
Abstract
The present study aimed to investigate in vitro biological activities of extract of Eugenia punicifolia leaves (EEP), emphasizing the inhibitory activity of enzymes related to metabolic syndrome and its antioxidant effects. The antioxidant activity was analyzed by free radicals scavengers in vitro assays: DPPH·, ABTS·+, O2 ·−, and NO· and a cell-based assay. EEP were tested in inhibitory colorimetric assays using α-amylase, α-glucosidase, xanthine oxidase, and pancreatic lipase enzymes. The EEP exhibited activity in ABTS·+, DPPH·, and O2 ·− scavenger (IC50 = 10.5 ± 1.2, 28.84 ± 0.54, and 38.12 ± 2.6 μg/mL), respectively. EEP did not show cytotoxic effects, and it showed antioxidant activity in cells in a concentration-dependent manner. EEP exhibited inhibition of α-amylase, α-glucosidase, and xanthine oxidase activities in vitro assays (IC50 = 122.8 ± 6.3; 2.9 ± 0.1; 23.5 ± 2.6), respectively; however, EEP did not inhibit the lipase activity. The findings supported that extract of E. punicifolia leaves is a natural antioxidant and inhibitor of enzymes, such as α-amylase, α-glucosidase, and xanthine oxidase, which can result in a reduction in the carbohydrate absorption rate and decrease of risks factors of cardiovascular disease, thereby providing a novel dietary opportunity for the prevention of metabolic syndrome.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Metabolic syndrome (MS) is characterized as metabolic abnormalities including risk factors for cardiovascular diseases such as obesity, hypertension, hyperglycemia, hypertriglyceridemia, and low high-density lipoprotein (HDL) cholesterol. According to Ervin [1], a little more than one third of the adults in the USA could be characterized as having MS. Studies showed that MS is associated with a higher fraction of oxidized low-density lipoprotein (LDL), which can result in an increased risk of future myocardial infarction. The LDL receptor deficient in obese rats was associated with increased oxidative stress and impaired antioxidant defense due to macrophage infiltration and accumulation of oxidized LDL in the aorta [2]. Degenerative diseases such as atherosclerosis, inflammatory injury, cancer, cardiovascular disease, and aging can be involved in pathogenicity of MS [3, 4]. Given the public health significance of MS, nutritional supplementation with botanicals that effectively address pathogenic mechanisms represents an attractive novel and potentially effective approach to the problem [5].
Eugenia punicifolia (Kunth) DC., (Mirtaceae), known as “pedra-ume caa”, is a shrub widely distributed in the Amazon region. This plant was selected based on preliminary data suggesting its hypoglycemic effects when used in traditional medicine to treat diabetes mellitus [6]. Due to these findings, extract of E. punicifolia leaves (EEP) could be used preventively to counter risk factors or treat subjects with MS. The aim of this work was to investigate the in vitro biological activities of EEP, emphasizing the inhibitory activity of enzymes related to MS and its antioxidant effects.
Methods
Chemicals and Reagents
Gallic acid, ascorbic acid, quercetin, resazurin sodium, Dulbecco's modified eagle's medium (DMEM), 2,2′-azinobis-3-ethylbenzothiazoline-6-sulfonic acid (ABTS), naphthyl ethylenediamine dihydrochloride, nitroblue tetrazolium (NBT), nicotinamide-adenine-dinucleotide (NADH), phenazine methasulfate (PMS), doxorubicin, orlistat, allopurinol, 2,2-diphenyl-1-picrylhydrazyl (DPPH·), 2′,7′-dichlorofluorescin diacetate (DCFH-DA), human salivary amylase (EC 3.2.1.1), α-glucosidase (EC 3.2.1.20), pancreatic lipase (EC 3.1.1.3), and xanthine oxidase (EC 1.17.3.2) were purchased from Sigma Chemical Company (St. Louis, USA). All remaining reagents were of the highest purity available (≥98 %). Amylase substrate and uric acid reagent were purchased from Labtest® (Belo Horizonte, MG, Brazil).
Herbal Material
The herbal material (leaves) was collected from Manaus-Amazonas (Brazil) and authenticated by the herbarium of the Instituto Federal de Educação Ciência e Tecnologia do Amazonas by Valdely Ferreira Kinupp, where a voucher specimen was deposited under the registration number 6789.
Cell Culture
3T3-L1 cells (Mouse embryonic fibroblast) were obtained from the Cell Bank of Rio de Janeiro and were maintained in DMEM, supplemented with 10 % fetal bovine serum (FBS), penicillin (100 U/mL), and streptomycin (100 U/mL). The cells were incubated at 37 °C in a humidified atmosphere containing 5 % CO2.
Raw Material Treatment
The leaves were dried for 7 days on circulating air oven at 40 ± 5 °C temperature. After drying the leaves, they were knifed by a knife mill (1 mm mesh) and stored in polystyrene bottle. The raw material was characterized by granulometric analysis [7] using sieves of 210, 300, 420, 500, 600, 710, and 1,000 μm and loss of drying by gravimetric method [8].
Preparation of Spray-Dried Extract of E. punicifolia
The aqueous EEP was prepared by infusion for 15 min using a proportion of 7.5 % (w/v) and was characterized through dry residue. The spray-dried extract was obtained by drying of the extractive solution in a Mini Spray Drier (MSD 1.0, Labmaq, São Paulo, Brazil) and was characterized through the total tannin content.
Determination of Dry Residue
Samples of 20.0 g from extractive solution were analyzed by gravimetric method according to German Pharmacopoeia [9]. The results represent the mean of three determinations.
Analysis of Total Tannin Content
The total tannin content analysis was performed considering the capability of precipitation with protein. For determination of total polyphenol (TP), a diluted aliquot of extractive solution (ES) was analyzed on spectrophotometer at 271 nm using water as compensation solution. For determination of non-tannin fraction (NTF), an amount of 0.150 g of casein (Merck) was stirred with 10.0 mL of the ES during 1 h. After filtration, the assay proceeded as described for the total polyphenols content determination. The total tannin content (TTC) was expressed as gram of gallic acid per 100 g of extractive solution according to equations 1, 2, and 3. The results represent the mean of three determinations.
Where TP is the total polyphenols (gram percent), NTF is the non-tannin fraction (gram percent), TTC is the total tannin content (gram percent), A is the absorbance (A.U.), DF is the dilution factor, M is the drug weight (gram), and A 1%1cm is the specific absorption of gallic acid.
Chromatographic Profile
Liquid chromatography Shimadzu® model LC 20TA proeminence was employed and fitted with two 2LC-10Advp pumps, column oven model CTO 010ASvp, control system model CBM 20, degasser model DGU-20A5, automatic injector of samples model SIL-20A, and detector with photodiode array (DAD) model SPD-M20A. The oven temperature for column was maintained at 40 °C, and the chromatograms were observed at 275 nm. The control system was carried out by Lc solution® software. For the development of the chromatogram, pre-column (4 × 3 mm i.d.) and column (250 × 4.6 mm i.d.) C-18 Phenomenex®, model Gemini, porosity 5 μm was used. The chromatographic profile was obtained by using a isocratic method acetonitrile:phosphoric acid 1 % (24:76 v/v); the injected volume was 20 μL. The peaks of the chromatograms were identified and compared with the retention time and area of the gallic acid standard.
Antioxidant Activity Chemical Assays
2,2-Diphenyl-1-Picrylhydrazyl Radical-Scavenging Activity
The scavenger activity of the DPPH· was measured according to the method of Mahmoudi et al. [10]. Different concentrations of extract (5, 10, 25, 50, and 100 μg /mL) were added, at an equal volume, to the methanol solution of DPPH· (100 mM). After 15 min at room temperature, the absorbance was recorded at 517 nm. The experiment was repeated three times. Gallic acid and ascorbic acid were used as standard. Dimethyl sulfoxide (DMSO) was used as negative control. IC50 values denote the concentration of sample, which is required to scavenge 50 % of DPPH· free radicals. The antioxidant activity was calculated using the following equation: % inhibition = 100 − (sample abs/ABS control) × 100, where abs is the absorbance.
ABTS Assay
The ABTS assay was based on the method of Re et al. [11]. ABTS was dissolved in MiliQ water to a 7 mM concentration. ABTS·+ was produced by reacting ABTS stock solution with 5 mM potassium persulfate and kept in the dark at room temperature for 12–16 h. Once ABTS·+ is formed, MiliQ water was added to the solution (dilution 1:7). A 96-well plate flat-bottomed was added by 270 μL of ABTS solution with 30 μL of water. This solution was monitored by reading at 714 nm in a microplate reader (DTX 800, Beckman, CA, USA) to obtain absorbance of approximately 1.00 (control). Then, 30 μL of EEP from different concentrations (5, 10, 25, 50, and 100 μg/mL) were added to 270 μL of ABTS and the reaction was incubated for 15 min in the dark at room temperature. After incubation, the absorbance at 714 nm was measured. Gallic acid and ascorbic acid in the same EEP concentrations were measured following the same procedures described above and were used as positive controls. The antioxidant activity was calculated using the following equation: % inhibition = 100 − (abs sample/Abs control) × 100, where Abs is the absorbance.
Anion Superoxide Radical-Scavenging Assay
The anion superoxide radical (O2 ·−) scavenging activity was measured by NBT method [12]. Into each well of the microplate, 100 μL of NADH (390 μM), 100 μl of NBT (250 μM) in phosphate buffer (0.1 M, pH 7.4), and 50 μL of different concentrations of extract (5, 10, 25, 50, and 100 μg / mL) were added. The reaction was incubated at room temperature for 5 min, and the first reaction was measured at 560 nm against a blank (DTX 800, Beckman, CA, USA). Then, 100 μL of PMS solution (10 μM) in phosphate buffer (0.1 M, pH 7.4) was added to the mixture. The reaction mixture was incubated for 5 min at room temperature, and the absorbance at 560 nm was measured again. Decreased absorbance of the reaction mixture indicates increased superoxide anion scavenging activity. The percentage inhibition of superoxide anion radical generation of three parallel measurements was calculated using the following formula: % inhibition = 100 − (Abs end of the sample−Abs starting sample) × 100 abs control, where Abs is the absorbance. Gallic acid and ascorbic acid were used as positive controls.
Scavenging of Nitric Oxide In Vitro
A modified protocol from Govindarajan et al. [13] was used to measure the sequestration (nitric oxide, NO·) in which the Griess reagent was modified using naphthyl ethylenediamine dihydrochloride (0.1 % w/v) instead of 1-napthilamine (5 %). A reaction was made in a mixture containing 2 mL of sodium nitroprusside (10 mM), 0.5 mL of phosphate buffered saline (10 mM, pH 7.2), and 0.5 mL of EEP at concentrations of 5, 10, 25, 50, and 100 mg/ mL, and the standard solution of ascorbic acid in the same concentration was incubated at 25 ° C for 150 min. After incubation, 0.5 mL of the reaction mixture was mixed with 1 mL of sulphanilic acid (0.33 in 20 % glacial acetic acid). The reaction was allowed to rest for 10 min to complete diazotization of nitrite with sulfanilamide. Then, 1 mL of ethylenediamine naphthyl dihydrochloride was homogenized and incubated for 30 min at 25 °C. The absorbance of these solutions was measured in a microplate reader (DTX 800, Beckman, CA, USA) at 540 nm.
Cell Viability Assay
E. punicifolia extract cytotoxicity in fibroblasts cells was determined by the Alamar Blue method according to Nakayama and coworkers [14]. The Alamar Blue assay is a colorimetric assay involving the cellular reduction of resazurin to resorufin. Briefly, adherent cells (5 × 103 cells/well) were grown in 96-well tissue culture plates and exposed to EEP (50, 25, and 12.5 μg/mL) for 24, 48, and 72 h. After incubation, 10 μL of the Alamar Blue solution [0.4 % Alamar Blue (Resazurin) in PBS] was added and the cells were incubated for 2 h at 37 °C. Fluorescence was measured in microplate reader (DTX 800, Beckman, CA, USA) using excitation at 545 nm and emission at 595 nm and expressed as a percentage of the cells in control after background fluorescence were subtracted. Doxorubicin (5 μg/mL) was used as a positive control. The assays were done in quadruplicate.
Cellular Antioxidant Activity of E. punicifolia Extract
Cellular antioxidant activity was measured by intracellular reactive oxygen species (ROS) production using a non-fluorescent cell-permeating compound, DCFH-DA [15]. DCFH-DA is hydrolyzed by intracellular esterase and then oxidized by ROS into a fluorescent compound 2′-7′-DCF. 3T3-L1 cells were seeded at a density of 6 × 104 cell/well on a 96-well microplate in 100 μL of medium growth. Twenty-four hours after seeding, the medium growth was removed and the wells were washed with PBS. Next, 100 μL of 10 μM DCFH-DA dissolved in Hank's buffer were incubated for 30 min at 37 °C and 5 % CO2. The following cells were washed with 100 μL of PBS and 100 μL of EEP, and different concentrations were added. The fluorescence was immediately measured with excitation wavelength of 485 nm and emission wavelength of 520 nm using a microplate reader (DTX 800, Beckman, CA, USA). Controls with/without DCFH-DA were made. Quercetin was used as positive control.
Enzyme assays
α-Glucosidase Inhibitory Activity In Vitro
The α-glucosidase inhibitory activity was determined according to Andrade-Cetto et al. [16] by measuring the release of 4-nitrophenol from 4-nitrophenyl α-d-glucopyranoside (4-NPGP). The reaction started by adding 20 μL of EEP (12.5 to 0.78 μg/mL), DMSO, or control drug (quercetin), with 180 μL of the α-glucosidase enzyme from Saccharomyces cerevisiae (Sigma) were incubated for 2 min at 37 °C. Then, after the addition of 150 μL of the color reagent NPGP (Sigma), samples were incubated for more than 15 min at 37 °C. The assay media contained 10 mM of potassium phosphate buffer, pH 6.9, 5 mM of 4-NPGP, and 2 U of α-glucosidase. Quercetin also was used as positive control at the same concentration of extract. The reading of samples was performed by using a microplate reader at 405 nm. After the test was completed, the calculation of inhibition was applied with the equation: 100 − (A2 sample−A1 sample/A2 control−A1 control) × 100 where A1 is the absorbance of the initial reading (time 0), A2 is absorbance of the final reading (time 15 min), control is the absorbance of the test using DMSO. IC50 values were determined by nonlinear regression using the program Microcal ™ Origin ®, version 6.0 (Microcal Software Inc.).
α-Amylase Inhibitory Activity In Vitro
The α-amylase inhibitory activity was determined according to Subramaniam et al. [17]. In this colorimetric test, 10 μL of α-amylase enzyme, 3.3 U (Sigma) was incubated with 10 μL of EEP (250 to 31.2 μg/mL), DMSO, or control drug (quercetin) for 5 min at 37 ° C. After adding 180 μL for the Amylase Substrate (Labtest®), samples were incubated for 8 min and the first reaction was measured at 620 nm (DTX 800, Beckman, CA, USA). Then, 100 μL of the reactive α-amylase (Labtest®) and 150 μL of distilled water were added in the microplate and then incubated for more 5 min at 37 ° C and the second reaction was measured again. IC50 values were determined by nonlinear regression using the program Microcal™ Origin ®, version 6.0 (Microcal Software Inc.). The reagent of the α-amylase (Labtest) was diluted in distilled water (1:1) before being added to the microplate. The quercetin was used as positive control at the same EEP concentrations. After the test, the α-amylase inhibition was calculated using the equation: % inhibition = 100 − (A2 sample–A1 sample/A2 control−A1 control) × 100 where A1 is the absorbance of the initial reading and A2 is the absorbance of the final reading.
Xanthine Oxidase Inhibitory Activity In Vitro
Xanthine oxidase activity was determined by measuring the formation of uric acid from xanthine. The reagent 1 was prepared by mixture of xanthine oxidase (667 mM), EDTA (0.1 mM), and hidroxilamine (0.2 mM) in 50 mM phosphate buffer solution (pH 7.5). Into each microplate well, 40 μL of xanthine oxidase enzyme and 15 μL of extract (200 to 12.5 μg/mL), DMSO, or control drug (allopurinol) were added and incubated at 37 ° C for 5 min. Then, 95 μL of reagent 1 was added in the reaction and incubated again for 30 min at 37 ° C. The absorbance at 295 nm was measured in a microplate reader (DTX 800, Beckman, CA, USA). After, 150 μL of uric acid reagent was added in the mixture and the absorbance was measured again. DMSO was used as negative control and allopurinol as positive control. The inhibition percentage of xanthine oxidase activity was calculated using the following formula = % inhibition = 100 − (A2 sample−A1 sample/A2 control−A1 control) × 100 where A1 is the absorbance of the initial reading and A2 is the absorbance of the final reading [18].
Pancreatic Lipase Inhibitory Activity In Vitro
The pancreatic lipase inhibitory activity was determined according to Slanc et al. [19] by incubation of 20 μL of the EPP, diluent, or control drug (orlistat) with 180 μL of the enzyme lipase from porcine pancreas type II (SIGMA) for 2 min at 37 °C. After the addition of 200 μL of Tris buffer (Trizma® Hydrochloride) was made the first reading at 415 nm. The second reading was made 15 min after addition of 20 μL of PNP of color reagent (4-nitrophenyl palmitate) (Sigma), and the absorbance was measured at 405 nm. The assay media contained 75 mM Tris buffer, pH 8.5, 2.5 mM PNP, and 250 U of pancreatic lipase. The EEP or orlistat were diluted five times in sequence, with the following concentrations: 200 to 12.5 μg/mL. The measurements were performed in triplicate, and the IC50 value, the concentration of the extract that results in 50 % inhibition of maximal activity was determined.
Statistical Methods
Results were expressed as the means and standard deviations triplicate/quadruplicate measurements. Differences between groups were assessed by one-way analysis of variance (ANOVA) followed by the Tukey post hoc test. The 50 % inhibitory concentration (IC50) values were obtained by nonlinear regressions of concentration-response curves using the program Microcal™ Origin ® version 6.0 (Microcal Software Inc.). A value of p < 0.05 indicated significance.
Results and Discussion
The raw material of E. punicifolia presented a particle mean diameter of 623.5 μm and on drying loss of 10.22 %. These properties are important for the standardization of the extraction process since particles mean diameter has an influence on the extraction efficiency, and the loss on drying is important for conservation of the raw material. The aqueous extract presented a dry weight of 1,150 ± 0.004 g%, and the spray-dried extract presented 18.25 ± 0.03 g% of total tannin content represented as gallic acid which was a significant value. The presence of gallic acid in the spray-dried extract was confirmed through high-performance liquid chromatography as demonstrated in Fig. 1. Moreover, the extract presented total phenolics and flavonoids content of 21.60 ± 1.05 GAE mg/g and 2.62 ± 0.48 QE mg/g and showed a large amount of phenolic compounds greater than flavonoids content.

HPLC chromatographic profile of spray-dried extract of E. punicifolia (a) showing gallic acid identification (peak 1) and spectrum of gallic acid (b)
Products derived from medicinal plants can be used as new therapeutic agents due to the presence of secondary metabolites including groups such as polyphenols [20]. According to Cai et al. [21], there is a correlation between polyphenolic content and antioxidant activities in medicinal herbs. Several studies demonstrated that medicinal plants are a rich source of antioxidant compounds [22]. As presented in Table 1, EEP can be considered a free radical scavenger, and therefore, an antioxidant. ABTS·+, DPPH·, and O2 ·− scavenger assays showed concrete results with IC50 = 10.5 ± 1.2, 28.84 ± 0.54, and 38.12 ± 2.6 μg/mL, respectively, as compared with gallic acid and ascorbic acid in the same concentrations. Furthermore, radical-scavenging activities of EEP increased in a concentration-dependent manner. The free radical scavenging activity of E. punicifolia was less than known antioxidants, such as gallic acid 1.03 ± 0.1, 1.14 ± 0.2, and 11.67 ± 0.5 μg/mL and ascorbic acid 4.84 ± 0.3, 2.74 ± 0.3, and 89.5 ± 5.3 μg/mL; however, EEP is a crude extract containing a mixture of substances, which is difficult to direct compare with an isolated substance. Antioxidant capacity detected by ABTS assay was significantly higher in EEP compared to that by DPPH. These data suggest that ABTS assay may be more useful than DPPH for detecting antioxidant capacity in a variety of plants. This fact can be explained by the ABTS assay that is based on the generation of a blue/green ABTS·+ which is applicable to both hydrophilic and lipophilic antioxidant systems, whereas DPPH assay uses a radical dissolved in organic media and is therefore applicable to hydrophobic systems. As the EEP is a hydrophilic extract, its higher effects on ABTS·+ scavenger activity should be expected [23]. Also, this extract presented a lower nitric oxide-scavenging activity showing IC50 value of 2.36 ± 0.56 mg/mL against 0.9 ± 0.06 mg/mL and 0.2 ± 0.01 mg/mL for gallic acid and ascorbic acid, respectively.
Study using DPPH· radical scavenging from Artemisia absinthium extract showed IC50 value of 612 ± 30.6 μg/mL compared with the IC50 values for ascorbic acid and Quercetin 1.26 ± 0.11 and 1.32 ± 0.07 μg/mL, respectively [24]. Other studies have also been conducted to investigate the potential radical scavenging activities of lyophilized aqueous extract of propolis, where IC50 values for ABTS·+, DPPH·, and O2 ·− were found as 14.29, 31.81, and 9.89 μg/mL, respectively [25]. Thus, we can conclude that the EEP had a significant antioxidant activity when compared with other natural products.
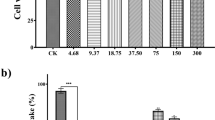
In the present study, the cytotoxic and antioxidant activity were measured in cell culture. Fibroblast line (3T3-L1 cell) treated with the EEP showed low toxicity against healthy mouse fibroblasts compared with cells treated with doxorubicin, an anti-cancer chemotherapy drug. Our results showed that after 24, 48, and 72 h, the doxorubicin at a concentration of 5 μg/mL promoted 47.3, 71, and 78.5 % lethality, respectively (p < 0.001 vs. control), while the EEP in the concentrations (50, 25, and 12 μg/mL) did not cause significant mortality (Fig. 2) when compared to doxorubicin (p > 0.05). These results corroborated with other studies of E. punicifolia in which it did not show hepatobiliary, microvascular, muscular, or pancreatic toxic effects in diabetic rats [6].

Cytotoxicity of EEP in 3T3-L1 cells. Adherent cells (5 × 105) grown in 96-well tissue culture plates. 3T3 cells were treated with varying concentrations of E. punicifolia extract (50, 25, and 12.5 μg/mL) for 24, 48, and 72 h, respectively. The values are means ± SD of three replicates. *p < 0.05 (indicates significant statistical difference by two-way ANOVA followed by Bonferroni comparison test)
The present study showed that EEP could protect mouse fibroblasts from oxidative damage. This antioxidant activity in cell indicated that the EEP reduced the concentration-dependent manner of the levels of intracellular ROS. Dichlorofluorescein is a probe that is trapped within cells and is easily oxidized to fluorescent dichlorofluorescein (DCF). The decrease in cellular fluorescence when compared to the control cells indicates the antioxidant capacity of the extract [15]. The oxidation inhibition values of the quercetin (5 μg/mL) were not significantly different compared with the EEP (Fig. 3). The low cytotoxicity effect of EEP and the antioxidant protection might be mediated by substances present such as gallic acid in other phenolic compounds.

Antioxidant activity measured by DCF assay of E. punicifolia extract. 3T3-L1 cells were preloaded for 30 min with 10 μM of DCFH-DA and washed with PBS. After 3T3-L1 cells were treated (25, 6.25, and 1.12 μg/mL) and then fluorescence levels were measured with an excitation wavelength of 485 nm and emission wavelength of 520 nm. Results were expressed in oxidation inhibition (%) and *p < 0.05 (indicates significant statistical difference by one-way ANOVA followed by Tukey's multiple comparison test). The values are means ± SD of three replicates
It is known that the quercetin is an important dietary flavonoid present in different vegetables, fruits, seeds, nuts, tea, and red wine, promoting robust antioxidant activity against oxidative stress [26, 27]. A study recommended that quercetin be used as a standard in assays for quantifying cellular antioxidant activity as it is a pure compound that is easily and economically obtained, as well as stable [15]. In the same study, the increase in fluorescence from DCF formation was inhibited by pure phytochemical compounds as the quercetin and fruit extracts in a concentration-dependent manner. The IC50 value of quercetin was 5.55 μM.
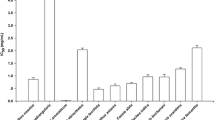
Various studies have reported inverse correlation between consumption of plant foods and the incidence of some degenerative diseases [28]. One practical approach for controlling postprandial plasma glucose rise, which is associated with diabetes, is to slow down glucose absorption through the inhibition of carbohydrate-hydrolyzing enzymes throughout the digestive tract [29]. The possible inhibition of α-amylase, α-glucosidase, pancreatic lipase, and xanthine oxidase enzymes using in vitro assays from the EEP (Fig. 4) was also evaluated. As shown in Fig. 4a, EEP inhibited pancreatic α-amylase enzyme. Thus, plants with inhibitory properties of digestive enzymes may have potential to reduce postprandial hyperglycemia allowing new approaches for alternative adjuvant therapy in combination with other oral hypoglycemic [30].

Inhibition of α-amylase, α-glucosidase, and xanthine oxidase by E. punicifolia extract. Inhibition of α-amylase by EEP (a) and quercetin (b); inhibition of α-glucosidase by EEP (c) and quercetin (d); inhibition of xanthine oxidase by EEP (e) and allopurinol (f). Data are expressed as IC50 (mean ± SD) values for triplicate determinations
The IC50 value of pancreatic α-amylase was better than in quercetin (Fig. 4b). In regard to the pancreatic α-glucosidase enzyme (Fig. 4c, d), it should be noted that EEP inhibited the enzyme and showed identical values found for quercetin. The comparison of inhibition activities of quercetin against α-glucosidase and α-amylase was also showed in Kim et al. [31] studies. Here, IC50 values for quercetin were 0.15 mg/mL and >0.60 mg/mL for α-glucosidase and α-amylase, respectively. In the present study, the EEP inhibited α-amylase and α-glucosidase activity in vitro assays; however, their α-glucosidase inhibitory activity was higher than that of α-amylase and did not inhibit significantly pancreatic lipase when tested in the concentration of 200 μg/mL inhibited only 17.23 ± 1.27 % of enzyme activity. These results are similar with studies of Ahmed et al. [32] in which the Eugenia jambolana extract, belonging also to the family Myrtaceae, significantly inhibited α-amylase and α-glucosidase activities in a concentration-dependent manner (IC50 values of 2.4 and 66 g/mL), respectively. Adyanthaya et al. [33] also reported that some foods and herbs have potential beneficial effects on diabetic glycemic control by inhibiting these enzymes. Hence, retardation of starch digestion by inhibition of digestive enzymes plays a key role in the control of diabetes and metabolic syndrome. Herbal medicines are getting more importance in the treatment of diabetes by inhibit the digestive enzymes as α-amylase and α-glucosidase [32]. So, they can be good way to identify possible hypoglycemic effect. The concentration-dependent effect was observed on increasing the concentrations of the extract solution, suggesting a competitive type of inhibition. Plots of percent inhibition vs. concentration of extract showed typical sigmoidal concentration- response curves (Fig. 4).
Regarding the inhibition of xanthine oxidase (XO), EEP showed a lower activity (Fig. 4e) compared to the allopurinol (Fig. 4f). It has been shown that XO inhibitors may be useful for treating hepatic disease, gout, and chronic diseases such as type 2 diabetes, which are caused by the generation of uric acid and superoxide anion radical [34, 35]. XO is considered an important biological source of superoxide radicals that contributes to oxidative stress in organisms and is involved in many pathological processes such as inflammation, atherosclerosis, cancer, and aging. In the present study, the result of XO inhibition showed by EEP can be explained by O2 ·− inhibition once this radical is generated during the assay and this can be interfered in enzyme inhibition result [36]. Other studies report a potential inhibition of enzymes such as XO, α-amylase, α-glucosidase, and an angiotensin converting enzyme, which is attributed to the presence of tannin in raw plant material [37]. Among others, chemical compounds isolated from Eugenia genera can be cited flavonoids (myricetrin, quercetin, and quercetrin), steroids, mono-and triterpenoids, tannins, anthraquinones, and essential oils have also been found [37].
For the first time, it was shown that this species can be considered a natural antioxidant and may be useful in preventing the ill effects of excessive free radical generation in the human body as they are harmful to health. Also, E. punicifolia can inhibit digestives enzymes, such as α-amylase, α-glucosidase, and xanthine oxidase that justify its use by population to diabetes treatment. This study opens new paths in research that act in the treatment or prevention of metabolic syndrome from a natural resource of the Amazon region.
Conclusion
Leaves extract of E. punicifolia significantly inhibited in vitro α-amylase, α-glucosidase, and xanthine oxidase enzyme activity and presented antioxidant activity in free radical scavengers and cell-based assays. These results will contribute to understand the mechanism of action of folk uses of this plant and in the animal models of MS and may indicate favorable use of this species in preventing or treating conditions related to MS such as hyperglycemia.
Abbreviations
- EEP:
-
Standardized extract of Eugenia punicifolia leaves
- MS:
-
Metabolic syndrome
- DPPH·:
-
2,2-diphenyl-1-picrylhydrazyl
- ABTS+ :
-
2,2′-azinobis-3-ethylbenzothiazoline-6-sulfonic acid
- O2 ·− :
-
Anion superoxide radical
- NO·:
-
Nitric oxide
- HDL:
-
High-density lipoprotein
- LDL:
-
Low-density lipoprotein
- NBT:
-
Nitroblue tetrazolium
- NADH:
-
Nicotinamide-adenine-dinucleotide
- PMS:
-
Phenazine methasulfate
- DCFH-DA:
-
2′,7′-dichlorofluorescin diacetate
- TP:
-
Total polyphenol
- NTF:
-
Non-tannin fraction
- ES:
-
Extractive solution
- TTC:
-
Total tannin content
- DF:
-
Dilution factor
- DMEM:
-
Dulbecco's Modified Eagle Medium
- FBS:
-
Fetal bovine serum
- 4-NPGP:
-
4-nitrophenyl α-d-glucopyranoside
- XO:
-
Xanthine oxidase
- ROS:
-
Reactive oxygen species
References
Ervin B., Ph.D. R.D. (2009) National Health Statistic Report. 5 (13), 1–7.
Holvoet, P. (2008). Verhandelingen-Koninklijke Academie voor Geneeskunde van Belgie., 70(3), 193–219.
Halliwell, B. (1997). Nutrition Reviews, 55(1 Pt 2), S44–S49.
Hansel, B., Giral, P., Nobecourt, E., Chantepie, S., Brucker, E. J., & Kontush, A. (2004). The journal of Clinical Endocrinology & Metabolism, 89(10), 4963–4971.
Cefalu, W. T., Ye, J., Zuberi, A., Ribnicky, D. M., Raskin, I., Liu, Z., et al. (2008). American Journal Clinical Nutrition, 87(2), 481S–487S.
Brunetti, I. L., Vendramini, R. C., Januario, A. H., Franca, S. C., & Pepato, M. T. (2006). Pharmaceutical Journal, 44(1), 35–43.
Voigt, R. (2005). Pharmazeutische Technologie 10. überarb. Aufl., Ullstein Mosby, Berlin
The United States Pharmacopeia. (2000). 25th ed., Mack Printing Company, Easton, PA.
Hartke, K., Mutschler, E. (1987). Deutsches Arzneibuch-9-Kommentar. Ausgabe 1986. Suttgart, Wissenschaftliche.
Mahmoudi, M., Ebrahimzadeh, M., Ansaroudi, F., Nabavi, S. F., & Nabavi, S. M. (2009). Journal of Biotechnology, 8(24), 7170–7175.
Re, R., Pellegrini, N., Proteggente, A., Pannala, A., Yang, M., & Rice-Evans, C. (1999). Free Radical Biology & Medicine, 26(9/10), 1231–1237.
Öztürk, M., Aydogmus-Öztür, F., Duru, M. E., & Topçu, G. (2007). Food Chemistry, 103(2), 623–630.
Govindarajan, R., Rastogi, S., Vijayakumar, M., Shirwaikar, A., Ajay, K. S. R., Mehrotra, S., et al. (2003). Biological Pharmaceutical Bulletin, 26(10), 1424–1427.
Nakayama, G. R., Caton, M. C., Nova, M. P., & Parandoosh, Z. (1997). Journal of Immunological Methods, 204(2), 205–208.
Wolfe, L. L., & Liu, R. H. (2007). Journal of Agricultural Food Chemistry, 55(22), 8896–8907.
Andrade-Cetto, A., Becerra-Jiménez, J., & Cárdenas-Vázquez, R. (2008). Journal of Ethnopharmacology, 116(1), 27–32.
Subramaniam, R., Asmawi, M. Z., & Sadikun, A. (2008). Acta Biochimica Polonica, 55(2), 391–398.
Bondet, V., Brand-Williams, W., & Berset, C. (1997). Food Science and Technology, 30, 609–615.
Slanc, P., Doljak, B., Kreft, S., Lunder, M., Janes, D., & Strukelj, B. (2009). Phytotherapy Research, 23(6), 874–877.
Espín, J. C., García-Conesa, M. T., & Tomás-Barberán, F. A. (2007). Phytochemistry, 68, 2986–3008.
Cai, Y., Luo, Q., Sun, M., & Corke, H. (2004). Life Sciences, 74(17), 2157–2184.
Apel, K., & Hirt, H. (2004). Annual Review of Plant Biology, 55, 373–399.
Floegel, A., Kim, D., Chung, S., Koo, S., & Chun, O. K. (2011). Journal of food Composition and Analysis, 24, 1043–1048.
Mahmoudi, M., Ebrahimzadeh, M., Ansaroudi, F., Nabavi, S. F., & Nabavi, S. M. (2009). Journal of Biotechnology, 8(24), 7170–7175.
Ilhami, G., Bursal, E., Sehitoglu, M. H., Bilsel, M., & Goren, A. C. (2010). Food and Chemical Toxicology, 48(8–9), 2227–2238.
Formica, J. V., & Regelson, W. (1995). Food and Chemical Toxicology, 33(12), 1061–1080.
Rice-evans, C. A., & Miller, N. J. (1996). Biochemical Society Transactions, 24(3), 790–795.
Hung, H. C., Joshipura, K. J., Jiang, R., Hu, F. B., Hunter, D., Smith-Warner, S. A., et al. (2004). Journal of the National Cancer Institute, 96(21), 1577–1584.
Shim, Y. J., Doo, H. K., Ahn, S. Y., Kim, Y. S., Seong, J. K., Park, I. S., et al. (2003). Journal of Ethnopharmacology, 85, 283–287.
Van de Laar, F., Lucassen, P. L., Akkermans, R. P., Van de Lisdonk, E. H., Rutten, G. E., & Van Weel, C. (2005). Diabetes Care, 28, 154–163.
Kim, S. H., Sung-Hoon, J. O., Young-In, K., & Jae-Kwan, H. (2011). International Journal of Molecular Science, 12(6), 3757–3769.
Ahmed, F., Chandra, J. N. N. S., & Timmaiah, N. V. (2009). Pharmacognos, 1(4), 317–321.
Adyanthaya, I., Kwon, Y. I., Apostolidis, E., & Shetty, K. (2010). Journal of food Biochemistry, 34(1), 31–49.
Lin, C. C., Huang, P. C., & Lin, J. M. (2000). The American Journal Chinese Medicine, 28(1), 87–96.
Heber, D., Seeram, N., Wyatt, H., Henning, S. M., Zhang, Y., Ogden, L. G., et al. (2007). Journal of Agricultural and Food Chemistry, 55(24), 0050–10054.
Zajácz, A., Gyémánt, G., Vittori, N., & Kandra, L. (2007). Carbohydrate Research, 342, 717–723.
Consolini, A. E., & Sarubio, M. G. (2002). Journal of Ethnopharmacology, 81(1), 57–63.
Acknowledgments
The authors are grateful to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado do Amazonas (FAPEAM) for the financial support of this research. ESL is member of the INCT of Processes Redox in Biomedicina-Redoxoma (MCT/CNPq). APAB received a grant from DCR/CNPq/FAPEAM. Thanks to Célio Maia Chaves from EMBRAPA for the Eugenia punicifolia plant material donation and to Jim Hesson of AcademicEnglishSolutions.com for proofreading the English.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Galeno, D.M.L., Carvalho, R.P., de Araújo Boleti, A.P. et al. Extract from Eugenia punicifolia is an Antioxidant and Inhibits Enzymes Related to Metabolic Syndrome. Appl Biochem Biotechnol 172, 311–324 (2014). https://doi.org/10.1007/s12010-013-0520-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-013-0520-8