGTCGGCATCAACGGG-3′ containing XbaI and GPD-R 5′-CCCGGG CTACTGCGCACCGTCCTTCTCCG-3′ containing SmaI; ArCspA, CSP-F 5′-TCTAGAATGGCATTGGGAACCGTCAAG-3′ containing XbaI and CSP-R 5′-CCCGGGCTA CTCTGCAGGCCGGACGTTTTC-3′ containing SmaI; BrTSR15, BrTSR15-F 5′-TCTAGAATGCAGATCAAACTCCATGAG-3′ containing XbaI and BrTSR15-R 5′-GGATCCCTAAATATATGAATATTCCAC-3′ containing BamHI; BrTSR53, BrTSR53-F 5′-TCTAGAATGACTTCATGGAAGAAAAC-3′ containing XbaI and BrTSR53-R 5′-GGATCCTTAAGAAGACAGATTGTTTG-3′ containing BamHI (restriction sites are underlined). The resulting constructs were used for the rice transformation.
Agrobacterium-mediated rice transformation
The over-expression constructs 35S::PsGPD, 35S::ArCspA, 35S::BrTSR15 and 35S::BrTSR53 were introduced into Agrobacterium tumefaciens strains LB4404 to generate transgenic rice. Rice plants were transformed by Agrobacterium infection as described by Toki et al. (2006). Transgenic rice plants were regenerated from transformed calli on selection medium containing 4 mg/L phosphinothricin and 500 mg/L cefotaxime and subsequently grown in a greenhouse.
Extraction of genomic DNA and PCR analysis
Plant genomic DNA was extracted and purified from leaves using Inclone™ Genomic DNA Prep Kit (Inclone) according to the manufacturer’s protocol. PCR analysis to detect the Bar gene was performed using Ex Taq DNA polymerase (TaKaRa) with the primer Bar-F 5′-ATGAGCCCAGAACGACGCCCG-3′ and Bar-R 5′-GATCTCGGTGACGGGCAGGAC-3′.
Determination of relative copy numbers by quantitative real-time PCR
To determine copy numbers using quantitative real-time PCR, leaf samples from transgenic and wild-type plants were used. PCR reactions were multiplexed to simultaneously amplify the tub and nos genes. Quantitative multiplex real-time PCR assays were carried out Applied Biosystems StepOnePlus™ Real-Time PCR System (StepOnePlus™ system) in 96-well plates using a TaqMan system with a final volume of 20 µl. The reaction mixture contained 2 × TaqMan Master Mix (Applied Biosystems), 30 ng genomic DNA sample, and an optimal concentration of each gene-specific primer and probe (900 nM each primer and 200 nM each probe). PCR primers and probes were designed using the Primer Express software (Applied Biosystems). As the endogenous control, the primers and probes were obtained from a pre-designed TaqMan® copy number assay of the rice tubulin alpha-1 chain gene (AK102560). For the transgene, the primers and probe were designed specific to the terminator of nos gene. The primers used for the terminator of nos were as follows: NOS, NOS-F 5′-GCATGACGTTATTTATGAGATGGGTTT-3′, NOS-R 5′-TGCGCGCTATATTTTGTTTTCTATCG-3′ and NOS-probe 5′-TAGAGTCCCGCAATTAT-3′. The amplification conditions consisted of one cycle of 2 min at 50 °C and 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. In the reaction plate, each sample was measured in triplicate.
To calculate gene copy number, the relative quantitation analysis of genomic DNA targets using the real-time PCR data was analyzed using the Applied Biosystems CopyCaller® Software v2.0 (Applied Biosystems) according to the manufacturer’s protocol. The Applied Biosystems CopyCaller™ Software v1.0 was used to determine the copy number status of each target region, and calculation was performed according to a maximum-likelihood algorithm of the software. The raw copy value (RCV) represents a non-integer number of the calculated copy, whereas the predicted copy number (PCN) is defined as an integer for the copy that is determined by the algorithm (0, 1, 2, or 3+).
Analysis of flanking T-DNA sequences by adapter ligation PCR
To isolate flanking sequences of T-DNA, we used the adapter ligation PCR method. First, 0.5 µg of genomic DNA was digested with 2 U restriction enzyme and ligated with 5 U of T4 DNA ligase (Takara) in a 20 µl reaction volume. The reaction mixture contained T4 DNA ligase buffer and 50 pmol of the adapters and was incubated at 37 °C for 1 h. The first PCR was conducted in a 20 µl reaction containing PCR pre-mix, 0.5 pmol each primer (Ada1 and LB1 or RB), and 1 µl digestion/ligation product. PCR was performed in a PTC-200 thermal cycler (MJ Research) and conducted as follows (Ada2 and LB2 or RB2): first denaturation step at 95 °C for 5 min, 20 cycles of 30 s at 94 °C and 1 min at 72 °C, followed by a final elongation step at 72 °C for 10 min. The second PCR was conducted with 5 µl of the first PCR product under the conditions of first denaturation step at 94 °C for 5 min, 40 cycles of 30 s at 94 °C, 30 s at 60 °C, and 1 min at 72 °C, followed by a final elongation step at 72 °C for 10 min. Amplified products were loaded on a 1 % agarose gel, and the PCR products were purified using the HiYield™ Gel/PCR DNA Extraction Kit (RBC) and sequenced by ABI3730XL (Applied Biosystems) using LB2 or RB2 primer.
In silico mapping of T-DNA insertion sites
To determine the positions of the T-DNA insertion sites in the single copy transgenic rice plants, FST sequences were used to search the FSTVAL (Flanking Sequence Tags Validator) software (http://bioinfo.mju.ac.kr/fstval/), an open access tool, and to manage the bulk Flanking Sequence Tag (FST) mapping via the Internet. Significance was indicated when matched sequences had E values <10−5 for the FSTs in the BLASTN searches (Kim et al. 2012).
Extraction of total RNA and quantitative real-time PCR analysis
Total RNA was isolated from the leaves of wild-type and transgenic rice plants using an RNeasy plant mini kit (Qiagen). For real-time quantitative RT-PCR analysis, first-strand cDNA was synthesized from 1 µg of DNase-treated total RNA using AmfiRivert II cDNA Synthesis Master Mix (GenDEPOT). All reactions were performed using the SYBR Premix Ex Taq (TaKaRa) and carried out in an Applied Biosystems StepOnePlus™ Real-Time PCR System (StepOnePlus™ system) according to the manufacturers’ instructions. The gene-specific primers used for the quantitative RT-PCR were as follows: PsGPD, GPD-qPCR-F 5′-AAGCCCGCTGATTACAAAGAGA-3′ and GPD-qPCR-R 5′-GGCGAGAGTATCCGTATTCGTT-3′; ArCspA, Csp-qPCR-F 5′-ATGTCTTTGGGAACCGTCAAG-3′ and Csp-qPCR-R 5′-CTCTGCAGGCCGGACGTTTTC-3′; BrTSR15, BrTSR15-qPCR-F 5′-CAGATCAAACTCCATGAGATAGGG-3′ and BrTSR15-qPCR-R 5′-TTCCACCTTTATCTCGAAAATCCT-3′; BrTSR53, BrTSR53-qPCR-F 5′-TGGAAGAAAACAATTGCAACGCCA-3′ and BrTSR53-qPCR-R 5′-AAGAATTGACCACATGACGAGGAC-3′; OsActin1 (LOC_Os03g50890), OsActin1-qPCR-F 5′-ACAGGTATTGTGTTGGACTCTGG-3′ and OsActin1-qPCR-R 5′-AGTAACCACGCTCCGTCAGG-3′. The reaction cycle was: 1 cycle of 95 °C for 30 s followed by 40 cycles of 95 °C for 5 s and 60 °C for 34 s. In the reaction plate, each sample was measured in triplicate. The expression level of OsActin1 was used for the normalization of real-time PCR results. Changes in gene expression were analyzed by StepOne software v.2.2.2 (Applied Biosystems).
Protein extraction and immunoblot analysis
Approximately 0.1 g rice leaf tissue of wild-type, myc-tagged 35S::BrTSR15 and 35S::BrTSR53 plants was harvested and homogenized with an extraction buffer (125 mM Tris–HCl pH8.8, 1 % (w/v) SDS, 10 % (v/v) glycerol, 50 mM Na2S2O5) that contained protease inhibitor mixture (Roche Applied Science). Crude protein extracts (5 µg) were resolved on a 15 % SDS–polyacrylamide gel. Following electrophoresis, the proteins were transferred onto a Hybond-ECL Nitrocellulose membrane (GE Healthcare). The membranes were incubated with a 1:1,000 dilution of antibody raised against Anti-myc Tag (Upstate). HRP-conjugated anti-mouse IgG (Sigma) was used as the secondary antibody, and the signals were visualized using ECL western blotting detection reagents (GE Healthcare).
Results
Generation of T-DNA insertion lines of transgenic rice T0 plants
To construct T-DNA inserted transgenic rice, the desired targets were selected from 4 genes that were derived from edible mushroom, brassicaceae and microbes. Diverse genes were selected to eliminate any genotype-related differences that might arise when selecting the transformant. Transgenic rice plants were generated by introducing the over-expression construct that contained the full-length cDNA of the genes that were under the control of the CaMV 35S promoter, PsGPD(35S::PsGPD), ArCspA(35S::ArCspA), BrTSR15(35S::BrTSR15) and BrTSR53(35S::BrTSR53) (Fig. 1a). Over-expression transgenic rice plants containing PsGPD, ArCspA, BrTSR15 and BrTSR53 were generated through Agrobacterium-mediated transformation. T0 transgenic plants were screened by PPT (phosphinothricin) resistance selection on medium, PCR amplification analysis and Bar immune strip test (Fig. 1b–d). We generated a total of 383 transgenic lines (149 PsGPD, 118 ArCspA, 44 BrTSR15 and 72 BrTSR53 transformants).

Schematic representation of the expression vector constructs for rice transformation. a Constructs used for the over-expression of PsGPD, ArCspA, BrTSR15 and BrTSR53 in rice, for which each full-length cDNA was cloned into the plant expression vector that harbored a phosphinothricin (PPT) resistance gene. b Examples of T0 generation transgenic plants. Left panel, shoot and root formation in MS media; right panel, acclimatization. c PCR analysis of 35S::PsGPD transgenic T0 plants using the bar specific primer. Lanes: V vector, NC non-transformant plants. d Detection of bar gene expression of 35S::PsGPD transgenic rice plants using immune strip
Isolation of single copy T-DNA insertion lines
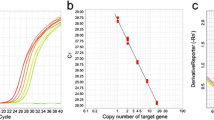
Many primary transformants must be analyzed for molecular features, i.e., copy number and transcript level, because the transgene copy number can influence the expression level, which often results in gene silencing in the transgenic plants in which multiple transgene copies are integrated into one or more chromosomal locations (Flavell 1994; Vaucheret et al. 1998). The transgene copy numbers of all transgenic rice plants were investigated by TaqMan copy number assay. The frequency of the transgene copy numbers was very diverse in PsGPD, ArCspA, BrTSR15 and BrTSR53 transformants (Fig. 2). A total of 383 lines (149 PsGPD (35S::PsGPD) transformants, 118 ArCspA (35S::ArCspA) transformants, 44 BrTSR15 (35S::BrTSR15) transformants and 72 BrTSR53 (35S::BrTSR53) transformants) were identified by transgenic copy number assay (Fig. 2). The single copy frequency for each target gene ranged from 44.1 to 65.9 %, and the average single copy frequency of transformants was 53 %. The copy number of integrated genes varied from one to 13 copies. The average copy number of the T-DNA insertion was 1.81 (Table 1).

TaqMan copy number assay of GPD(35S::PsGPD), ArCspA(35S::ArCspA), BrTSR15(35S::BrTSR15) and BrTSR53(35S::BrTSR53) for transgene copy determination. The sample CN data were calculated using the Applied Biosystems CopyCaller Software v2.0
Distribution of T-DNA insertion sites within genes and intergenic regions
FSTs analysis of T0 plants was carried out only for those transgenic plants in which a single copy T-DNA insertion was confirmed. Transgenic plants with the T-DNA insert in an intergenic region are less likely to be affected by other genes; therefore, single copy/intergenic transgenic plants were selected using FST analysis. We obtained 157 FSTs from 203 lines of single copy transformants. The isolated FST sequences were analyzed by the BLASTN homology search program using the rice genome sequence database of RAP3. Among the FSTs, acceptable FSTs were matched to genome by BLAST and defined the locations of the T-DNA insertion sites. To facilitate the analysis, the DNA sequence from ATG to the stop codon, together with the 5′ upstream-1000 and 3′ downstream-300 sequences, was defined as a gene. The FTS segments that were not included in any gene were identified as intergenic insertions. To manage bulk FSTs in statistics, flanking sequences were automatically evaluated using FSTVAL, and the genome positions of the FSTs were mapped. FST mapping showed that the T-DNA integration frequency was not proportional to chromosome size. Of the 157 transgenic plants for which FST analysis was performed, 5 FSTs (3 %) were in the exon region, 14 FSTs (9 %) were in the intron region, 32 FSTs (20 %) were in the 5′ upstream region, and 7 FSTs (4 %) were in the 3′ downstream region; therefore, 58 FSTs (36 %) of the T-DNA insertion sites were in the genic regions (Fig. 3b). Conversely, T-DNA insertion in the intergenic region was found in 97 FSTs (62 %) (Table 2). The insertion sites and frequency along the chromosomes are graphically presented in Fig. 3. The assignment of these 157 FSTs on chromosomes revealed that FSTs are distributed throughout the chromosomes (Table S1).

Chromosomal positions of T-DNA insertions in the rice genome. Of the 203 sequences from T-DNA insertion batches, 157 sequences passed through the filtration and chromosome mapping steps. a Frequency graphs of T-DNA insertion. b Distribution maps of the T-DNA in the rice genome. Each number under the chromosome means the drawing scale (kb/px)
Expression of the transgenes in transgenic rice plants
To screen homozygous lines of intergenic and single copy insertion, transgenic rice plants were analyzed using TaqMan copy number assay. Heterozygous lines were single copies, and homozygous lines were two copies from T1 generation plants (Fig. S1). The mRNA transcripts of homozygous lines were examined by quantitative real-time PCR analysis from leaf tissue. A total of 55 lines, including 13 BrTSR15, 15 BrTSR53, 12 PsGPD and 15 ArCspA transformants, were analyzed. The transcript levels were significantly increased in all of the transgenic plants compared with wild-type (Fig. 4a). In several lines of myc-tagged BrTSR15 and BrTSR53 transgenic plants, target proteins were also detected by western blot analysis using anti-myc tag antibody. The BrTSR15 and BrTSR53 proteins were highly expressed in mature leaves (Fig. 4b).

Expression of the transgenes in the transgenic plants. a Expression levels of the PsGPD, ArCspA, BrTSR15 and BrTSR53 genes in rice. Quantitative real-time PCR was analyzed via the 2−ΔΔCT method using the OsActin1 gene as an internal control. Each data point represents the mean (±SD) of three separate experiments. b Western blot analysis of expression of the BrTSR15-myc and BrTSR53-myc fusion protein in wild-type, 35S::BrTSR15 and 35S::BrTSR53 transgenic plants. Total soluble proteins were extracted in the 4-week-old leaf tissues of rice plants. Each lane was loaded with 20 μg total protein on two gel at same time. One gel was used to perform western blot using anti-myc antibody, and the other was stained with coomassie blue
Discussion
GM crops are alternatives that can contribute to solving issues related to climate change, global warming, and a lack of food and animal feed. Since GM crops were first cultivated in 1996, they have been put to practical use, and the total area of cultivation has increased dramatically every year (James 2012). Because the development of GM crops is essential, we attempted to effectively construct a GM rice event. In particular, we saved time and labor by performing analyses on as many first-generation transgenic plants as possible. The flow diagram that we describe is summarized in Fig. S2. Using our GM event candidate process, we were able to significantly increase the transformation efficiency and reduce the time required to generate transgenic plants. Here, we discuss the most efficient method to produce genetically modified plants. To construct a GM plant that can lead to the mass development of transgenic plants, a suitable transformation strategy that can save time, be less labor-intensive, and involve the appropriate efforts is required. Reducing the number of plants to handle in a short period can be accomplished if molecular biological analysis is largely completed on the T0 generation. We generated transgenic plants that harbored single copy T-DNA insertions for GM rice. In total, we identified 383 T-DNA insertion sites in the rice genome. Using our optimized protocol, we achieved an average T-DNA insertion number of 1.81 and a single copy T-DNA insertion rate of 53 %. The criteria for a candidate plant include a line with single copy insertion of T-DNA, intergenic insertion and homozygous genotype with suitable expression. The transformant status, copy number of T-DNA insertions, and insertion sites should be confirmed in the T0 generation. In this study, the probability of single copy T-DNA insertion was 53 %, and the intergenic insertion probability was 62 %. These results might aid in the development of strategies to construct transgenic plants. Furthermore, the selected single copy/intergenic lines were propagated after homozygous screening and confirmation of the transcription or translation level in the T1 generation. In this study, over-expression was confirmed in the majority of the single copy/intergenic lines.
Significantly, this highly efficient protocol allowed for a minimization of labor and space per transgenic line production because the transformations are conducted on a large scale. The generation of a large number of rice T-DNA lines is no longer a limiting factor because of the establishment of highly efficient transformation procedures (Sallaud et al. 2003). The present results indicate that the T-DNA tagged rice population and the database of rice will be useful for GM event research and development. The system could also be applied to the genetic breeding of other GM rice strains.
References
Cho JI, Lim HM, Siddiqui ZS, Park SH, Kim AR, Kwon TR, Lee SK, Park SC, Jeong MJ, Lee GS (2014) Over-expression of PsGPD, a mushroom glyceraldehyde-3-phosphate dehydrogenase gene, enhances salt tolerance in rice plants. Biotechnol Lett 36:1641–1648
De Preter K, Speleman F, Combaret V, Lunec J, Laureys G, Eussen BH, Francotte N, Board J, Pearson AD, De Paepe A, Van Roy N, Vandesompele J (2002) Quantification of MYCN, DDX1, and NAG gene copy number in neuroblastoma using a real-time quantitative PCR assay. Mod Pathol 15:159–166
Flavell RB (1994) Inactivation of gene expression in plants as a consequence of specific sequence duplication. Proc Natl Acad Sci USA 91:3490–3496
Fuse T, Sasaki T, Yano M (2001) Ti-plasmid vectors useful for functional analysis of rice genes. Plant Biotechnol 18:219–222
Halpin C (2005) Gene stacking in transgenic plants-the challenge for 21st plant biotechnology. Plant Biotechnol J 3:141–155
Hiei Y, Komari T (2008) Agrobacterium-mediated transformation of rice using immature embryos or calli induced from mature seed. Nat Protoc 3:824–834
Ingham DJ, Beer S, Money S, Hansen G (2001) Quantitative real-time PCR assay for determining transgene copy number in transformed plants. Biotechniques 31:132–140
James C (2012) Global status of commercialized biotech/GM crops: 2012. ISAAA brief No. 44. ISAAA: Ithaca, NY
Kim JS, Kim J, Lee TH, Jun KM, Kim TH, Kim YH, Park HM, Jeon JS, An G, Yoon UH, Nahm BH, Kim YK (2012) FSTVAL: a new web tool to validate bulk flanking sequence tags. Plant Methods 8:19–27
Lee SC, Lim MH, Kim JA, Lee SI, Kim JS, Jin M, Kwon SJ, Mun JH, Kim YK, Kim HU, Hur Y, Park BS (2008) Transcriptome analysis in Brassica rapa under the abiotic stresses using Brassica 24K oligo microarray. Mol Cells 26:595–605
Li D, Fu Q, Wang F, Yao Q, Lai F, Wu JC, Zhang ZT (2004) Resistance of transgenic rice containing both sck and cry1Ac genes against Chilo suppressalis and Cnaphalocrocis medinalis. Chin J Rice Sci 18:43–47
Mason G, Provero P, Vaira AM, Accotto GP (2002) Estimating the number of integrations in transformed plants by quantitative real-time PCR. BMC Biotechnol 24:20–31
Rommens CM (2007) Intragenic crop improvement: combining the benefits of traditional breeding and genetic engineering. J Agric Food Chem 55:4281–4288
Sallaud C, Meynard D, van Boxtel J, Gay C, Bès M, Brizard JP, Larmande P, Ortega D, Raynal M, Portefaix M, Ouwerkerk PB, Rueb S, Delseny M, Guiderdoni E (2003) Highly efficient production and characterization of T-DNA plants for rice (Oryza sativa L.) functional genomics. Theor Appl Genet 106:1396–1408
Song P, Cai CQ, Skokut M, Kosegi BD, Petolino JF (2002) Quantitative real-time PCR as a screening tool for estimating transgene copy number in WHISKERS™ derived transgenic maize. Plant Cell Rep 20:948–954
Toki S, Hara N, Ono K, Onodera H, Tagiri A, Oka S, Tanaka H (2006) Early infection of scutellum tissue with Agrobacterium allows high-speed transformation of rice. Plant J 47:969–976
Travella S, Ross SM, Harden J, Everet C, Snape JW, Harwoord WA (2005) A comparison of transgenic barley lines produced by particle bombardment and Agrobacterium-mediated techniques. Plant Cell Rep 23:780–789
Tu J, Datta K, Alam MF, Fan Y, Khush GS, Datta SK (1998) Expression and function of a hybrid Bt toxin gene in transgenic rice conferring resistance to insect pests. Plant Biotechnol 15:195–203
Tu J, Zhang G, Datta K, Xu C, He Y, Zhang Q, Khush GS, Datta SK (2000) Field performance of transgenic elite commercial hybrid rice expressing Bacillus thuringiensis delta-endotoxin. Nat Biotechnol 18:1101–1104
Vaucheret H, Beclin C, Elmayan T, Feuerbach F, Godon C, Morel JB, Mourrain P, Palauqui JC, Vernhettes S (1998) Transgene-induced gene silencing in plant. Plant J 16:651–659
Wu G, Cui H, Ye G, Xia Y, Sardana R, Cheng X, Li Y, Altosaar I, Shu Q (2002) Inheritance and expression of the cry1Ab gene in Bt (Bacillus thuringiensis) transgenic rice. Theor Appl Genet 104:727–734
Acknowledgments
This study was supported by a grant from the Next-Generation Biogreen 21 Program (Project No. PJ011257) and a grant from National Academy of Agricultural Science (NAAS) Agenda Program (Project No.PJ010086) in Rural Development Administration, Jeonju, Republic of Korea and National Marine Biodiversity Institute Research Program (2015M00500).
Author information
Authors and Affiliations
Corresponding author
Additional information
H. Lim and H.-J. Hwang equally contributed to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lim, H., Hwang, HJ., Kim, AR. et al. A simple, rapid and systematic method for the developed GM rice analysis. Plant Biotechnol Rep 10, 25–33 (2016). https://doi.org/10.1007/s11816-015-0384-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-015-0384-1