
Abstract
Catalytic properties of two glucoamylases, AmyC and AmyD, without starch binding domains from Rhizopus oryzae strain 99-880 are determined using heterologously expressed enzyme purified to homogeneity. AmyC and AmyD demonstrate pH optima of 5.5 and 6.0, respectively, nearly one unit higher than the Rhizopus AmyA glucoamylase enzyme. Optimal initial activities are at 60 and 50 °C for AmyC and AmyD, respectively. Inactivation of both enzymes occurs at 50 °C following 30 min pre-incubation. The two enzymes demonstrate substantially slower catalytic rates toward soluble starch relative to AmyA. AmyC has similar k cat and K m for oligosaccharides to other Rhizopus and Aspergillus glucoamylases; however, the enzyme has a 2-fold lower K maltosem . AmyD has a 3-fold higher K m and lower k cat for maltooligosaccharides than AmyC and other glucoamylases. AmyC (but not AmyD) exhibits substrate inhibition. K i for substrate inhibition decreases with increasing length of the oligosaccharides. Data from pre-steady-state binding of AmyC to maltose and maltotriose and pre-steady-state to steady-state catalytic turnover experiments of AmyC acting on maltotriose were used to interrogate models of substrate inhibition. In the preferred model, AmyC accumulates an enzyme-maltose-maltotriose dead-end complex in the steady state.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glucoamylase (α-1,4-glucan glucohydrolase, amyloglucosidase; EC 3.2.1.3; GA) is of considerable importance to the fermentation and food industries for saccharification of starch and amylopectin. GA enzymes catalyze hydrolysis of α(1-4) glycosidic bonds to release d-glucose residues from the non-reducing ends of starch and oligosaccharides. GA also has limited ability to release d-glucose residues by promoting hydrolysis of α(1-6) linkages of amylopectin. Rhizopus and Aspergillus GA enzymes are perhaps the most studied GA enzymes due to their industrial importance. Kinetic studies and X-ray crystal structures of the catalytic domain of the enzyme from these two species, in addition to several other fungal species, have revealed salient aspects of GA catalysis and enzyme structure [1–6].
GA enzymes generally consist of three distinct regions; a starch binding domain (SBD), a catalytic domain (CD), and an O-glycosylated linker region that provides stability and linking of the SBD and CD domains. There are, however, a number of GA enzymes that only possess the catalytic domain. The SBD and CD domains are independent folding units with the SBD located at the N terminus in Rhizopus sp. and at the C terminus in Aspergillus species [7]. Partial or complete deletion of the SBD renders the enzymes incapable of binding or degrading raw starch, but hydrolysis of soluble starch and maltooligosaccharides remains. The active site of glucoamylase found in the CD comprises several conserved loops which contain most of the conserved amino acids found among GAs and may contain up to seven subsites for binding substrate according to substrate specificity determinations, with structural evidence for at least four subsites [3, 7].
The GA-catalyzed reaction proceeds via an acid-base mechanism with product glucose leaving the enzyme with inverted anomeric configuration implicating a single transition state. Steady-state kinetic constants, pH dependencies, and distances between amino acid residues are consistent with two glutamic acid residues serving as the acid and base catalysts in fungal GA enzymes [8]. In addition, pre-steady-state experiments monitoring intrinsic fluorescence implicate involvement of more than one tryptophan residue in the GA kinetic mechanism. Rates of intrinsic protein fluorescence change exhibit hyperbolic dependence on substrate concentration, consistent with the minimal 3-step reaction mechanism of Eq. 1 where E is enzyme, S is substrate, and P and Q are products [2, 4, 6].
The fluorescence change upon mixing enzyme and substrate is firmly established as reporting substrate binding events and not substrate hydrolysis [6]. The initial binding step, forming ES, is too rapid to monitor by stopped flow spectroscopy, whereas the second step, isomerization of ES to form *ES is readily measured. Conversion of ES and *ES, in which the substrate is considered correctly positioned for hydrolysis, causes quenching of intrinsic fluorescence [1, 2].
Much of the GA structure-function studies to date are on GA enzymes with SBD’s isolated from a number of fungal species; the exception is the crystal structure determination and biochemical characterization of the GA from Saccharomycopsis fibuligera [9, 10]. A number of additional GA enzymes and genes lacking SBD have been isolated from Aspergillus and Rhizopus oryzae species [11–13]. However, to date, the GA lacking a SBD from Aspergillus has not been biochemically characterized, and the AmyB enzyme from R. oryzae strain NRRL 395 has no apparent GA activity despite having high homology to the catalytic domain of the SBD-containing AmyA GA from the same strain [13]. Two additional genes encoding active GA enzymes lacking SBDs, amyC and amyD, have been isolated from R. oryzae strain 99-880 [12]. The two enzymes encoded, AmyC with 46% and AmyD with 48% identity to the Rhizopus AmyA catalytic domain, are actively transcribed with multiple carbon sources, including soluble starch [12]. Although there are some minor differences in gene expression of the two genes relative to the amyA gene, the role these two enzymes, or any of the GA enzymes without an SBD identified in Rhizopus and Aspergillus sp., play in overall carbohydrate metabolism is unclear. It is also not clear how prevalent the AmyC and AmyD enzymes are in Rhizopus; as previous work has demonstrated that AmyA is the predominant enzyme form at nearly 80% [14]. Until the relatively recent discovery of the additional Rhizopus GA genes, it was thought that the remainder of GA activity was attributable to GA isoforms resulting from proteolytic cleavage of the AmyA enzyme [14]. Rhizopus strain 99-880 underwent whole genome duplication, which may to some extent explain the existence of these two genes without an SBD [15]; but it does not explain their maintenance in the genome. Detailed biochemical studies of these enzymes may offer some clues to the biological significance and therefore the maintenance of these genes in the Rhizopus 99-880 genome.
In the work presented here, we recombinantly expressed the Rhizopus 99-880 AmyC and AmyD GA enzymes in Pichia pastoris since it would be difficult to ensure a pure form of the individual enzymes if isolated from the native host due to their similar molecular mass. We reveal catalytic properties of the AmyC and AmyD GA enzymes and show that the catalytic properties of these two enzymes demonstrate some differences from each other and from the more abundant AmyA enzyme.
Materials and Methods
GA Expression and Purification
P. pastoris strain X-33 was transformed with amyC or amyD cDNA as previously described [12]. Single colony isolates containing the desired construct were used to heterologously produce the respective AmyC and AmyD enzymes in 1 L fermentation cultures. Briefly, a frozen stock was used to inoculate a 250 mL baffled Erlenmeyer flask containing 50 mL buffered minimal glycerol medium which was then used to inoculate the fermentor containing 1 L of media after overnight growth [16]. Dissolved oxygen in the fermentation culture was maintained at 30%. Temperature was maintained at 30 °C, while pH was maintained at 5.5 with ammonium hydroxide (28%). The glycerol batch, glycerol fed-batch and transition to methanol phases were performed as previously described [16]. The methanol fed batch phase was carried out for 17 to 20 h with the methanol feed rate increased steadily over the first 10 h as described by the vendor (Invitrogen; Carlsbad, CA). Cultures were harvested by centrifugation and culture supernatant frozen at −80 °C for later use.
The GAs were purified from the culture supernatant by adjusting the pH to 7.0 before application to a His-Prep 16/10 IMAC column (GE Healthcare; Uppsala, Sweden) equilibrated with 50 mM sodium phosphate, 10 mM imidazole, pH 7.0. The column was washed with 3 column volumes of equilibration buffer and then eluted with 50 mM sodium phosphate, 250 mM imidazole, pH 7.0. Fractions containing activity were pooled, buffer exchanged, and concentrated into 50 mM succinate pH 5.5 using Biomax 10 K molecular weight cutoff centrifugal filters (Millipore, Bedford, MA). Protein concentrations were determined at 280 nm using extinction coefficients of εm = 8.17 × 104 M−1 cm−1 and εm = 8.63 × 104 M−1 cm−1 for AmyC and AmyD, respectively [17]. The GAs were deglycosylated with endoglycosidase H and PNGase F (New England Biolabs; Ipswich, MA) according to supplier instructions to determine if the enzymes were glycosylated in the Pichia expression system. The purity, molecular mass and extent of glycosylation of the enzymes were assessed by SDS-PAGE using 12% gels (BioRad; Hercules, CA). Deglycosylated enzymes were not used for determination of kinetic parameters.
Temperature and pH Optima Assays
pH optima and stabilities for both GA enzymes were determined by measuring activity of 3 μg enzyme acting on 1% soluble starch (Sigma; St. Louis, MO) in the following buffers set to constant ionic strength [I = 300 mM] adjusted with NaCl: 100 mM citrate, pH 3.0; 100 mM succinate, pH 3.5–6.0; 100 mM sodium phosphate, pH 6.5–7.5; and 30 mM sodium pyrophosphate, pH 8.0. For the pH stability assays, the enzyme was incubated without substrate for 30 min in buffers listed above ranging from pH 3–8 at 30 °C. Care was taken to ensure that the pH of the activity assay buffer, 100 mM succinate (I = 300 mM) was not altered from the desired pH of 5.5 (AmyC) or 6.0 (AmyD) upon addition of preincubated enzyme to the assay. The pH stability and optima assays were performed at 40 °C in a final volume of 550 μL. For both stability and optima assay sets, the assay was started with the addition of enzyme and 120 μL aliquots removed at desired time points and quenched with 20 μL 2 M HCl. The maximum duration of the activity assay was 5 min with a minimum of four time points used to calculate the initial rate. Assay sets were performed in triplicate. Product, glucose, was detected using a glucose oxidase reagent kit (Megazyme; Wicklow, Ireland) according to supplied instructions and quantified using a standard curve with glucose as the standard.
Temperature optima and stabilities were determined in a manner similar to the pH assays, with detection of glucose using the glucose oxidase method. Optima reactions (550 μL) contained 1% soluble starch in 100 mM succinate [I = 300 mM] pH 5.5 (buffer SI5.5) for AmyC and 100 mM succinate [I = 300 mM] pH 6.0 (buffer SI6.0) for AmyD at temperatures ranging from 20 to 80 °C. For temperature stability determinations, enzyme was incubated without substrate in buffer SI5.5 (AmyC) or buffer SI6.0 (AmyD) at temperatures ranging from 20 to 80 °C for 30 min, placed on ice for 1 min, and assayed to determine the remaining activity using 3 μg enzyme acting on 1% soluble starch as substrate at 40 °C and buffer SI5.5 or buffer SI6.0 for AmyC and AmyD, respectively. A minimum of four time points were used to obtain the initial rate and ensure linearity over the course of the assay.
Steady-state Kinetics
Initial reaction rates with soluble starch and the maltooligosaccharides, maltose through maltohexaose (DP2 to DP6; Sigma, St. Louis, MO), were determined by monitoring the release of glucose using the glucose oxidase method outlined above. Reactions contained a minimum of eight substrate concentrations ranging from 0.15(K m) to 40(K m) and enzyme concentrations ranging from 18 to 250 nM in buffer SI5.5 (AmyC) or buffer SI6.0 (AmyD) at 40 or 8 °C. The amount of enzyme used and time points collected (minimum of four) were such that activity was linear over the course of the assay and less than 10% of the substrate was consumed. Kinetic parameters were determined by fitting the initial rate data to Eq. 2 (no substrate inhibition) and Eq. 3 (with substrate inhibition) using the computer program GraFit [18]: v is initial velocity, E t is total enzyme concentration, k cat is maximum rate of catalysis, S is substrate, K m is Michaelis constant, and K i(app) is apparent inhibitor dissociation constant. Rate parameters, v/E t and k cat, are expressed in moles glycoside bonds broken per second per mole enzyme active sites (s−1).
Reaction progress curves of the enzymes acting on maltotriose were performed at 8 °C. For AmyC reactions, 1 μM enzyme was incubated with 2.8 mM maltotriose in buffer SI5.5. At selected time points, aliquots of the reaction were quenched with HCl to pH 1.5 and analyzed for glucose, maltose, and maltotriose by HPLC with pulsed amperometric detection as described for d-xylose [19]. Reaction progress curves for AmyD were generated in a similar manner, except that 3.5 μM of enzyme was used and incubated with 2.8 mM maltotriose in buffer SI6.0.
Pre-steady-state Kinetics
Pre-steady-state binding experiments were performed by measuring the intrinsic protein fluorescence using an Applied Photophysics SX.18MV-R stopped-flow spectrofluorometer (Applied Photophysics; Surrey, UK). Reactions contained buffer SI5.5 at 8 °C and a final enzyme concentration of 13 μM. Maltose and maltotriose concentrations ranged from 0.05 to 60 mM. Excitation wavelength was 280 nm and emission of the reaction was monitored above 320 nm. Data of time-dependent intrinsic fluorescence were fitted to the single exponential decay of Eq. 4 using PC Pro-K global analysis software (Applied Photophysics; Surrey, UK): F obs is molar fluorescence of enzyme at a specific substrate concentration and time t, A is molar fluorescence amplitude of the reaction, k obs is the observed first order rate constant, and C is the endpoint molar fluorescence of enzyme at a particular substrate concentration.
Initial fluorescence (F init) was determined from reactions in absence of substrate. ΔF represents the overall fraction change in fluorescence at equilibrium caused by incubating enzyme with substrate as calculated through Eq. 5.
ΔF max of Eq. 6 is the maximum ΔF occurring at saturating S and \( {K_d} = \left[ {\hbox{E}} \right]\left[ {\hbox{S}} \right]/\left( {\left[ {\hbox{ES}} \right] + \left[ { * {\hbox{ES}}} \right]} \right) \) is the indicated dissociation constant.
Rate constants were determined from fitting k obs data to Eq. 7 where k on is the rate for ES isomerization to *ES of Eq. 1, K 1 (k -1/k 1) is the rapid equilibrium dissociation constant of S from ES of Eq. 1, and k off is the rate of isomerization of *ES to ES of Eq. 1.
The pre-steady-state to steady-state progression of maltotriose hydrolysis was performed using a rapid quenched flow apparatus model RQF-4 (KinTek; Austin, TX). Contents of one loop, holding 15 µL of 180 µM AmyC in buffer SI5.5 at 8 °C, were mixed with the contents of a second loop, holding 15 µL 5.6 mM maltotriose in buffer SI5.5 at 8 °C. Reactions, at times after mixing of 4 ms to 3 s, were quenched by lowering the pH to 1.5 with HCl. Quenched samples were analyzed for glucose by HPLC with amperometric detection as described for d-xylose [19]. Glucose production versus time data were fitted through KinTek Explorer [20] as described in the text.
Results and Discussion
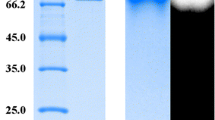
AmyC and AmyD were heterologously expressed in 1 L fermentations followed by purification from their respective culture supernatants in a single step via the C-terminal His-Tag using nickel chelate affinity chromatography. The purified enzymes were homogenous as judged by SDS-PAGE, with AmyC migrating near the expected molecular mass of 52 kD and AmyD slightly below the expected molecular mass of 51 kD (Fig. 1). Although the AmyD enzyme appears to have a mass near 47 kD on the SDS-PAGE gel, matrix assisted laser desorption ionization-time of flight spectrometry gave a molecular mass of 52.3 kD for the AmyD enzyme as produced (data not shown). Both enzymes were glycosylated as treatment with endoglycosidase H and PNGase F resulted in slight reductions in molecular mass (Fig. 1). Glycosylation of the Rhizopus GAs in the Pichia system is not unexpected as an Aspergillus awamori GA previously shown to be glycosylated when isolated from the native species also resulted in a glycosylated enzyme when recombinantly expressed in P. pastoris [21]. AmyC and AmyD have two putative N-glycosylation sites each, with one of the glycosylation sites identical in the two enzymes, as well as conserved among the Rhizopus AmyA and Aspergillus GAs based on sequence alignment (data not shown). The other putative glycosylation sites are a short distance apart and are not common to AmyA or the Aspergillus GAs. Direct measurement to determine location and extent of glycosylation of the two recombinant GA enzymes was not pursued.

SDS-PAGE analysis of Pichia-expressed AmyC and AmyD. Purified enzyme (3 μg) and enzyme treated with either endoglycosylase H or PNGase F per manufacturer's instructions (3 μg), were run on a 12% SDS-PAGE gel. Lane 1, rainbow marker; lane 2, AmyC, untreated; lane 3, AmyC, PNGase treated; lane 4, AmyC Endo H treated; lane 5, PNGase only; lane 6, AmyD, untreated; lane 7, AmyD, PNGase treated; lane 8, AmyD, Endo H treated
The pH optimum curve for AmyC activity shows a peak at pH 5.5, while optimal pH range is somewhat broader for AmyD, ranging from pH 5.5 to 6.5 (Fig. 2). pH optima for AmyC and AmyD are about one pH unit above previously characterized Rhizopus AmyA GAs which are reported to be optimal at pH 4.5 to 5.0 [14, 22, 23] as well as a number of Aspergillus SBD-containing GA enzymes that were found to be optimal between pH 3.5 and 5 [24]. Both enzymes are reasonably stable across a pH range of 4–8, however; AmyD demonstrates greatly reduced stability below pH 4 (Fig. 2). Temperature stabilities of the two enzymes are similar to that previously reported for the AmyA GA [14, 23] with both enzymes stable for 30 min up to 40 °C, followed by a steep decline in stability above 40 °C (Fig. 3). AmyC demonstrates optimal activity at 60 °C, similar to that found for the Rhizopus AmyA GA enzyme [23], however; the enzyme is only stable at this temperature for about 5 min, the duration of the assay as performed, since incubation at this temperature for 30 min results in a completely inactive enzyme (Fig. 3). The same temperature optima trend applies to AmyD with optimal activity somewhat lower at 50 °C for the 5 min duration of the assay, but completely inactive after 30 min incubation at 50 °C (Fig. 3).

pH stabilities and optima of Pichia-expressed AmyC and AmyD. pH stabilities of AmyC and AmyD were determined by pre-incubating the enzymes for 30 min at the indicated pH and 25 °C followed by activity assays (1% soluble starch) at 40 °C and pH 5.5 (AmyC) or pH 6.0 (AmyD). pH optima of AmyC and AmyD were determined by performing activity assays (1% soluble starch) at the indicated pH and 40 °C. For the pH optima, 100% activity corresponds to rate of 45.3 and 31.4 s−1 for AmyC and AmyD, respectively

Temperature stabilities and optima of AmyC and AmyD. Temperature stabilities of AmyC at pH 5.5 and AmyD at pH 6.0 were determined by pre-incubating the enzymes for 30 min at 20 to 80 °C followed by activity assays (1% soluble starch) at 40 °C. Temperature optima of AmyC and AmyD were determined by performing activity assays (1% soluble starch) at temperatures ranging from 20 to 80 °C. For temperature optima, 100% activity corresponds to rate of 94.9 and 49.7 s−1 for AmyC and AmyD, respectively
The reaction progression of AmyC, acting on maltotriose, indicates similar rates of glucose and maltose accumulation, initially, followed by greater rates of glucose accumulation than maltose accumulation as maltose is acted on by the enzyme (Fig. 4). Similar results are found with AmyD (Electronic supplementary material Fig. 1). The pattern is fully consistent with the enzymes acting on substrate nonprocessively, catalyzing hydrolysis of a single glycosidic bond followed by release of products before initiating another catalytic cycle. Therefore, nonprocessivity is established and it is taken into account in our analysis of steady-state initial-rate data and determined parameters.

Reaction progress curve of 1 µM AmyC acting on 2.8 mM maltotriose at pH 5.5 and 8 °C. At indicated time points, aliquots of the reaction were quenched with HCl to pH 1.5 and analyzed by HPLC with amperometric detection
Steady-state kinetic parameters were determined, using initial-rate data, for the enzymes acting on soluble starch and a series of oligosaccharides (DP2-DP6) to more directly compare with previously characterized glucoamylases. With soluble starch as the substrate, AmyC shows a k cat of 58.2 ± 2.0 s−1 and a K m of 1.39 ± 0.11 mg/ml. AmyD demonstrated a lower affinity towards soluble starch than AmyC with a K m of 2.11 ± 0.15 mg/ml and a slower rate of catalysis with a k cat of 35.1 ± 1.6 s−1. AmyC and AmyD are not nearly as effective towards soluble starch hydrolysis as the SBD-containing AmyA isolated from R. oryzae strain NRRL 395 [23] with a reported k cat value of 152 s−1. The K m value obtained for AmyC is comparable to AmyA, reported to be 1.34 mg/ml [23], while the AmyD K m is nearly 2-fold higher.
With regard to the oligosaccharide series, DP2–DP6, AmyD follows the trend of decreasing K m and increasing k cat with increasing oligomer size (Table 1), as recognized with other fungal GAs. k cat increases and K m decreases from DP2 through DP5 for AmyC. k cat/K m values of AmyC acting on DP2 through DP6 suggest five subsites, while those of AmyD suggest six or more subsites. AmyD has an approximately 6-fold higher K m and nearly 2-fold lower k cat for the oligosaccharide substrates in comparison to AmyC and the previously characterized Rhizopus AmyA GA [1, 25]. As a result, the second order rate constant, k cat/K m, is substantially less than that of AmyC and the previously characterized Rhizopus and Aspergillus GAs [1, 21, 25]. AmyC has k cat and K m values similar to the previously characterized Aspergillus GAs [21], however AmyC has 2-fold lower K m and similar k cat towards maltose but 2-fold higher K m and similar k cat values towards DP3-DP6. A similar pattern emerges in comparing AmyC with the reported kinetic constants for Rhizopus AmyA [25], except the k cat values are much higher for AmyC, leading to much higher k cat/K m values. Relative k cat/K m values (AmyC/AmyD) decrease with the progression (DP2, 10.9), (DP3, 10.8), (DP4, 9.7), (DP5, 7.0), and (DP6, 4.0).
AmyC is subject to substrate inhibition by the maltooligosaccharide substrates (Table 1, Fig. 5). At 40 °C, as substrate concentration increases above K m, AmyC-catalyzed hydrolysis activity is noticeably inhibited by oligosaccharides DP3 to DP6 as substrates. There is no detectable substrate inhibition by maltose or, perhaps surprisingly, soluble starch since inhibition increases with longer maltooligosaccharide chain length. For DP3 to DP6, K i(app) values are 54- to 96-fold higher than the corresponding K m values. At 8 °C, AmyC exhibits substrate inhibition by DP2, DP3, and DP6, and the respective K i(app) values are 132-, 18-, and 22-fold higher than the corresponding K m values. Clearly, inhibition is more pronounced at the lower temperature. Optimum substrate concentrations for producing maximum observable velocities can be calculated from the first derivative of Eq. 3. The substrate concentration where the maximum observable velocity occurs simplifies to the relationship: (K m*K i(app))0.5. Maximum observable velocities are calculated by substitution into Eq. 3. Calculated values are listed in Table 1. Notably, for substrates DP3 through DP6 at 40 °C, optimum substrate concentrations are 0.10- to 0.14-fold the corresponding K i(app) values and 7.4- to 9.8-fold the corresponding K m values. Maximum observable velocities (v mo) are 0.79- to 0.83-fold the corresponding k cat values. At 8 °C, for DP3 and DP6, optimum substrate concentrations are respectively 0.24- and 0.22-fold the corresponding K i(app) values and 4.2- and 4.7-fold the corresponding K m values. Maximum observable velocities (v mo) are 0.68- to 0.70-fold the corresponding k cat. There was no indication of inhibition of AmyD under similar reaction conditions at 40 °C. It has been noted in previous reports that very high substrate concentrations can lead to inhibition of GAs [26]; however, substrate inhibition was not analyzed and K i values were not reported. High substrate concentrations have been employed to study reversion reactions catalyzed by certain GAs [27]. Production of higher order DPs was not detected in reactions of AmyC acting on 20 mM DP3 at 8 °C and pH 5.5 and monitored by HPLC with amperometric detection (data not shown), indicating that reversion reactions do not contribute to the observed substrate inhibition.

Dependence of AmyC activity on maltotriose concentration. Steady-state kinetic reactions were at 40 °C (empty circles) and 8 °C (filled squares) in 100 mM succinate (I = 300 mM), pH 5.5. Glucose was monitored by the glucose oxidase method to determine initial rates. Curves are drawn from fits of the initial rate data to Eq. 3. Determined parameter values are listed in Table 1
In an effort to gain more understanding of the apparent substrate inhibition, we chose the two models of Eqs. 8 and 9, which include substrate inhibition, to examine with experimental data of AmyC (or AmyD) acting on maltotriose. In the model of Eq. 8, substrate inhibition occurs when two equivalents of substrate bind to the same active site to form *ESS as the dead-end complex. In the model of Eq. 9, substrate inhibition occurs due to accumulation of an enzyme-products intermediate (*EPQ) in the steady-state, which can bind substrate to form *EPQS as the dead-end complex. In the models, S is maltotriose, P is glucose and Q is maltose. Glucose binds weakly to the enzyme and its release from the enzyme is considered to be rapid [6]. Therefore, the rate of release of glucose (P) is encompassed in the rate constant for maltose (Q) release.
To obtain experimental data for the models, we first set out to determine substrate binding rates by stopped-flow experiments monitoring intrinsic fluorescence as has been done for other GAs [1, 2, 4, 25, 26, 28, 29]. Stopped-flow reactions were run at 8 °C since inhibition is more severe at the lower temperature and because stopped-flow reaction rates of substrate binding were too fast for accurate determination at higher temperature (e.g., 25 °C). Binding of maltose and maltotriose to AmyC exhibits a single exponential kinetic of decline in intrinsic fluorescence as demonstrated by the reaction of 0.5 mM maltotriose with AmyC at pH 5.5 and 8 °C (Fig. 6). In the mixing time of the reaction apparatus (1 ms) there was a 3.8% decline of intrinsic fluorescence. In the observable time of the binding reaction there was a further 16.7% decline of intrinsic fluorescence. This is consistent with a rapid initial binding phase followed by a slower isomerization of ES to *ES in accord with Eqs. 1, 8, and 9. Similarly conducted binding reactions of AmyD with maltose (and maltotriose) at pH 6.0 and 8 °C exhibit a very different kinetic pattern with a 20 ms lag phase with little change of intrinsic fluorescence followed by an exponential phase of small amplitude (data not shown). The latter kinetic pattern is suggestive of two opposing changes of intrinsic fluorescence upon binding substrate, which overlap transiently to produce the lag through cancellation. The observable changes are too small to pursue until large amounts of AmyD become available or until a superior method is developed.

Kinetic of maltotriose binding to AmyC at pH 5.5 and 8 °C. An aliquot (50 µL) of AmyC (26 μM) was mixed with an equal volume of maltotriose (1 mM) in 100 mM succinate [I = 300 mM], pH 5.5 at 8 °C. Internal fluorescence (excitation 280 nm) was monitored above 320 nm
Binding reactions of AmyC containing several different concentrations of maltose provided data to determine binding rates and equilibrium constants. In Fig. 7a, ΔF, the fraction of intrinsic fluorescence quenched upon binding maltose, is plotted versus maltose concentration. Fitting the data to Eq. 6 provides K maltosed = 0.127 ± 0.009 mM (Table 2). In Fig. 7b, k maltoseobs , the rate constant for the observable quenching of intrinsic fluorescence, is plotted versus maltose concentration. Clearly, the binding data fit the hyperbola of Eq. 7 instead of a line, the latter of which would be seen in single step binding. Fitting the data to Eq. 7 provides values for k maltoseon and k maltoseoff (k -4 and k 4 of Eqs. 8 and 9) and K maltoseinitial (k 5/k -5 of Eqs. 8 and 9), which are listed in Table 2.

Dependence of AmyC intrtinsic fluorescence decrease and observed first-order rate constant on maltose concentration at pH 5.5 and 8 °C. Reactions contained AmyC (13 μM) and varied maltose concentrations in 100 mM succinate pH 5.5 (I = 300 mM). Internal fluorescence (excitation 280 nm) was monitored above 320 nm. Reactions were run in triplicate for each maltose concentration. Standard errors are shown. a ΔF (%) versus maltose concentration. Percent change in fluorescence, ΔF (%), was calculated through Eq. 5. The curve is drawn from fitting the data to Eq. 6. Determined parameters are indicated and listed in Table 2. b k obs versus maltose concentration. k obs values were determined by fitting the data to Eq. 4. The curve is drawn from fitting the data to Eq. 7. Determined parameters are indicated and listed in Table 2
Binding reactions of AmyC to several different concentrations of maltotriose at pH 5.5 and 8 °C provided data to analyze through Eqs. 6 and 7 (Fig. 8). Determined binding parameters (K 1, k 2, k -2) are listed in Table 2. Notably, values for k maltotrioseon (k 2 = 131 s−1) and k maltoseon (k -4 = 295 s−1) are substantially larger than k maltotriosecat (4.91 s−1) and k maltosecat (1.31 s−1) values of AmyC acting on the substrates at 8 °C, supporting the idea that fluorescence quenching reports binding events rather than substrate hydrolysis as has been clearly established for the Aspergillus niger GA [6]. k on values are 5-fold (maltose) to 10-fold (maltotriose) lower than those reported for Rhizopus and Aspergillus sp. GAs [1, 2, 4, 25, 26, 28, 29]. In addition, k off/k on of AmyC binding maltose is similar to that of previously characterized GAs, however, this ratio is 4 to 7-fold higher for binding maltotriose than those of the other GAs [1, 2, 4, 25, 26, 28, 29]. The k on values demonstrate that isomerization from ES to *ES occurs slower for AmyC than for previously characterized GAs, and it is not immediately clear why AmyC is substantially slower at this step of the reaction pathway. One possibility lies in the Trp120 loop that has been implicated in substrate binding in the Aspergillus GAs [28]. AmyC does possess the conserved Trp residue, but there are a number of amino acid differences in this loop, with the most prominent being a proline residue rather than a serine residue immediately upstream of the conserved Trp. Proline residues tend to create rigidity in the structure and this residue could impede the mobility of the loop, which may be needed for substrate binding. It should also be noted that similar to the Aspergillus W120F mutant, the k on (k 2) rates for AmyC decrease with substrate length, and is the opposite of wild-type Aspergillus enzyme, where k on increases with substrate length [2].

Dependence of AmyC internal fluorescence decrease and observed first-order rate constant on maltotriose concentration at pH 5.5 and 8 °C. Reactions contained AmyC (13 μM) and varied maltotriose concentrations in 100 mM succinate pH 5.5 (I = 300 mM). Intrinsic fluorescence (excitation 280 nm) was monitored above 320 nm. Reactions were run in triplicate for each maltotriose concentration. Standard errors are shown. a ΔF (%) versus maltotriose concentration. Percent change in fluorescence, ΔF (%), was calculated through Eq. 5. The curve is drawn from fitting the data to Eq. 6. Determined parameters are indicated and listed in Table 2. b k obs versus maltotriose concentration. k obs values were determined by fitting the data to Eq. 4. The curve is drawn from fitting the data to Eq. 7. Determined parameters are indicated and listed in Table 2
Our next step in the analysis of AmyC inhibition was to test our binding parameters against experimental progress curve data of 90 µM AmyC acting on 2.8 mM maltotriose at pH 5.5 and 8 °C. Reactions were conducted in a quenched flow apparatus with samples quenched with acid from time 4 ms to 3 s. Concentrations of d-glucose were determined in the quenched samples and plotted in the progress curve of Fig. 9. The data of glucose produced in the reaction versus time clearly exhibit a burst phase followed by a linear phase (burst to steady-state) (Electronic supplementary material Fig. 2). To fit the glucose produced versus time data in KinTek Explorer, we used the binding parameters listed in Table 2 (K 1, K 5, k 2, k -2, k 4, and k -4). Because the reaction proceeded into the steady-state where some of the maltose produced from maltotriose serves as substrate, it is necessary to include Eq. 10 with Eq. 8 or Eq. 9 to account for maltose as substrate in fitting of the data.

Progress curve of 90 µM AmyC acting on 2.8 mM maltotriose at pH 5.5 and 8 °C. Glucose concentrations of quenched flow samples were determined by HPLC with amperometric detection. The glucose concentration at time zero is attributed mainly to glucose release from the glycosylated AmyC upon HCl quenching. The solid curve is drawn from the fit of the data to the models of Eqs. 9 and 10 through KinTek Explorer. The dashed curve is drawn from the fit of the data to the models of Eqs. 8 and 10. Values from Table 2 were used as inputs. Fitted parameter values are indicated
The value of k 6 was taken as equal to k maltosecat (1.31 s−1). The fit was charged with determining values for the parameters k 3 and K maltotriosei . First the data were fitted to Eqs. 8 and 10 (dashed curve of Fig. 9). The resulting fit is unsatisfactory because the fitted curve ignores the burst phase of the reaction and K maltotriosei is very large. The fit of the data to Eqs. 9 and 10 is satisfactory (solid curve of Fig. 9). The fitted curve follows the data and the determined values for K maltotriosei and k 3 are reasonable (Table 2). The large error associated with the value of K maltotriosei (2.86 ± 37) can be lowered by calculating the value from the relationship of Eq. 11 where K maltotriosei(app) (6.2 ± 2.1 mM) is the substrate inhibition constant determined by steady-state initial-rate data collected at 8 °C (Table 1). The calculated K maltotriosei value is 3.17 ± 1.15 mM (Table 2).
In addition to the above, models of Eqs. 8 and 9 were modified by eliminating the step defined by k 4 and k -4, making the release of products rapid. When combined with the unmodified Eq. 10, the models were fitted, as before, through KinTek Explorer. These fits also performed poorly in describing the data of AmyC acting on maltotriose (Electronic supplementary material Fig. 3), as the fits again ignore the burst phase.
Clearly, the model of Eqs. 9 and 10 provide the best fit to the progress data of AmyC acting on maltotriose. The steady-state velocity equation of AmyC acting on maltotriose according to the model of Eq. 9, in the absence of products, is described by Eq. 12 (derived in Electronic supplementary material). Associated values for k cat and K m are described by Eqs. 13 and 14, respectively.
Using the microscopic kinetic and equilibrium constants of Table 2 as inputs for Eq. 13, the calculated k cat is 5.03 ± 0.65 s−1. This compares with the value of 4.91 ± 0.61 s-1 determined from steady-state data (Table 1, Fig. 5). The K m, calculated from Eq. 14, is 0.098 ± 0.001 mM. This compares with the value of 0.347 ± 0.090 mM, determined from steady-state data (Table 1, Fig. 5). Differences between parameter values calculated through Eqs. 13 and 14, and those calculated through Eq. 3 may be attributed to the sensitivity of Eq. 3 fits to small differences in initial-rate data. For example, predicted initial velocities of AmyC using Eq. 12 and parameter values of Table 2 appear quite similar to the observed values (Electronic supplementary material Fig. 4), yet the determined parameters from fitting the data to Eq. 3 differ as indicated above.
Microscopic and steady-state rate constants have been reported for pre-steady state binding and steady-state catalysis reactions conducted at 8 °C for the GA from A. niger acting on maltotriose and maltose [2]: using the same definitions as those of Eq. 9, K maltotriose1 = 2.0 mM, k maltotriose_on2 = 1630 s-1, k maltotriose_off-2 = 23 s-1, k maltose_off4 = 33 s-1, k maltose_on-4 = 1170 s-1, k maltotriosecat = 1.75 s-1, and K maltotriosem = 0.057 mM. The microscopic kinetic constant, k maltotriose3 , was not determined, but it can be calculated from the other rate constants (k maltotriose_on2 , k maltotriose_off-2 , k maltose_off4 , k maltose_on-4 ) and Eq. 13 as k maltotriose3 = 1.9 s-1. If the A. niger GA had the same intrinsic K maltotriosei as AmyC (2.86 mM, Table 2), then its value for K maltotriosei(app) would equal approximately K maltotriosei (1+k 4/k 3) or 53 mM, 930-fold larger than its K maltotriosem , and considerably higher than the K maltotriosei(app) = 6.2 mM (13-fold K maltotriosem ) determined for AmyC. Substrate inhibition by maltotriose has not been reported for the A. niger GA.
Conclusions
There are three GAs encoded by the genome of R. oryzae 99-880. One of these, AmyA, is equipped with a starch binding domain, giving it an advantage over the other two GAs, AmyC and AmyD, which lack SBDs, in acting on insoluble starch. The two GAs without SBDs have similar pH and temperature profiles which differ from the AmyA profiles. The catalytic rates of AmyC and AmyD toward soluble starch are considerably lower than that of AmyA and activity of all three enzymes toward maltooligosaccharide substrates demonstrate considerable differences. AmyC has higher k cat and lower K m values than AmyD for the five maltooligosaccharides examined (DP2 to DP6). This advantage of AmyC over AmyD dissipates to some extent with increasing length and concentration of substrate, the latter because AmyC is subject to substrate inhibition for substrates DP3 and larger, whereas AmyD is not. AmyC is not inhibited when soluble starch is the substrate and has a higher affinity for maltose, suggesting that this enzyme’s interaction with substrate differs from previously characterized GA enzymes. The catalytic properties of these two enzymes, while different from AmyA, make it difficult to discern exactly how these two GA enzymes increase the fitness of this Rhizopus species. There are a number of possible roles that can be suggested, however. It is possible that the AmyC/D enzymes play a much greater role in the absence of starch and the presence of oligosaccharides, low carbon availability, or perhaps, under other stress conditions as suggested for the Aspergillus GlaB enzyme [11]. Despite the unsettled role of the AmyC and AmyD enzymes, substrate inhibition exhibited by AmyC does afford an additional means to grasp the kinetic profile of GAs.
References
Hiromi, K., Ohnishi, M., & Tanaka, A. (1983). Molecular and Cellular Biochemistry, 51, 79–95.
Olsen, K., Christensen, U., Sierks, M. R., & Svensson, B. (1993). Biochemistry, 32, 9686–9693.
Aleshin, A. E., Firsov, L. M., & Honzatko, R. B. (1994). Journal of Biological Chemistry, 269, 15631–15639.
Christensen, U., Olsen, K., Stoffer, B. B., & Svensson, B. (1996). Biochemistry, 35, 15009–15018.
Sierks, M. R., & Svensson, B. (1996). Biochemistry, 35, 1865–1871.
Christensen, U. (2000). Biochemical Journal, 349, 623–628.
Coutino, P. M., & Reilly, P. J. (1997). Proteins, 29, 334–347.
Sauer, J., Sigurskjold, B. W., Christensen, U., Frandsen, T. P., Mirgorodshaya, E., Harrison, M., et al. (2000). Biochimica et Biophysica Acta, 1543, 275–293.
Ševčík, J., Solovicová, A., Hostinová, E., Gašperik, J., Wilson, K. S., & Dauter, Z. (1998). Acta Crystallographica Section D Biological Crystallography, 54, 854–866.
Solovicová, A., Christensen, T., Hostinová, E., Gašperik, J., Ševčík, J., & Svensson, B. (1999). European Journal of Biochemistry, 264, 756–764.
Ishida, H., Hata, Y., Ichikawa, E., Kawato, A., Suginami, K., & Imayasu, S. (1998). Journal of Fermentation and Bioengineering, 86, 301–307.
Mertens, J. A., & Skory, C. D. (2006). Current Microbiology, 54, 462–466.
Mertens, J. A., & Skory, C. D. (2007). Enzyme and Microbial Technology, 40, 874–880.
Takahashi, T., Tsuchida, Y., & Irie, M. (1978). Journal of Biochemistry, 84, 1183–1194.
Ma, L., Ibrahim, A. S., Skory, C., Grabherr, M. G., Burger, G., Butler, M., et al. (2009). PLoS Genetics, 5(7), e1000549. doi:10.371/journal.pgen.1000549.
Zhang, W., Liu, C., Inan, M., & Meagher, M. M. (2004). Journal of Industrial Microbiology and Biotechnology, 31, 330–334.
Gill, S. C., & von Hippel, P. H. (1989). Analytical Biochemistry, 182, 319–26.
Leatherbarrow, R. J. (2001). Grafit Version 5. Horley: Erithacus Software Ltd.
Jordan, D. B. (2008). Applied Biochemistry and Biotechnology, 146, 137–149.
Johnson, K. A., Simpson, Z. B., & Blom, T. (2009). Analytical Biochemistry, 387, 20–29.
Fierobe, H. P., Mirgorodskaya, E., Frandsen, T. P., Roepstorff, P., & Svensson, B. (1997). Protein Expression and Purification, 9, 159–170.
Wang, H. L. (1988). Biotechnology and Applied Biochemistry, 10, 191–200.
Yu, Y., & Hang, Y. D. (1991). Food Chemistry, 40, 301–308.
Norouzian, D., Akbarzadeh, A., Scharer, J. M., & Young, M. M. (2006). Biotechnology Advances, 24, 80–85.
Hiromi, K., Nitta, Y., Numata, C., & Ono, S. (1973). Biochimica et Biophysica Acta, 302, 362–375.
Natarajan, S., & Sierks, M. R. (1996). Biochemistry, 35, 15269–15279.
Pazur, J. H., Cepure, A., Okada, S., & Forsberg, L. S. (1977). Carbohydrate Research, 58, 193–202.
Natarajan, S., & Sierks, M. R. (1996). Biochemistry, 35, 3050–3058.
Olsen, K., Svensson, B., & Christensen, U. (1992). European Journal of Biochemistry, 209, 777–784.
Acknowledgments
We thank Michael Bowman for MALDI-TOF analysis of the expressed AmyC and AmyD proteins.
Author information
Authors and Affiliations
Corresponding author
Additional information
The mention of firm names or trade products does not imply that they are endorsed or recommended by the U.S. Department of Agriculture over other firms or similar products not mentioned.
An erratum to this article can be found at http://dx.doi.org/10.1007/s12010-010-9030-0
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 495 kb)
Rights and permissions
About this article
Cite this article
Mertens, J.A., Braker, J.D. & Jordan, D.B. Catalytic Properties of Two Rhizopus oryzae 99-880 Glucoamylase Enzymes Without Starch Binding Domains Expressed in Pichia pastoris . Appl Biochem Biotechnol 162, 2197–2213 (2010). https://doi.org/10.1007/s12010-010-8994-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-010-8994-0