
Abstract
Purpose of review
This review aims to provide a summary of the pathophysiology, clinical presentation and management options for facioscapulohumeral dystrophy (FSHD). We discuss current management options and delve into updates about developments in targeted therapy.
Recent findings
New breakthroughs in FSHD research have led to a further understanding of aberrant DUX4 protein expression in the underlying pathophysiology of FSHD. This has paved the way for the development of targeted therapies aimed at targeting DUX4 expression or its downstream effects. Therapeutic strategies for FSHD primarily target DUX4 through three main avenues: small molecules, antisense oligonucleotide therapeutics and CRISPR-based approaches. This review discusses these strategies further. Presently, all prospective targeted therapies are in the pre-clinical phase, except for losmapimod, which is currently undergoing a phase 3 clinical trial.
Summary
Given the absence of approved disease-modifying treatments for FSHD, the primary approach for management currently involves multidisciplinary supportive measures which are limited. Recent developments in the form of targeted therapies and strategies for the definitive treatment of FSHD indicate a promising era.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Facioscapulohumeral dystrophy (FSHD) is a genetically acquired condition that is characterised by gradually progressive asymmetrical muscle weakness of the face, scapular region, upper limbs (humeral) and distal lower limbs (peroneal) [1]. It is the third most common adult-onset muscular dystrophy, and the estimated prevalence of FSHD is approximately 4 to 12 cases per 100,000 individuals [2,3,4]. At present, there is no disease-modifying treatment for FSHD. However, ongoing research in epigenetics has led to a deeper understanding of the underlying pathogenesis of FSHD, spurring the identification of potential therapeutic targets. In this article, we discuss the pathogenesis, clinical features and diagnosis of FSHD and review current management strategies as well as potential therapeutics for patients with FSHD.
Pathogenesis
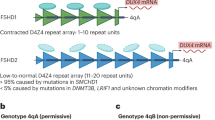
FSHD can be classified into 2 subtypes: FSHD1 and FSHD2. FSHD1 accounts for 95% of cases, whereas FSHD2 makes up the remaining 5% [2]. Both subtypes are clinically indistinguishable and arise due to inappropriate expression of the double homeobox protein 4 (DUX4) gene in the skeletal muscles [5••]. DUX4 encodes for a transcription factor that is involved in the regulation of genes for pre- and post-implantation embryogenesis [6•]. It is typically epigenetically suppressed in most somatic cells, except in the thymus and testis [7, 8]. When expressed in skeletal muscles, it can induce downstream effects like cell death, oxidative stress, inflammation and disrupted myogenesis, leading to the development of FSHD [6•, 9, 10].
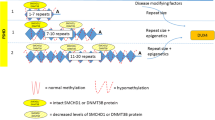
The DUX4 gene lies within a macrosatellite repeat array that comprises 3.3 kb D4Z4 repeat units, in the subtelomeric region of chromosome 4 at 4q35 [8, 11, 12]. In healthy individuals, this array is made up of 11–100 D4Z4 repeat units, which are normally highly methylated and exist as euchromatin in most cells. In FSHD1, this array is contracted to 1–10 repeat units [8, 13,14,15]. Contraction of D4Z4 repeat arrays leads to hypomethylation and chromatin relaxation, facilitating inappropriate DUX4 expression [5••, 16]. Additionally, expression of DUX4 requires polyadenylation of the DUX4 transcript, which only occurs with the 4qA but not the 4qB haplotype. Hence, FSHD is manifested in individuals with D4Z4 repeat contractions of 1–10 units on the permissive 4qA haplotype.
FSHD1 is inherited via an autosomal dominant pattern [17], although 10 to 30% of FSHD1 cases exist due to sporadic occurrences, from de novo pathogenic contraction of the D4Z4 locus [18]. In FSHD1, there is an inverse correlation between the size of the D4Z4 repeat and the severity of the disease. Patients with 1–3 repeat units are most severely affected and have an earlier disease onset, compared to those with 8–10 repeat units who appear to have a milder disease which is later in onset [19, 20].
Patients with FSHD2 exhibit contraction independent, DNA hypomethylation on both copies of D4Z4, due to pathogenic variants in chromatin-modifying genes [16, 21]. The inheritance of FSHD2 is digenic, requiring the inheritance of dysfunctional chromatin-modifying genes and a moderate repeat contraction of D4Z4 repeat number between 8 and 30 on the 4qA permissive haplotype [21]. Eighty-five percent of patients with FSHD2 carry a variant in the SMCHD1 (structural maintenance of chromosomes flexible hinge domain containing 1) gene on chromosome 18 [17, 21]. SMCHD1 serves as an epigenetic repressor that binds to the D4Z4 repeat to maintain a repressed chromatin state in somatic cells via methylation, and its reduced activity in FSHD2 leads to hypomethylation of the D4Z4 array, enabling the aberrant expression of the DUX4 protein [22,23,24]. Variants in DNMT3B (de novo DNA methyltransferase gene) and LRIF1 genes similarly lead to chromatin relaxation and inappropriate DUX4 expression [15, 25, 26], manifesting as FSHD.
Clinical characteristics
Muscle weakness
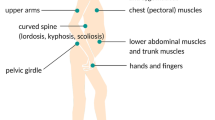
FSHD is characterised by progressive muscle weakness that develops in a rostro-caudal pattern, involving the face, scapular stabilisers, upper arm, abdomen, lower leg (peroneal muscles) and hip girdle [5••]. In contrast to other dystrophies, FSHD often has asymmetric muscle involvement [27]. The disease onset varies from infancy to middle age, although most affected patients develop symptoms by the second decade [28]. The clinical progression is usually slow, and patients typically have a normal or near-normal lifespan. Disease severity is highly variable amongst individuals, and in general, patients who develop symptoms at an earlier onset have more severe disease [1, 27, 29]. In the long run, approximately 20% of the patients become wheelchair-dependent [30].
Weakness of the facial muscles, especially the orbicularis oculi and orbicularis oris, develops in the initial stages [27, 31]. This results in difficulties with closing the eyes tightly, smiling, pursing the lips and whistling [1]. In FSHD, facial weakness can be absent or mild early in the course of the disease, and may remain mild for many years [1].
Scapular winging is commonly noted early in the course of the disease. During abduction of the arms, there is characteristic upward and lateral riding of the scapula, due to preferential weakness of the lower trapezius muscles [28]. The deltoid muscles typically remain largely unaffected until the later stages of the disease. In contrast, the pectoral muscles, biceps and triceps are often affected early on, resulting in marked weakness and atrophy of the upper arm [32]. The forearm muscles are commonly spared, giving rise to the appearance of a “Popeye-arm” appearance [33]. In individuals with more severe disease, distal upper extremity weakness can be present, affecting the wrist and finger extensors as well [33].
In the abdomen, the lower abdominal muscles are selectively involved, resulting in a protuberant abdomen, exaggerated lumbar lordosis and a positive Beevor’s sign [34, 35]. A positive Beevor’s sign is characterised by upward movement of the umbilicus upon flexion of the neck in a supine position, and it occurs due to lower abdominal muscle weakness [34]. It has been extensively described in patients with FSHD and has a sensitivity and specificity of approximately 90% [36].
Lower limb weakness manifests with peroneal muscle weakness predominantly, leading to foot drop [37]. In some patients, weakness of the hip girdle muscles may be present as well.
Other systemic manifestations
Beyond skeletal muscle manifestations, FSHD can also lead to the involvement of other systems, causing respiratory dysfunction, retinal vasculopathy, hearing loss and pain.
Respiratory insufficiency in FSHD is predominantly related to weakness of the expiratory abdominal muscles, diaphragmatic dysfunction and chest wall deformities [38, 39]. In 10 to 39% of the FSHD population, a restrictive ventilatory pattern can be seen on spirometry testing [39, 40]. However, only approximately 1 to 3% of patients require respiratory support with chronic non-invasive ventilation [38, 41].
Retinal vasculopathy can occur in up to 50 to 75% of patients with FSHD, resulting in increased vascular tortuosity, telangiectatic blood vessels and microaneurysms [42]. The changes are usually bilateral and subtle and can only be demonstrated via fluorescein angiography [43]. While vision is generally unaffected in FSHD, a small percentage of patients may experience a Coats-like syndrome [43]. This syndrome occurs due to retinal telangiectasia and exudative retinopathy that can progress to retinal detachment, causing visual loss [44]. Sensorineural hearing loss may also be present in individuals with FSHD and is usually gradual and progressive [45]. The risks of hearing loss and/or exudative retinopathy are postulated to be higher in patients with larger D4Z4 repeat contraction sizes and those with early-onset disease [44, 46, 47].
Chronic pain is a significant, troubling and under-recognised symptom in patients with FSHD and has been reported to be present in up to 82% of patients [48,49,50]. It commonly affects the shoulders and lower back [51]. The pain is likely multifactorial, stemming from factors such as hyperlordosis of the lumbar spine, and muscle weakness and atrophy resulting in a restricted range of motion and discomfort [51].
FSHD does not typically result in cardiomyopathy. However, cardiac arrhythmias have been reported in patients with FSHD, though the majority of patients are asymptomatic. An incomplete right bundle branch block is most commonly described and was shown to be present in approximately 23 to 33% of patients [52], followed by supraventricular tachycardia in approximately 10% of patients [53].
Diagnosis
Genetic testing confirms the diagnosis of FSHD and should be obtained in patients with typical presentations and no first-degree relatives with genetic confirmation of the disease, or in patients with atypical presentations. First-degree relatives of a genetically confirmed proband who present with a classical FSHD phenotype may be diagnosed without further genetic testing [54].
The Southern blot method is typically used for the diagnosis of FSHD1. This procedure involves cleaving genomic DNA into specific fragments using restriction enzymes, separation of fragments by size using gel electrophoresis and, subsequently, hybridization with a p13E-11 probe [55, 56]. A reduction in fragment size of less than 10 D4Z4 repeats on the 4q35 chromosome, on a permissive 4qA allele, is consistent with a diagnosis of FSHD1.
Despite Southern blotting being the standard diagnostic tool for FSHD1, it has its limitations. It requires large amounts of high-quality molecular weight DNA, is labour-intensive and time-consuming and may require the use of radioactive material. It estimates the number of D4Z4 repeats based on the size of detected bands, which can lead to inaccuracies [57, 58]. To distinguish between 4qA, 4qB and 10q haplotypes, multiple restriction enzymes and probes are required [56, 59, 60]. Cases with somatic mosaicism or rearrangements may be undetected with standard gel electrophoresis, although this can be mitigated by using pulsed-field gel electrophoresis (PFGE) [60].
Optical genomic mapping (OGM), which maps locations of restriction enzymes in DNA molecules, is emerging as a valuable tool in genetic testing of FSHD as it addresses certain limitations associated with Southern blotting. Studies have shown that OGM can measure the number of D4Z4 repeats with higher precision, distinguish between DNA segments from 4q35 and 10q26 and accurately identify cases with mosaicism [58, 61,62,63]. In addition, it is more cost-effective and has a shorter turnaround time [62]. However, it is unable to detect rearrangements as it cannot differentiate the 4q35 and 10q26 D4Z4 repeats and telomere ends [61].
In individuals who display the classical phenotype of FSHD but do not have the D4Z4 repeat contraction typically seen in FSHD1, FSHD2 should be considered. The diagnosis of FSHD2 requires the identification of a pathogenic variant in chromatin modifier genes (SMCHD1, DNMT3B, LRIF1) with the identification of decreased 4q35 methylation on the permissive 4qA haplotype [64]. It is advisable to first evaluate for mutations in the SMCHD1 gene, as they account for approximately 80–85% of all FSHD2 cases [21, 65]. If available, whole-exome sequencing (WES) should be offered, as it can evaluate SMCHD1, DNMT3B and LRIF1 concurrently [66, 67].
It has been proposed that OGM in conjunction with WES can help provide a comprehensive approach to the detection of both FSHD1 and FSHD2. However, it is important for healthcare providers to interpret genetic testing outcomes with caution due to inherent test limitations. In addition, the length of D4Z4 repeat does not reliably predict the disease course or severity, due to phenotypic variability and incomplete penetrance [68, 69].
Current management
The present approach to managing FSHD is primarily supportive in nature, since disease-modifying therapy has not yet progressed beyond clinical trials. This includes exercise and rehabilitation, optimization of pain control and conducting longitudinal surveillance for extra-skeletal systemic manifestations. Certain patients also benefit from orthopaedic interventions, such as scapular fixation surgery.
Clinical trials have shown that aerobic exercises may help improve the patient’s exercise performance and cardiovascular fitness, without damaging muscle tissue [70,71,72]. The physiotherapist can tailor exercises based on the individual’s physical status, with the aim to enhance range of motion and alleviate pain [5••, 54]. Orthotic devices, such as ankle-foot orthoses and lumbar corsets, are commonly recommended. It is notable that approximately 20% of patients may require a wheelchair for mobility after reaching the age of 50 [30, 73]. As upper limb weakness may restrict the use of a manual wheelchair, a motorised wheelchair is the preferred option for patients with FSHD. Along with exercises, nonsteroidal anti-inflammatory drugs can be used for managing acute pain, while chronic pain can be addressed with anti-depressants or anti-seizure medications [51, 74••].
All individuals with FSHD should undergo a baseline pulmonary function test, and those with kyphoscoliosis, lumbar hyperlordosis, chest wall deformities, co-existing chronic lung or cardiac conditions, severe disease leading to wheelchair dependence or severe proximal weakness should have annual testing [54, 75]. Approximately 1% of FSHD patients require nocturnal non-invasive ventilatory support, and this usually occurs only decades after the onset of the disease [41]. Sleep-disordered breathing such as obstructive sleep apnea and nocturnal hypoventilation can also be present in patients with FSHD [39, 76, 77]. As such, clinicians should also screen patients for symptoms such as early-morning headaches and non-restorative sleep and, if present, consider polysomnography for further evaluation [76, 77]. It is recommended to initiate nocturnal non-invasive ventilation in FSHD patients with a forced vital capacity of less than 60% on lung function tests or those with sleep-disordered breathing disorders [54]. Routine cardiac screen is not required unless the patient is symptomatic [54].
In terms of surveillance for ophthalmic manifestations, all patients should undergo a baseline fundoscopy and dilated retinal examination [54]. There is a higher risk of retinal complications in patients with early-onset FSHD or those with D4Z4 repeat array fragments that are less than 15 kb in size, suggesting a need for closer monitoring in these patients [44]. If signs of retinal vasculopathy are detected, prompt intervention with photocoagulation can help to prevent further retinal damage [78]. Additionally, some patients with FSHD have weakness of the orbicularis oculi, resulting in difficulties with eyelid closure and lagophthalmos. Topical lubricants, ointments and eye patches can be used at night to prevent exposure keratopathy from developing as a consequence of this.
Individuals with FSHD have an increased risk of developing sensorineural hearing loss. Much like the risk associated with retinal complications, the risk of developing hearing loss is greater in those with shorter D4Z4 repeat arrays and those with FSHD characterised by an earlier onset (e.g. infantile or adolescent-onset) [47]. Regular evaluations are recommended for these specific groups [46]. In patients with adult-onset FSHD, routine hearing assessments are not necessary unless symptoms are present [79].
Scapular fixation surgery involves surgical fixation of the scapula to the posterior thorax. Apart from cosmetic improvements, scapulothoracic arthrodesis has resulted in functional improvements in shoulder flexion and abduction, for patients with severe scapular winging and preserved deltoid strength [80, 81]. The Horwitz manoeuvre, a bedside manual scapular fixation test which imitates the post-surgical mechanics, can help predict post-surgical improvement [80, 81]. However, physicians should carefully evaluate the potential complications of surgery in comparison to its benefits, taking into consideration the patient’s disease progression rate and the need for prolonged post-surgical bracing [82].
Future therapies
Previously, clinical trials involving albuterol, salbutamol, diltiazem, corticosteroids and certain myostatin inhibitors (MYO-029 and ACE-083) did not demonstrate clinical benefit for individuals with FSHD [74••, 83,84,85,86,87,88,89,90,91]. However, recent advancements in research have provided further insight into the fundamental pathophysiology of FSHD, particularly the aberrant expression of the DUX4 protein in skeletal muscles. This understanding has paved the way for the development of targeted therapies directed at suppressing DUX4 expression or mitigating its downstream effects. This is done via the following: (i) epigenetic silencing of the D4Z4 repeats, (ii) blocking DUX4 mRNA production and (iii) targeting downstream pathways triggered by DUX4 expression [92, 93••].
In this segment, we discuss targeted therapies that are currently being investigated in clinical and/or pre-clinical studies. The main avenues for targeting DUX4 include small molecules, oligonucleotide therapeutics and CRISPR-based approaches. At present, all potential therapies are in the pre-clinical stage, with the exception of losmapimod, which is currently undergoing a phase 3 clinical trial [94••]. Although none of these novel therapies have been approved yet, they represent a pivotal change in the treatment landscape of FSHD, moving beyond traditional supportive and symptomatic therapies to treatments that directly target the fundamental root cause.
Small molecules
Small molecule drugs are developed through chemical synthesis that bind to cellular targets to affect disease processes [95]. In contrast to biologics, they are non-immunogenic and have low molecular weight, allowing for oral administration with favourable cellular uptake, and are generally more cost-effective [95].
Losmapimod is an oral, selective, small molecular inhibitor of the P38 mitogen-activated protein kinase (MAPK) pathway, which is a modulator of DUX4 expression and a mediator of inflammation [96]. Previous pre-clinical studies done with mice models showed a significant reduction of DUX4 levels of approximately 80% with losmapimod [9, 96]. It has been shown to be well tolerated with no serious adverse events in a phase 1 study [97•].
A randomised, double-blind, placebo-controlled multicentre phase 2b clinical trial on losmapimod (ReDUX4) involving 80 patients with FSHD1 was recently completed [98]. The patients were randomised 1:1 to receive either losmapimod 15mg, twice daily or a placebo for 48 weeks. The primary endpoint, which was a reduction in DUX4-driven gene expression in skeletal muscle, was not achieved. However, there was a statistically significant benefit in the secondary endpoints in terms of structural, functional and patient-reported outcomes [98,99,100]. After 48 weeks of treatment, patients who received losmapimod showed reduced progression of muscle fat infiltration (MFI) on MRI (0.03% vs. 0.52%; disparity, − 0.49; 95% CI, − 0.86 to − 0.12; p = 0.01) compared to those who received placebo [101]. Reachable workspace (RWS), used as a performance measure of the shoulder and proximal arm function, also showed that patients who received losmapimod performed better than those who received a placebo in the RWS measure with weights. Analysis of reachable surface area (RSA) showed that the annualised rate of change (%/year) in total RSA for losmapimod versus placebo in the dominant arm was − 0.44 versus − 8.42, p = 0.07; in the non-dominant arm, this was 4.88 versus − 4.02, p = 0.01 [98]. In the losmapimod group, assessment of maximum voluntary isometric contraction (MVICT) via hand-held dynamometry also showed stabilisation across various parameters [94••]. Additionally, relative to placebo, these patients also reported significant improvement in the Patients’ Global Impression of Change (PGIC) assessment as well (difference, − 0.58; p = 0.02) [98].
An open-label extension of the above trial was conducted, and preliminary data was presented at the 2023 AAN Annual Meeting [102]. Participants who were on losmapimod continued to receive the drug (LOS/LOS) and, at week 96, were assessed for durability of treatment response, via assessment of RWS. Participants who received a placebo were converted to losmapimod at week 48 (PBO/LOS) and received the drug for another 48 weeks. Annualised total RSA showed stability in the LOS/LOS group in the 2nd year (0.18%/year) compared to the 1st (− 0.77%/year) [102]. In the PBO/LOS group, participants exhibited trends of slowing or halting of disease progression based on RWS, as shown by improvement in annualised total RSA in the 2nd versus the 1st year (4.07%/year versus − 9.96%/year, respectively) [102]. Throughout the extended duration, no drug-related serious adverse events or discontinuation due to adverse events were reported.
At present, Fulcrum Therapeutics has just completed the enrolment of 260 patients in a double-blind, multi-national, placebo-controlled phase 3 trial (REACH) (ClinicalTrials Identifiers: NCT05397470), to further evaluate the efficacy and safety of losmapimod for the treatment of FSHD [103, 104]. The primary endpoint involves evaluation of change from baseline RWS, along with secondary endpoints such as analysis of MFI using whole-body MRI, quality of life in the neurological disorders upper extremity scale (Neuro-QoL UE) and PGIC [104]. Preliminary data is anticipated to be reported in the fourth quarter of 2024.
Antisense oligonucleotides
Antisense oligonucleotides (ASOs) are modified single-stranded DNA or RNA sequences that bind to complementary targeted mRNA sequences, thereby preventing or altering the translation of protein. DUX4 expression can be targeted by ASOs that bind to specific DUX4 mRNA sequences [105]. Pre-clinical studies with ASOs have proven efficacy of reduction of DUX4 and DUX4 target genes in cultured FSHD myocytes and FSHD mouse models [105,106,107,108,109]. The drawback with ASOs, however, is limited bioavailability and poor cellular uptake, which limits the effectiveness of delivery to the target muscle tissue [110].
AOC 1020, developed by Avidity Biosciences, comprises a unique monoclonal antibody that binds to the transferrin receptor 1 (TfR1) combined with a siRNA designed to specifically target DUX4 mRNA [111, 112]. FORTITUDE, a randomised, placebo-controlled, double-blind phase 1/2 clinical trial (ClinicalTrials.gov Identifiers: NCT05747924), is currently ongoing, to evaluate AOC 1020 in 72 participants with FSHD, with the aim to evaluate the safety and tolerability of the drug when administered intravenously [112, 113]. The primary objective of this study is to evaluate the safety and tolerability of AOC 1020, whereas secondary objectives include analysing the pharmacokinetics and pharmacodynamics of the drug. There are three parts to this study—part A consists of dose titration to evaluate the safety of the drug at two low doses, whereas part B involves ascending doses of the drug to study two presumably effective doses [112]. Finally, part C aims to evaluate clinical outcomes. The study will assess measures of mobility and muscle strength, including the use of MRI to measure muscle volume and composition [112]. There will also be an open-label extension study, whereby eligible participants will be given the option to enrol in. Avidity intends to share data from a preliminary assessment of approximately half of the study participants in the first half of 2024.
Myostatin inhibitors
Myostatin is a growth differentiation factor that plays an essential role in regulating skeletal muscle growth [114]. As such, myostatin inhibition has been postulated to help increase muscle mass and, in turn, muscle strength [115]. GYM329 (RO7204239) is an investigational monoclonal anti-myostatin antibody which targets inactive latent myostatin, preventing its conversion to active myostatin, thus reducing the levels of myostatin in muscle and blood [116].
MANOEUVRE (ClinicalTrials.gov Identifiers: NCT05548556) is a multi-centre, randomised, placebo-controlled, double-blind phase 2 trial that aims to evaluate the safety, tolerability, efficacy, pharmacokinetics and pharmacodynamics of GYM329 (RO7204239) in adult patients with genetically confirmed FSHD1 or FSHD2 [116, 117]. It is currently in the recruitment phase, with the aim of enrolling 48 participants. The trial involves participants receiving subcutaneous RO7204239 or placebo injection every 4 weeks, over a treatment period of 52 weeks. Primary outcome measures include assessing percentage change from baseline in contractile muscle volume (CMV) of quadriceps femoralis via MRI bilaterally as well as analysis of adverse effects experienced by participants, whereas secondary outcome measures include assessment of motor function and strength and change from baseline in CMV in other muscle groups as assessed by MRI as well as changes in serum myostatin levels [116]. After completion, there will be an open-label extension, and participants will be given the option to participate and receive RO7204239 for another 52 weeks.
Gene therapy (CRISPR)
CRISPR/CAS9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9) gene editing techniques are in development for various genetic diseases, including FSHD. The technique involves combining a guide RNA sequence complementary to the target DNA, with the CAS9 enzyme, forming the CRISPR/CAS9 complex. The complex can target DNA sequences complementary to the guide RNA, allowing the CAS9 enzyme to make targeted double-stranded DNA breaks. This can be used to disrupt the cut genes or utilise DNA repair to insert new DNA template sequences.
Applications of CRISPR/CAS9 techniques for FSHD are in various pre-clinical stages of development. One application involves the use of an inactivated form of the CAS9 enzyme (dCAS9-KRAB system) to induce epigenetic silencing of the DUX4 gene (instead of creating double-stranded DNA cuts), resulting in decreased production of the DUX4 transcripts and downregulation of target genes [118, 119]. Other studies have targeted the DUX4 polyadenylation signal, required to stabilise the DUX4 transcript [120, 121••]. In FSHD2, a study targeted the intronic variant of the methylation regulation gene SMCHD1 with CRISPR/CAS9 gene editing, which restored SMCHD1 expression and suppression of DUX4 [122]. There is potential progress made towards human studies, with an announcement of plans in 2024 for a first-in-human trial for a CRISPR/CAS9 treatment targeting epigenetic silencing of DUX4 expression, delivered by an adeno-associated virus vector [123].
Conclusion
FSHD is one of the most common muscular dystrophies in the adult population that manifests with disabling skeletal muscle weakness and multi-systemic complications. At present, the mainstay of management is limited to supportive management to preserve and optimise functional independence, with the aim to improve quality of life. In recent years, further understanding of the underlying molecular pathophysiology of FSHD has led to advances in pre-clinical and clinical trials for targeted therapy. This holds a promising potential for disease-modifying management in the foreseeable future, which may alter the disease trajectory for this condition.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Tawil R, Van Der Maarel SM. Facioscapulohumeral muscular dystrophy. Muscle Nerve. 2006;34(1):1–15.
Deenen JC, Arnts H, van der Maarel SM, Padberg GW, Verschuuren JJ, Bakker E, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83(12):1056–9.
Flanigan KM, Coffeen CM, Sexton L, Stauffer D, Brunner S, Leppert MF. Genetic characterization of a large, historically significant Utah kindred with facioscapulohumeral dystrophy. Neuromuscul Disord. 2001;11(6–7):525–9.
Mostacciuolo ML, Pastorello E, Vazza G, Miorin M, Angelini C, Tomelleri G, et al. Facioscapulohumeral muscular dystrophy: epidemiological and molecular study in a north-east Italian population sample. Clin Genet. 2009;75(6):550–5.
•• Mul K. Facioscapulohumeral muscular dystrophy. Continuum (Minneap Minn). 2022;28(6):1735–51. A concise review that discusses the pathophysiology, clinical features, genetic testing and current management options for FSHD and provides updates on targeted therapies.
• Mocciaro E, Runfola V, Ghezzi P, Pannese M, Gabellini D. DUX4 role in normal physiology and in FSHD muscular dystrophy. Cells. 2021;10(12):2–6. A summary of the cellular and molecular processes of DUX4 and its pivotal role in the pathophysiology of FSHD.
Snider L, Geng LN, Lemmers RJ, Kyba M, Ware CB, Nelson AM, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6(10):e1001181.
van der Maarel SM, Frants RR. The D4Z4 repeat-mediated pathogenesis of facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2005;76(3):375–86.
Oliva J, Galasinski S, Richey A, Campbell AE, Meyers MJ, Modi N, et al. Clinically advanced p38 inhibitors suppress DUX4 expression in cellular and animal models of facioscapulohumeral muscular dystrophy. J Pharmacol Exp Ther. 2019;370(2):219–30.
Lim KRQ, Nguyen Q, Yokota T. DUX4 signalling in the pathogenesis of facioscapulohumeral muscular dystrophy. Int J Mol Sci. 2020;21(3):4–7.
Himeda CL. The genetics and epigenetics of facioscapulohumeral muscular dystrophy. 2019;266.
Wijmenga C, Brouwer OF, Moerer P, Wijmenga C, Frants RR, Weber JL. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4 The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. 1990;863–5.
Wijmenga C, Frants RR, Brouwer OF, Moerer P, Weber JL, Padberg GW. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet. 1990;336(8716):651–3.
Wijmenga C, Hewitt JE, Sandkuijl LA, Clark LN, Wright TJ, Dauwerse HG, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet. 1992;2(1):26–30.
Himeda CL, Jones PL. The genetics and epigenetics of facioscapulohumeral muscular dystrophy. Annu Rev Genomics Hum Genet. 2019;20:265–91.
van Overveld PG, Lemmers RJ, Sandkuijl LA, Enthoven L, Winokur ST, Bakels F, et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. 2003;35(4):315–7.
Lemmers RJ, Goeman JJ, van der Vliet PJ, van Nieuwenhuizen MP, Balog J, Vos-Versteeg M, et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet. 2015;24(3):659–69.
Köhler J, Rupilius B, Otto M, Bathke K, Koch MC. Germline mosaicism in 4q35 facioscapulohumeral muscular dystrophy (FSHD1A) occurring predominantly in oogenesis. Hum Genet. 1996;98(4):485–90.
Sacconi S, Briand-Suleau A, Gros M, Baudoin C, Lemmers RJLF, Rondeau S, et al. FSHD1 and FSHD2 form a disease continuum. Neurology. 2019;92(19):e2273–85.
Zernov N, Skoblov M. Genotype-phenotype correlations in FSHD. BMC Med Genomics. 2019;12(Suppl 2):43.
Lemmers RJ, Tawil R, Petek LM, Balog J, Block GJ, Santen GW, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44(12):1370–4.
Lemmers RJ, van den Boogaard ML, van der Vliet PJ, Donlin-Smith CM, Nations SP, Ruivenkamp CA, et al. Hemizygosity for SMCHD1 in facioscapulohumeral muscular dystrophy type 2: consequences for 18p deletion syndrome. Hum Mutat. 2015;36(7):679–83.
Hamanaka K, Goto K, Arai M, Nagao K, Obuse C, Noguchi S, et al. Clinical, muscle pathological, and genetic features of Japanese facioscapulohumeral muscular dystrophy 2 (FSHD2) patients with SMCHD1 mutations. Neuromuscul Disord. 2016;26(4–5):300–8.
van den Boogaard ML, Lemmers RJ, Camaño P, van der Vliet PJ, Voermans N, van Engelen BG, et al. Double SMCHD1 variants in FSHD2: the synergistic effect of two SMCHD1 variants on D4Z4 hypomethylation and disease penetrance in FSHD2. Eur J Hum Genet. 2016;24(1):78–85.
van den Boogaard ML, Lemmers RJLF, Balog J, Wohlgemuth M, Auranen M, Mitsuhashi S, et al. Mutations in DNMT3B modify epigenetic repression of the D4Z4 repeat and the penetrance of facioscapulohumeral dystrophy. Am J Hum Genet. 2016;98(5):1020–9.
Hamanaka K, Šikrová D, Mitsuhashi S, Masuda H, Sekiguchi Y, Sugiyama A, et al. Homozygous nonsense variant in. Neurology. 2020;94(23):e2441–7.
Kilmer DD, Abresch RT, McCrory MA, Carter GT, Fowler WM, Johnson ER, et al. Profiles of neuromuscular diseases. Facioscapulohumeral muscular dystrophy. Am J Phys Med Rehabil. 1995;75(5):S131-9.
Orrell RW. Facioscapulohumeral dystrophy and scapuloperoneal syndromes. Handb Clin Neurol. 2011;101:167–80.
Goselink RJM, Mul K, van Kernebeek CR, Lemmers RJLF, van der Maarel SM, Schreuder THA, et al. Early onset as a marker for disease severity in facioscapulohumeral muscular dystrophy. Neurology. 2019;92(4):e378–85.
Statland JM, Tawil R. Risk of functional impairment in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2014;49(4):520–7.
Goselink RJM, Schreur V, van Kernebeek CR, Padberg GW, van der Maarel SM, van Engelen BGM, et al. Ophthalmological findings in facioscapulohumeral dystrophy. Brain Commun. 2019;1(1)2–3.
Wicklund MP. The muscular dystrophies. Continuum (Minneap Minn). 2013;19(6 Muscle Disease):1535–70.
Statland J, Tawil R. Facioscapulohumeral muscular dystrophy. Neurol Clin. 2014;32(3):721–8, ix.
Sharma C, Acharya M, Kumawat BL, Nath K. Beevor’s sign in facioscapulohumeral muscular dystrophy. BMJ Case Rep. 2014;2014.
Awerbuch GI, Nigro MA, Wishnow R. Beevor’s sign and facioscapulohumeral dystrophy. Arch Neurol. 1990;47(11):1208–9.
Shahrizaila N, Wills AJ. Significance of Beevor’s sign in facioscapulohumeral dystrophy and other neuromuscular diseases. J Neurol Neurosurg Psychiatry. 2005;76(6):869–70.
Gambelli CN, Bredin J, Doix AM, Garcia J, Tanant V, Fournier-Mehouas M, et al. The effect of tibialis anterior weakness on foot drop and toe clearance in patients with facioscapulohumeral dystrophy. Clin Biomech (Bristol, Avon). 2023;102:105899.
Santos DB, Boussaid G, Stojkovic T, Orlikowski D, Letilly N, Behin A, et al. Respiratory muscle dysfunction in facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2015;25(8):632–9.
Moreira S, Wood L, Smith D, Marini-Bettolo C, Guglieri M, McMacken G, et al. Respiratory involvement in ambulant and non-ambulant patients with facioscapulohumeral muscular dystrophy. J Neurol. 2017;264(6):1271–80.
Scully MA, Eichinger KJ, Donlin-Smith CM, Tawil R, Statland JM. Restrictive lung involvement in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2014;50(5):739–43.
Wohlgemuth M, van der Kooi EL, van Kesteren RG, van der Maarel SM, Padberg GW. Ventilatory support in facioscapulohumeral muscular dystrophy. Neurology. 2004;63(1):176–8.
Osborne RJ, Welle S, Venance SL, Thornton CA, Tawil R. Expression profile of FSHD supports a link between retinal vasculopathy and muscular dystrophy. Neurology. 2007;68(8):569–77.
Ghorbanian S, Jaulim A, Chatziralli IP. Diagnosis and treatment of coats’ disease: a review of the literature. Ophthalmologica. 2012;227(4):175–82.
Statland JM, Sacconi S, Farmakidis C, Donlin-Smith CM, Chung M, Tawil R. Coats syndrome in facioscapulohumeral dystrophy type 1: frequency and D4Z4 contraction size. Neurology. 2013;80(13):1247–50.
Brouwer OF, Padberg GW, Ruys CJ, Brand R, de Laat JA, Grote JJ. Hearing loss in facioscapulohumeral muscular dystrophy. Neurology. 1991;41(12):1878–81.
Lutz KL, Holte L, Kliethermes SA, Stephan C, Mathews KD. Clinical and genetic features of hearing loss in facioscapulohumeral muscular dystrophy. Neurology. 2013;81(16):1374–7.
Chen TH, Lai YH, Lee PL, Hsu JH, Goto K, Hayashi YK, et al. Infantile facioscapulohumeral muscular dystrophy revisited: expansion of clinical phenotypes in patients with a very short EcoRI fragment. Neuromuscul Disord. 2013;23(4):298–305.
Jensen MP, Hoffman AJ, Stoelb BL, Abresch RT, Carter GT, McDonald CM. Chronic pain in persons with myotonic dystrophy and facioscapulohumeral dystrophy. Arch Phys Med Rehabil. 2008;89(2):320–8.
Hamel J, Johnson N, Tawil R, Martens WB, Dilek N, McDermott MP, et al. Patient-reported symptoms in facioscapulohumeral muscular dystrophy (PRISM-FSHD). Neurology. 2019;93(12):e1180–92.
Padua L, Aprile I, Frusciante R, Iannaccone E, Rossi M, Renna R, et al. Quality of life and pain in patients with facioscapulohumeral muscular dystrophy. Muscle Nerve. 2009;40(2):200–5.
Morís G, Wood L, FernáNdez-Torrón R, González Coraspe JA, Turner C, Hilton-Jones D, et al. Chronic pain has a strong impact on quality of life in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2018;57(3):380–7.
van Dijk GP, van der Kooi E, Behin A, Smeets J, Timmermans J, van der Maarel S, et al. High prevalence of incomplete right bundle branch block in facioscapulohumeral muscular dystrophy without cardiac symptoms. Funct Neurol. 2014;29(3):159–65.
Trevisan CP, Pastorello E, Armani M, Angelini C, Nante G, Tomelleri G, et al. Facioscapulohumeral muscular dystrophy and occurrence of heart arrhythmia. Eur Neurol. 2006;56(1):1–5.
Tawil R, Kissel JT, Heatwole C, Pandya S, Gronseth G, Benatar M, et al. Evidence-based guideline summary: evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy: report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2015;85(4):357–64.
Rieken A, Bossler AD, Mathews KD, Moore SA. CLIA laboratory testing for facioscapulohumeral dystrophy: a retrospective analysis. Neurology. 2021;96(7):e1054–62.
Deak KL, Lemmers RJ, Stajich JM, Klooster R, Tawil R, Frants RR, et al. Genotype-phenotype study in an FSHD family with a proximal deletion encompassing p13E–11 and D4Z4. Neurology. 2007;68(8):578–82.
Vasale J, Boyar F, Jocson M, Sulcova V, Chan P, Liaquat K, et al. Molecular combing compared to Southern blot for measuring D4Z4 contractions in FSHD. Neuromuscul Disord. 2015;25(12):945–51.
Dai Y, Li P, Wang Z, Liang F, Yang F, Fang L, et al. Single-molecule optical mapping enables quantitative measurement of D4Z4 repeats in facioscapulohumeral muscular dystrophy (FSHD). J Med Genet. 2020;57(2):109–20.
Lemmers RJ, Osborn M, Haaf T, Rogers M, Frants RR, Padberg GW, et al. D4F104S1 deletion in facioscapulohumeral muscular dystrophy: phenotype, size, and detection. Neurology. 2003;61(2):178–83.
Lemmers RJ, van der Wielen MJ, Bakker E, Padberg GW, Frants RR, van der Maarel SM. Somatic mosaicism in FSHD often goes undetected. Ann Neurol. 2004;55(6):845–50.
Stence AA, Thomason JG, Pruessner JA, Sompallae RR, Snow AN, Ma D, et al. Validation of optical genome mapping for the molecular diagnosis of facioscapulohumeral muscular dystrophy. J Mol Diagn. 2021;23(11):1506–14.
Guruju NM, Jump V, Lemmers R, Van Der Maarel S, Liu R, Nallamilli BR, et al. Molecular diagnosis of facioscapulohumeral muscular dystrophy in patients clinically suspected of FSHD using optical genome mapping. Neurol Genet. 2023;9(6): e200107.
Efthymiou S, Lemmers RJLF, Vishnu VY, Dominik N, Perrone B, Facchini S, et al. Optical genome mapping for the molecular diagnosis of facioscapulohumeral muscular dystrophy: advancement and challenges. Biomolecules. 2023;13(11):2–8.
Erdmann H, Scharf F, Gehling S, Benet-Pagès A, Jakubiczka S, Becker K, et al. Methylation of the 4q35 D4Z4 repeat defines disease status in facioscapulohumeral muscular dystrophy. Brain. 2023;146(4):1388–402.
Vincenten SCC, Van Der Stoep N, Paulussen ADC, Mul K, Badrising UA, Kriek M, et al. Facioscapulohumeral muscular dystrophy-reproductive counseling, pregnancy, and delivery in a complex multigenetic disease. Clin Genet. 2022;101(2):149–60.
Mitsuhashi S, Boyden SE, Estrella EA, Jones TI, Rahimov F, Yu TW, et al. Exome sequencing identifies a novel SMCHD1 mutation in facioscapulohumeral muscular dystrophy 2. Neuromuscul Disord. 2013;23(12):975–80.
Strafella C, Caputo V, Bortolani S, Torchia E, Megalizzi D, Trastulli G, et al. Whole exome sequencing highlights rare variants in. Front Genet. 2023;14:1235589.
Ruggiero L, Mele F, Manganelli F, Bruzzese D, Ricci G, Vercelli L, et al. Phenotypic variability among patients With D4Z4 reduced allele facioscapulohumeral muscular dystrophy. JAMA Netw Open. 2020;3(5):e204040.
Wohlgemuth M, Lemmers RJ, Jonker M, van der Kooi E, Horlings CG, van Engelen BG, et al. A family-based study into penetrance in facioscapulohumeral muscular dystrophy type 1. Neurology. 2018;91(5):e444–54.
Olsen DB, Ørngreen MC, Vissing J. Aerobic training improves exercise performance in facioscapulohumeral muscular dystrophy. Neurology. 2005;64(6):1064–6.
Bankolé LC, Millet GY, Temesi J, Bachasson D, Ravelojaona M, Wuyam B, et al. Safety and efficacy of a 6-month home-based exercise program in patients with facioscapulohumeral muscular dystrophy: a randomized controlled trial. Medicine (Baltimore). 2016;95(31):e4497.
Andersen G, Prahm KP, Dahlqvist JR, Citirak G, Vissing J. Aerobic training and postexercise protein in facioscapulohumeral muscular dystrophy: RCT study. Neurology. 2015;85(5):396–403.
Katz NK, Hogan J, Delbango R, Cernik C, Tawil R, Statland JM. Predictors of functional outcomes in patients with facioscapulohumeral muscular dystrophy. Brain. 2021;144(11):3451–60.
•• Aguirre AS, Astudillo Moncayo OM, Mosquera J, Muyolema Arce VE, Gallegos C, Ortiz JF, et al. Treatment of facioscapulohumeral muscular dystrophy (FSHD): a systematic review. Cureus. 2023;15(6):e39903. A concise review about results from different studies for previous treatments for FSHD, summarising statistically significant results and those without an effect.
Teeselink S, Vincenten SCC, Voermans NC, Groothuis JT, Doorduin J, Wijkstra PJ, et al. Long-term follow-up of respiratory function in facioscapulohumeral muscular dystrophy. J Neurol. 2022;269(7):3682–9.
Runte M, Spiesshoefer J, Heidbreder A, Dreher M, Young P, Brix T, et al. Sleep-related breathing disorders in facioscapulohumeral dystrophy. Sleep Breath. 2019;23(3):899–906.
Della Marca G, Frusciante R, Dittoni S, Vollono C, Buccarella C, Iannaccone E, et al. Sleep disordered breathing in facioscapulohumeral muscular dystrophy. J Neurol Sci. 2009;285(1–2):54–8.
Matos R, Beato J, Silva M, Silva S, Brandão E, Falcão-Reis F, et al. Combined treatment with intravitreal bevacizumab and laser photocoagulation for exudative maculopathy in facioscapulohumeral muscular dystrophy. Ophthalmic Genet. 2017;38(5):490–3.
Rogers MT, Zhao F, Harper PS, Stephens D. Absence of hearing impairment in adult onset facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2002;12(4):358–65.
Giannini S, Faldini C, Pagkrati S, Grandi G, Digennaro V, Luciani D, et al. Fixation of winged scapula in facioscapulohumeral muscular dystrophy. Clin Med Res. 2007;5(3):155–62.
Eren İ, Gedik CC, Kılıç U, Abay B, Birsel O, Demirhan M. Management of scapular dysfunction in facioscapulohumeral muscular dystrophy: the biomechanics of winging, arthrodesis indications, techniques and outcomes. EFORT Open Rev. 2022;7(11):734–46.
Eren İ, Erşen A, Birsel O, Atalar AC, Oflazer P, Demirhan M. Functional outcomes and complications following scapulothoracic arthrodesis in patients with facioscapulohumeral dystrophy. J Bone Joint Surg Am. 2020;102(3):237–44.
Payan CA, Hogrel JY, Hammouda EH, Lacomblez L, Ollivier G, Doppler V, et al. Periodic salbutamol in facioscapulohumeral muscular dystrophy: a randomized controlled trial. Arch Phys Med Rehabil. 2009;90(7):1094–101.
van der Kooi EL, Vogels OJ, van Asseldonk RJ, Lindeman E, Hendriks JC, Wohlgemuth M, et al. Strength training and albuterol in facioscapulohumeral muscular dystrophy. Neurology. 2004;63(4):702–8.
van der Kooi EL, Kalkman JS, Lindeman E, Hendriks JC, van Engelen BG, Bleijenberg G, et al. Effects of training and albuterol on pain and fatigue in facioscapulohumeral muscular dystrophy. J Neurol. 2007;254(7):931–40.
Kissel JT, McDermott MP, Natarajan R, Mendell JR, Pandya S, King WM, et al. Pilot trial of albuterol in facioscapulohumeral muscular dystrophy. FSH-DY Group Neurology. 1998;50(5):1402–6.
Kissel JT, McDermott MP, Mendell JR, King WM, Pandya S, Griggs RC, et al. Randomized, double-blind, placebo-controlled trial of albuterol in facioscapulohumeral dystrophy. Neurology. 2001;57(8):1434–40.
Elsheikh BH, Bollman E, Peruggia M, King W, Galloway G, Kissel JT. Pilot trial of diltiazem in facioscapulohumeral muscular dystrophy. Neurology. 2007;68(17):1428–9.
Tawil R, McDermott MP, Pandya S, King W, Kissel J, Mendell JR, et al. A pilot trial of prednisone in facioscapulohumeral muscular dystrophy. FSH-DY Group Neurology. 1997;48(1):46–9.
Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63(5):561–71.
Statland JM, Campbell C, Desai U, Karam C, Díaz-Manera J, Guptill JT, et al. Randomized phase 2 study of ACE-083, a muscle-promoting agent, in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2022;66(1):50–62.
Wang LH, Tawil R. Current therapeutic approaches in FSHD. J Neuromuscul Dis. 2021;8(3):441–51.
•• Salsi V, Vattemi GNA, Tupler RG. The FSHD jigsaw: are we placing the tiles in the right position? Curr Opin Neurol. 2023;36(5):455–63. A thorough review which analyses and summarises 121 literature reports published between 2021 and 2023 regarding advances in FSHD clinical and molecular research.
•• Jagannathan S, de Greef JC, Hayward LJ, Yokomori K, Gabellini D, Mul K, et al. Meeting report: the 2021 FSHD International Research Congress. Skelet Muscle. 2022;12(1):1. A summary of updates about disease mechanism in FSHD, interventional strategies and refinement of clinical outcome measures and results from the ReDUX4 trial, a phase 2b clinical trial of losmapimod in FSHD.
Danladi MF. Biologics vs. small molecules: drug costs and patient access. Medicine in Drug Discovery: Elsevier. 2021; p 2–4.
Rojas LA, Valentine E, Accorsi A, Maglio J, Shen N, Robertson A, et al. p38 α regulates expression of DUX4 in a model of facioscapulohumeral muscular dystrophy. J Pharmacol Exp Ther. 2020;374(3):489–98.
Mellion ML, Ronco L, Berends CL, Pagan L, Brooks S, van Esdonk MJ, et al. Phase 1 clinical trial of losmapimod in facioscapulohumeral dystrophy: safety, tolerability, pharmacokinetics, and target engagement. Br J Clin Pharmacol. 2021;87(12):4658–69 Results from a phase 1 clinical trial of losmapimod show the drug was well tolerated with no serious adverse events.
Tawil A. Annualized rates of change from a phase 2, randomized, double-blind, placebo-controlled, 48-week study of losmapimod in subjects with FSHD: ReDUX4 (S48.010) Neurology. 2023;100(17Supplement 2):S48.
Tawil R, Wagner K. Clinical research: O.5 A phase 2, randomized, double-blind, placebo-controlled, 48-week study of the efficacy and safety of losmapimod in subjects with FSHD: ReDUX4. Neuromuscul Dis. 2021;S48.
Ghasemi M, Emerson CP, Hayward LJ. Outcome measures in facioscapulohumeral muscular dystrophy clinical trials. Cells. 2022;11(4):4–13.
R T, J S, L W. A phase 2, randomized, double-blind, placebo-controlled, 48-week study of the efficacy and safety of losmapimod in subjects with FSHD:ReDUX4. AAN Annual Meeting. Washington: Seattle; 2022. p. 2022.
Wang L, Han J, Shoskes J, Jiang J, Tawil A. Results from 96 weeks open-label extension of a phase 2 trial of losmapimod in subjects with FSHD: ReDUX4. 2023 AAN Annual Meeting; Boston. 2023;100(17Supplement 2).
Efficacy and safety of losmapimod in treating participants with facioscapulohumeral muscular dystrophy (FSHD) (REACH). Clinicaltrials.gov: National Library of Medicine. 2023.
Tawil R, Han J, Wang L, Vissing J, Engelen Bv, Statland J, et al. P.136 design of reach: phase 3 randomized, double-blind, placebo-controlled, 48-week study of the efficacy and safety of losmapimod in FSHD. Neuromuscul Dis. 2022;32:S104.
Bouwman LF, den Hamer B, van den Heuvel A, Franken M, Jackson M, Dwyer CA, et al. Systemic delivery of a DUX4-targeting antisense oligonucleotide to treat facioscapulohumeral muscular dystrophy. Mol Ther Nucleic Acids. 2021;26:813–27.
Ansseau E, Vanderplanck C, Wauters A, Harper SQ, Coppée F, Belayew A. Antisense oligonucleotides used to target the DUX4 mRNA as Therapeutic approaches in faciosscapulohumeral muscular dystrophy (FSHD). Genes (Basel). 2017;8(3):270–80.
Lim KRQ, Bittel A, Maruyama R, Echigoya Y, Nguyen Q, Huang Y, et al. DUX4 Transcript knockdown with antisense 2′-O-methoxyethyl gapmers for the treatment of facioscapulohumeral muscular dystrophy. Mol Ther. 2021;29(2):848–58.
Chen JC, King OD, Zhang Y, Clayton NP, Spencer C, Wentworth BM, et al. Morpholino-mediated knockdown of DUX4 toward facioscapulohumeral muscular dystrophy therapeutics. Mol Ther. 2016;24(8):1405–11.
Marsollier AC, Ciszewski L, Mariot V, Popplewell L, Voit T, Dickson G, et al. Antisense targeting of 3′ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: a new gene-silencing approach. Hum Mol Genet. 2016;25(8):1468–78.
McClorey G, Banerjee S. Cell-penetrating peptides to enhance delivery of oligonucleotide-based therapeutics. Biomedicines. 2018;6(2):1–9.
Halseth A, Ackermann E, Brandt T, Chen C, Cho H, Stahl M, et al. P51 Phase 1/2 study to evaluate AOC 1020 for adult patients with facioscapulohumeral muscular dystrophy: FORTITUDE trial design Neuromuscul Dis. 2023;33(Supplement 1):S71.
Amy H, Elizabeth A, Teresa B, Chao-Yin C, Mark S, Kelly D, et al. Phase 1/2 study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamic effects of AOC 1020 administered intravenously to adult patients with facioscapulohumeral muscular dystrophy (FORTITUDE) trial design (P3–8.007). Neurology. 2023;100(17Supplement 2):3–8.007.
Phase 1/2 study of AOC 1020 in adults with facioscapulohumeral muscular dystrophy (FSHD) (FORTITUDE). ClinicalTrials.gov: National Library Of Medicine. 2023.
Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A. 2001;98(16):9306–11.
Han HQ, Mitch WE. Targeting the myostatin signaling pathway to treat muscle wasting diseases. Curr Opin Support Palliat Care. 2011;5(4):334–41.
Vissing J, Eichinger K, Morrow J, Statland J, Tasca G, Dodman A, et al. P.141 Manoeuvre study design: a study of GYM329 (RO7204239) in patients with facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul Dis. 2022;32(Supplement 1):S105.
A study to evaluate RO7204239 in participants with facioscapulohumeral muscular dystrophy (MANOEUVRE). ClinicalTrials.gov: National Library of Medicine. 2024.
Himeda CL, Jones TI, Jones PL. CRISPR/dCas9-mediated transcriptional inhibition ameliorates the epigenetic dysregulation at D4Z4 and represses DUX4-fl in FSH muscular dystrophy. Mol Ther. 2016;24(3):527–35.
Himeda CL, Jones TI, Jones PL. Targeted epigenetic repression by CRISPR/dSaCas9 suppresses pathogenic. Mol Ther Methods Clin Dev. 2021;20:298–311.
Joubert R, Mariot V, Charpentier M, Concordet JP, Dumonceaux J. Gene editing targeting the DUX4 polyadenylation signal: a therapy for FSHD? J Pers Med. 2020;11(1).
•• Das S, Chadwick BP. CRISPR mediated targeting of DUX4 distal regulatory element represses DUX4 target genes dysregulated in facioscapulohumeral muscular dystrophy. Sci Rep. 2021;11(1):12598. The article explores the use of CRISPR/Cas9 technology to target the DUX4 gene and provides a proof-of-concept of the effect of silencing the polyadenylation sequence on pathogenic DUX4 expression.
Goossens R, van den Boogaard ML, Lemmers RJLF, Balog J, van der Vliet PJ, Willemsen IM, et al. Intronic. J Med Genet. 2019;56(12):828–37.
Adhikari A, Boregowda S, Zheng H, Aguirre O, Norton A, Yang X, et al. P309 EPI-321: a promising gene therapy for facioscapulohumeral muscular dystrophy (FSHD) targeting D4Z4 epigenome. Neuromuscular Disorders: Elsevier; 2023. p. S120–S1.
Author information
Authors and Affiliations
Contributions
A.C, Q.Z.X and K.N wrote the main manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chin, A.X.Y., Quak, Z.X., Chan, Y.C. et al. Updates on Facioscapulohumeral Muscular Dystrophy (FSHD). Curr Treat Options Neurol 26, 261–275 (2024). https://doi.org/10.1007/s11940-024-00790-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11940-024-00790-x