
Abstract
Purpose of Review
Single-cell profiling, either in suspension or within the tissue context, is a rapidly evolving field. The purpose of this review is to outline recent advancements and emerging trends with a specific focus on studies in spondyloarthritis.
Recent Findings
The introduction of sequencing-based approaches for the quantification of RNA, protein, or epigenetic modifications at single-cell resolution has provided a major boost to discovery-driven research. Fluorescent flow cytometry, mass cytometry, and image-based cytometry continue to evolve. Spatial transcriptomics and imaging mass cytometry have extended high-dimensional analysis to cells in tissues. Applications in spondyloarthritis include the indexing and functional characterization of cells, discovery of disease-associated cell states, and identification of signatures associated with therapeutic responses. Single-cell TCR-seq has provided evidence for clonal expansion of CD8+ T cells in spondyloarthritis.
Summary
The use of single-cell profiling approaches in spondyloarthritis research is still in its early stages. Challenges include high cost and limited availability of diseased tissue samples. To harness the full potential of the rapidly expanding technical capabilities, large-scale collaborative efforts are imperative.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
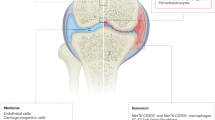
The diseases belonging to the spondyloarthritis (SpA) family are immune-mediated inflammatory conditions. Multiple lines of evidence, including data from genome-wide association studies, suggest the involvement of lymphocytes in SpA pathogenesis, though other cell types are likely to play significant roles as well [1]. Recent technological advances have revolutionized the way we study cells, permitting the simultaneous measurement of numerous parameters per cell. These studies have revealed remarkable heterogeneity within cell populations that were previously thought to be rather uniform. Single-cell profiling has emerged as a powerful tool enabling the identification of previously unknown functional cell states and characterization of differentiation trajectories. Sequencing-based approaches now allow the examination of transcriptome, proteome, and epigenome of individual cells either alone or in combination (multi-omics) with thousands of cells per experiment. High-throughput cytometry approaches such as fluorescent flow cytometry, which analyze millions of cells in a typical experiment, have also seen an increase in the number parameters that can be measured per cell. The expanding spectrum of single-cell profiling techniques holds great promise for providing fresh insight into the underlying biology of both health and disease (Fig. 1). In this review, we outline recent technological advances and explore their application in studies of SpA pathogenesis.

Single-cell profiling—overview. Created with https://www.biorender.com/
Methods for the Profiling of Single Cells in Suspension
Fluorescent Flow Cytometry
Cells in suspension are fluorescently labeled, typically using fluorochrome-conjugated antibodies. In the flow cytometer, the cells are directed in a single file line past a series of lasers which excite the fluorochromes to emit photons with a characteristic spectrum of wavelengths. Current high-end cytometers contain up to six lasers and can process up to 30,000 cells per second. In conventional fluorescent flow cytometry, the intensity of the emitted light is measured in a narrow window around the peak emission wavelength of each fluorochrome. Most fluorochromes have relatively broad emission spectra resulting in fluorescent spillover between the optical channels of the cytometer. This necessitates mathematical data correction, referred to as compensation, which becomes increasingly complex as the number of fluorochromes increases. In spectral flow cytometry, instead of measuring emission peaks using discrete optical channels (one channel for each fluorochrome), the full spectrum of the emitted light is analyzed [2]. The contribution of individual fluorochromes to the total emitted light signal is then determined by “spectral unmixing.” This approach has been shown to provide better resolution between closely related dyes, thereby increasing the number of parameters that can be measured simultaneously [3]. Recent publications reported staining panels with > 40 parameters per cell [4•, 5•]. A major advantage of fluorescent flow cytometry, both conventional and spectral, is that the analysis of cells can be combined with a sorting mechanism that permits the collection of phenotypically defined viable cells for subsequent in vitro studies Table 1.
Image-Based Flow Cytometry
Image-based flow cytometry differs from fluorescent flow cytometry in that a picture is taken from each cell so that cell morphology can be assessed along with the fluorescent signals [6]. While cell throughput and fluorescent options have been lagging behind fluorescent flow cytometry, a recent study introduced an image-enabled cell sorter that could sort cells at a rate of ~ 15,000 cells/s [7]. Examples for the use of image-based flow cytometry include the study of apoptosis (by examining changes in cellular morphology), signal transduction, and protein-protein and cell-cell interactions [6].
Mass Cytometry
The term CyTOF, cytometry by time-of-flight, is often used synonymously with mass cytometry but is really a trademark for a commercial application of this technology. In mass cytometry, the cells are stained with antibodies that are labeled with elemental metal tags [8]. Individual cells are vaporized by an argon torch which ionizes the metal atoms. The identity and number of the metal ions are then determined using time-of-flight mass spectrometry. The number of detected metal ions is proportional to the abundance of the antigen. Mass cytometry eliminates spectral overlap by differentiating probes based on discrete atomic weights. Typical mass cytometry experiments measure 30–40 parameters simultaneously. While forward and side scatter are used to distinguish lymphocytes, monocytes, and granulocytes in fluorescent flow cytometry, these optical parameters do not exist in mass cytometry. To address this problem, Tsai et al. introduced “scatterbodies,” antibodies that target surrogates of physical cell properties such as cytoskeletal and granule components [9]. The physics of mass cytometry limit cellular throughput. As the cells pass through the mass cytometer, many are lost and only 30 to 40% of cells can be analyzed. The ion clouds generated by the torch also require a finite spacing that limits the rate of analysis to ~ 1,000 cells per second. Moreover, cells get destroyed during analysis, and cell sorting for subsequent experiments is impossible [10].
Sequencing-Based Cytometry
Cytometry by sequencing, the most recent addition to the single-cell profiling tool box, includes single-cell RNA-sequencing (scRNA-seq) and a variety of related applications that rely on next-generation sequencing (NGS) for target quantification. scRNA-seq enables unbiased, transcriptomic analysis of individual cells [11]. Using droplet-based approaches, investigators can now perform single-cell transcriptomic analysis of heterogenous cell populations with tens of thousands of cells [12]. Droplet-based scRNA-seq works by encapsulating an individual cell in a lipid droplet containing beads attached to a barcode and unique molecular identifiers. The cell is lysed and its RNA binds to the beads. During reverse transcription, which is performed inside the droplet, each cDNA molecule gets tagged with the cell-specific barcode and the unique molecular identifier. All subsequent steps can then be done with pooled cDNA. While fluorescent and mass cytometry depend on prior knowledge of the cellular phenotypes expected in a sample in order to design an informative antibody staining panel, scRNA-seq overcomes this limitation by providing an unbiased assessment of the entire transcriptome. The ability to do this for tens of thousands of cells at the same time has ushered in a new era of hypothesis-generating research studies. scRNA-seq has some disadvantages though. Lowly transcribed genes and very short transcripts may not be picked up by current sequencing methods. Moreover, multiplets, multiple cells trapped in the same droplet, or ambient RNA from lysed cells may confound the experimental output if not accounted for using appropriate technical or bioinformatic strategies to eliminate them from the downstream data analysis [13,14,15,16]. Similar to mass cytometry, cells are destroyed during sample processing. In addition, the workflow is significantly longer compared to non-sequencing methods, the bioinformatic requirements for data analysis are higher, and costs remain substantial.
In 2017, two groups independently described methods to combine RNA-seq with protein quantification at the single-cell level, which they called cellular indexing of transcriptomes and epitopes (CITE-seq) and RNA expression and protein sequencing (REAP-seq), respectively [17, 18]. Similar to other cytometric methods, antibodies specific for protein targets of interest are conjugated to a reporter which in this case is a unique oligonucleotide barcode. After staining, a droplet-based system is used to sequence the antibody barcodes and the cellular transcriptome simultaneously. The number of protein targets that can be measured with sequencing-based cytometry is limited only by the length of the barcode used to label each antibody clone. For example, using a barcode length of eight nucleotides provides over 65,000 unique identifiers. In the original REAP-seq study, 82 proteins were labeled with barcoded antibodies in addition to sequencing 20,000 genes [18]. In an analogous way, single-cell sequencing has been used to detect epigenetic modifications at the single-cell level. In the single-cell assay for transposase-accessible chromatin using sequencing (scATAC-seq), the prokaryotic Tn5 transposase inserts sequencing adapters into open chromatin regions, thereby allowing the genome-wide identification of open chromatin regions, often marking regulatory elements.[19] The combination of sequencing-based cytometry techniques for multi-omics analysis at the single-cell level is a very rapidly evolving field [20, 21, 22•, 23•].
Methods for the Profiling of Single Cells in Tissues
Traditional methods of immunohistochemistry (IHC), fluorescence microscopy, or in situ hybridization have been limited by the small number of cellular markers that can be measured simultaneously. Isolating cells from tissues for high-dimensional analysis of single-cell suspensions has therefore been used but is associated with a number of limitations. Fortunately, the technological advancements in profiling single cells in suspension have spilled over into the analysis of cells in tissues with a rapid growth of the diversity and power of available techniques (Table 2). An overview of major trends is provided below, and more detailed information can be found in dedicated reviews [24, 25•, 26, 27•].
Fluorescence Microscopy
Similar to fluorescent flow cytometry, spectral overlap is a major limiting factor of fluorescence microscopy [28]. Nevertheless, confocal microscopes using spectral array detectors in combination with sequential imaging have allowed measuring up to 14 colors per slide [29]. In sequential imaging, the tissue section is stained with a first set of antibodies and imaged. After removal of the antibodies (or just the fluorochromes), the section is stained with a second set of antibodies and imaged again. The obtained images are then combined in silico. In a variation of this approach, the monoclonal antibodies are labeled with a DNA barcode (rather than directly with fluorochromes). Tissue sections are first incubated with a panel of antibodies followed by hybridization with fluorescently labeled oligonucleotides. Co-detection by indexing (CODEX) using DNA-conjugated antibodies has been used to detect up to 60 markers on the same slide [30•]. Fluorescence microscopy has been helped by the development of tissue-clearing techniques which permit the penetration of staining reagents deep into tissues and imaging of three-dimensional structures using confocal microscopy [31, 32].
Imaging Mass Cytometry
Here, the tissue sections are stained with monoclonal antibodies labeled with metal tags. Two major variants have been described that differ in how the abundance of the metal tags is quantified: scanning mass cytometry (SMC) and multiplexed ion-beam imaging (MIBI) [24, 33, 34]. Imaging mass cytometry offers high sensitivity and dynamic range, and there is little to no channel cross-talk due to the discrete mass of the metal tags. However, data acquisition is slow and the area that can be scanned is small [35]. Segmentation algorithms can be applied to identify individual cells in the tissue section. These cells isolated in silico can then be analyzed using approaches developed for cells in suspension. Moreover, the generated two- or three-dimensional maps can be analyzed quantitatively to identify cell clusters or study cell-cell interactions between cells of different type or function [36].
Spatial Transcriptomics
This technique, also called spatially resolved transcriptomics, was highlighted as the Method of the Year 2020 [37]. Two principal approaches for the highly multiplexed quantification of gene transcripts in tissue sections have been described, NGS-based spatial transcriptomics and imaging-based spatial transcriptomics [25•]. In NGS-based spatial transcriptomics, mRNA is released from the tissue section and captured so that information about the localization of each molecule in two-dimensional space is retained. In the original description of this method [38], the tissue section is placed on a slide with a grid of dots (100 μm diameter, spaced 200 μm center-to-center) each coated with barcoded oligo-dT constructs. After reverse transcription is performed on the slide, the cDNA molecules are released and transcript frequency is measured by standard NGS. The origin of each transcript can be traced back to individual dots in the grid by means of the location-specific barcodes. The grid can then be overlaid onto a traditionally stained tissue section to correlate gene expression with histology. Using secondary data sources, it is possible to identify signatures of cell types in the bulk signal from each dot that permits (along with the histological appearance) drawing conclusion about the cells present in this area of the tissue section. NGS-based spatial transcriptomics approaches appeal to translational research for several reasons. They are relatively simple to implement due to their compatibility with standard laboratory instruments and established NGS workflows. They are also compatible with a variety of tissue and sample types. Recent advances have enabled the use of formalin-fixed paraffin-embedded (FFPE) tissues, which is ideal for applying this technology to previously collected clinical pathology specimens. Major limitations are a lack of customizability to detect specific transcripts and the relative low spatial resolution. NGS-based spatial transcriptomics is strictly speaking not currently single-cell analysis, since the barcoded area typically encompasses more than one cell. However, newer versions of this method, for instance, using barcoded beads, have substantially improved spatial resolution approaching cell size [39].
In imaging-based spatial transcriptomics, RNA-seq or multiplexed single-molecule FISH (smFISH) are performed on the tissue section and the results are read out using fluorescence microscopy. Examples include STARmap (spatially resolved transcript amplicon readout mapping), MERFISH (multiplexed error-robust fluorescence in situ hybridization), and seqFISH (sequential fluorescence in situ hybridization) [40,41,42]. Imaging-based spatial transcriptomics combines high sensitivity with high spatial resolution. One potential disadvantage of multiplexed smFISH is the limited number of mRNA probes, currently around 300 to 1000 targets, depending on the technology platform. Another challenge with this type of technology is the assignment of transcripts to segmented cells. As technology and bioinformatic methods continue to improve, one can expect that measurement of gene expression will continue to improve in terms of dimension, accuracy, and specificity.
Applications in Spondyloarthritis Research
In the following section, we highlight recently published studies as examples for the application of these techniques in SpA research that show both the promise and challenges in this field.
Indexing and Functional Characterization of Cells in a Tissue
A state-of-the-art example for this approach is provided by a recent study from the Accelerating Medicines Partnership (AMP) consortium which analyzed synovial tissue specimens from patients with rheumatoid arthritis. A total of 314,011 analyze cells from 79 patients were analyzed by CITE-seq. The combination of transcriptional profiling by RNA-seq with 58 surface protein markers identified 6 major cell types (B or plasma cells, T cells, NK cells, myeloid cells, fibroblasts, endothelial cells) with a total of 77 distinct cell states [43] which were then analyzed further. A similarly scaled study analyzed 528,253 cells from 19 skin samples with scRNA-seq revealing 34 cell types or states in 4 major groups (dermal non-immune cells, epidermal non-immune cells, lymphocytes and mast cells, antigen presenting cells) [44]. Sequencing-based cytometry is particularly well suited for cellular indexing because of the large number of parameters that can be measured at the same time. This avoids the need to make a priori restrictive choices that limit the analysis to predefined cell types. At least theoretically, an unbiased snapshot of all cells and their phenotypes in a given tissue can be obtained [45]. Practically, the snapshot will be incomplete though, as certain cell types (e.g., osteoclasts and adipocytes) are difficult to extract from the tissue. Moreover, the cell isolation procedure may impact the number and phenotype of the cells. Considerations of cost and feasibility often dictate to limit the analysis to subsets of cells such as CD45+ hematopoietically derived cells [46] or (even more restricted) distinct immune cells populations [47, 48].
Analysis of Samples with Very Few Cells
The unbiased measuring of many parameters with sequencing-based approaches is also useful when only a very small number of cells are available for analysis. Kasper et al. applied scRNA-seq to the analysis of cells in the aqueous humor of patients with acute anterior uveitis (AAU) [49]. The authors screened 4980 subjects and were ultimately able to obtain aqueous humor from the anterior chamber of the eye of 4 subjects with HLA-B27+ AAU, 2 with HLA-B27- AAU, and 1 patient with bacterial endophthalmitis. After quality control, they could analyze 12,305 total cells which they assigned to 13 distinct clusters. Not surprisingly, the bacterial endophthalmitis sample had more neutrophils, but there were also differences between the HLA-B27+ and HLA-B27- AAU patients. Unfortunately, a more detailed analysis was hampered by the small number and high-interindividual variability between samples. Batch variability is a well-recognized problem in scRNA-seq studies, and methods such as Harmony, LIGER, or Seurat 3 have been developed to adjust for this problem during data analysis [50,51,52]. However, a smart experimental design that minimizes the risk for batch effects in the first place remains essential.
Identification of Disease-Associated Immune Cell Subsets
Several studies have attempted to identify disease-associated subpopulations in SpA based on the premise that only a small fraction of inflammatory cells present in peripheral blood or diseased tissue is central to the disease process. For example, Qaiyum et al. designed a 36-marker mass cytometry panel enriched for integrins and cell adhesion molecules to analyze peripheral blood and synovial fluid from patients with axSpA. They found a distinct population of CD103+CD49a+CD8+ T cells (InEx cells) to be expanded in axSpA patients [53] that phenotypically corresponds to tissue-resident memory T cells [54]. Alber et al. studied PBMCs from patients with AS and healthy controls using CITE-seq and identified an NK cell population expressing CD16, CD161, and CD38 as well as cytotoxic genes that was overrepresented in AS patients [55••]. Steel et al. analyzed cytokine production of synovial fluid mononuclear cells from patients with PsA finding an enrichment of tissue-resident memory CD8+ T cells in the joints of PsA patients that were polyfunctional and expressed IL-17A as well as IFN-γ, TNF, and GM-CSF [56]. Using mass cytometry, Yager et al. described an expansion of monocytes and macrophages producing osteopontin, CCL2, and IL-8 in PsA synovial fluid [57].
Clonal Analysis of Lymphocyte Receptors
A recurring theme in cellular profiling studies in SpA and PsA has been a renewed focus on CD8+ T cells. Studies in the 1990s had demonstrated clonal expansion of CD8+ T cells in reactive arthritis and AS [58] consistent with an HLA-B27-restricted T cell response. However, technical limitations at the time prevented the identification of the antigenic targets of this immune response. Today’s single-cell technologies permit revisiting the arthritogenic peptide hypothesis by studying the TCR repertoire in individual cells isolated from target tissues. Using scTCR-seq, recent studies confirmed the clonal expansions of CD8+ T cells in synovial fluid in PsA and SpA [59,60,61,62••] as well as in aqueous humor in HLA-B27+ AAU patients [62••]. Importantly, these studies consistently identified cells with the same TRBV9-TRBJ2-3 sequence that was previously found in studies in cloned CD8+ T cells from reactive arthritis patients [63] and bulk CD8+ T cells [64]. Yang et al. then expressed recombinant TRCs and used yeast display to identify HLA-B27-restricted peptides recognized by TCRs with this conserved CDR3 sequence. These peptides mapped to disease associated microbes and human proteins [62••].
Predictive Biomarkers and Signatures of Therapeutic Response
Solberg et al. analyzed peripheral blood from patients with psoriasis using a 26-parameter mass cytometry panel searching for signatures associated with a response to three types of biologics. They found a reduction of CD8+ T cells and Th17 subsets that correlated with a reduction in PASI score. However, the number of analyzed patients was probably too small to identify a cellular biomarker predictive of response [65]. In a rare analysis of tissue specimens from patients enrolled in a phase 3 randomized controlled treatment trial, Mehta et al. compared skin biopsy samples from patients with plaque psoriasis treated with the IL-17A inhibitor secukinumab or the IL-23p19 inhibitor guselkumab. Tissue specimens collected at baseline and after 4 and 24 weeks of therapy were digested and analyzed by fluorescent flow cytometry. Interestingly, inhibition of IL-17A vs. IL-23 had differential effects on kinetics and type of observed changes in the inflammatory infiltrate. While both treatments resulted in a reduction of CD4+CD103-CD49a- T cells, only the IL-23p19 inhibitor reduced the frequency of CD8+ tissue-resident memory T cells at week 24 [66••].
Understanding Tissue Organization
In the first application of spatial transcriptomics in rheumatology, Carlberg et al. studied synovial tissue specimens from seropositive RA and SpA. Analyzing gene expression in lymphocyte aggregates, they found an enrichment of genes involved in adaptive immunity in RA and genes involved in extracellular matrix organization and tissue repair in SpA. While the resolution of the technique did not allow for the identification of individual cells, the authors could identify areas that were enriched for CD4+ and CD8+ memory T cells, class-switched B cells, and plasma cells, respectively [67]. Despite its limitations including small sample number and limited spatial resolution, the study highlights how spatial transcriptomics can provide insight in the organization of disease tissues and differences between different diseases.
Access to Diseased Tissue in SpA
Access to tissue samples has been a major challenge in studying SpA. While skin biopsies and joint aspirations are performed routinely in clinical practice and the feasibility of U/S-guided synovial biopsies for research purposes is established, the analysis of enthesial or spinal tissue is problematic. A single paper analyzed a small number of needle biopsy samples from the Achilles tendon enthesis, but there were no follow-up studies [68]. More recently, Pachowsky et al. reported the collection of tissue specimens suitable for IHC and gene expression analysis from the lateral epicondylus of the elbow using a low morbidity surgical procedure [69••]. The McGonagle group has published a series of papers analyzing discarded surgical specimens from non-AS patients undergoing laminectomy [70, 71]. A recent study from that group demonstrated the presence of MAIT cells at spinal entheses [72], but the value of these findings for understanding axSpA pathogenesis is diminished by the lack of tissue from axSpA patients. Appel et al. analyzed facet joint samples from AS patients undergoing corrective spine surgery in a series of papers [73,74,75]. Surgical specimens from axSpA patients are typically from late stage AS although it might be possible to distinguish lesions at various stages of evolution in the same patient [76]. Alternatively, the sacroiliac joint and subchondral bone might represent a valuable although currently underappreciated source of tissue for analysis [77]. There is a major unmet need for tissue specimens from the inflamed spine and SI joints from patients with axSpA and axial PsA.
Conclusions
The number of studies that have used single-cell profiling methodology for the analysis of cells in suspension or tissues in SpA is still small. Recent studies in RA and SLE from the AMP consortium have demonstrated the value of multi-center efforts for standardized sample collection and the critical importance of studying cells from diseased tissues rather than peripheral blood [78, 79]. AMP 2.0 includes patients with psoriatic disease, and one should hope that there will be similar efforts focusing on axSpA in the future.
Data Availability
Not applicable
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ermann J. Pathogenesis of axial spondyloarthritis - sources and current state of knowledge. Rheum Dis Clin North Am. 2020;46(2):193–206. https://doi.org/10.1016/j.rdc.2020.01.016.
Futamura K, Sekino M, Hata A, Ikebuchi R, Nakanishi Y, Egawa G, et al. Novel full-spectral flow cytometry with multiple spectrally-adjacent fluorescent proteins and fluorochromes and visualization of in vivo cellular movement. Cytometry A. 2015;87(9):830–42. https://doi.org/10.1002/cyto.a.22725.
Niewold P, Ashhurst TM, Smith AL, King NJC. Evaluating spectral cytometry for immune profiling in viral disease. Cytometry A. 2020;97(11):1165–79. https://doi.org/10.1002/cyto.a.24211.
Park LM, Lannigan J, Jaimes MC. OMIP-069: forty-color full spectrum flow cytometry panel for deep immunophenotyping of major cell subsets in human peripheral blood. Cytometry A. 2020;97(10):1044–51. https://doi.org/10.1002/cyto.a.24213. Description of a 40-color staining panel for human immune cells documenting the power of spectral flow cytometry.
Sahir F, Mateo JM, Steinhoff M, Siveen KS. Development of a 43 color panel for the characterization of conventional and unconventional T-cell subsets, B cells, NK cells, monocytes, dendritic cells, and innate lymphoid cells using spectral flow cytometry. Cytometry A. 2020; https://doi.org/10.1002/cyto.a.24288. Description of a 43-color staining panel for human immune cells documenting the power of spectral flow cytometry.
Zuba-Surma EK, Ratajczak MZ. Analytical capabilities of the ImageStream cytometer. Methods Cell Biol. 2011;102:207–30. https://doi.org/10.1016/b978-0-12-374912-3.00008-0.
Schraivogel D, Kuhn TM, Rauscher B, Rodriguez-Martinez M, Paulsen M, Owsley K, et al. High-speed fluorescence image-enabled cell sorting. Science. 2022;375(6578):315–20. https://doi.org/10.1126/science.abj3013.
Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81(16):6813–22. https://doi.org/10.1021/ac901049w.
Tsai AG, Glass DR, Juntilla M, Hartmann FJ, Oak JS, Fernandez-Pol S, et al. Multiplexed single-cell morphometry for hematopathology diagnostics. Nat Med. 2020;26(3):408–17. https://doi.org/10.1038/s41591-020-0783-x.
Bendall SC, Nolan GP, Roederer M, Chattopadhyay PK. A deep profiler’s guide to cytometry. Trends Immunol. 2012;33(7):323–32. https://doi.org/10.1016/j.it.2012.02.010.
Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–82. https://doi.org/10.1038/nmeth.1315.
Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202–14. https://doi.org/10.1016/j.cell.2015.05.002.
Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018;19(1):224. https://doi.org/10.1186/s13059-018-1603-1.
Wolock SL, Lopez R, Klein AM. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 2019;8(4):281–91 e9. https://doi.org/10.1016/j.cels.2018.11.005.
Kang HM, Subramaniam M, Targ S, Nguyen M, Maliskova L, McCarthy E, et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol. 2018;36(1):89–94. https://doi.org/10.1038/nbt.4042.
Young MD, Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience. 2020;9(12) https://doi.org/10.1093/gigascience/giaa151.
Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–8. https://doi.org/10.1038/nmeth.4380.
Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol. 2017;35(10):936–9. https://doi.org/10.1038/nbt.3973.
Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523(7561):486–90. https://doi.org/10.1038/nature14590.
Swanson E, Lord C, Reading J, Heubeck AT, Genge PC, Thomson Z, et al. Simultaneous trimodal single-cell measurement of transcripts, epitopes, and chromatin accessibility using TEA-seq. Elife. 2021:10. https://doi.org/10.7554/eLife.63632.
Mimitou EP, Lareau CA, Chen KY, Zorzetto-Fernandes AL, Hao Y, Takeshima Y, et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat Biotechnol. 2021;39(10):1246–58. https://doi.org/10.1038/s41587-021-00927-2.
Baysoy A, Bai Z, Satija R, Fan R. The technological landscape and applications of single-cell multi-omics. Nat Rev Mol Cell Biol. 2023:1–19. https://doi.org/10.1038/s41580-023-00615-w. State-of-the-art review of single-cell multi-omics.
Wang X, Fan D, Yang Y, Gimple RC, Zhou S. Integrative multi-omics approaches to explore immune cell functions: challenges and opportunities. iScience. 2023;26(4):106359. https://doi.org/10.1016/j.isci.2023.106359. State-of-the-art review of single-cell multi-omics
Bodenmiller B. Multiplexed epitope-based tissue imaging for discovery and healthcare applications. Cell Syst. 2016;2(4):225–38. https://doi.org/10.1016/j.cels.2016.03.008.
Rao A, Barkley D, Franca GS, Yanai I. Exploring tissue architecture using spatial transcriptomics. Nature. 2021;596(7871):211–20. https://doi.org/10.1038/s41586-021-03634-9. State-of-the-art review of spatial transcriptomics.
Hickey JW, Neumann EK, Radtke AJ, Camarillo JM, Beuschel RT, Albanese A, et al. Spatial mapping of protein composition and tissue organization: a primer for multiplexed antibody-based imaging. Nat Methods. 2022;19(3):284–95. https://doi.org/10.1038/s41592-021-01316-y.
Moffitt JR, Lundberg E, Heyn H. The emerging landscape of spatial profiling technologies. Nat Rev Genet. 2022;23(12):741–59. https://doi.org/10.1038/s41576-022-00515-3. State-of-the-art review of spatial transcriptomics.
Ranjit S, Lanzano L, Libby AE, Gratton E, Levi M. Advances in fluorescence microscopy techniques to study kidney function. Nat Rev Nephrol. 2021;17(2):128–44. https://doi.org/10.1038/s41581-020-00337-8.
Gerner MY, Kastenmuller W, Ifrim I, Kabat J, Germain RN. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity. 2012;37(2):364–76. https://doi.org/10.1016/j.immuni.2012.07.011.
Black S, Phillips D, Hickey JW, Kennedy-Darling J, Venkataraaman VG, Samusik N, et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat Protoc. 2021;16(8):3802–35. https://doi.org/10.1038/s41596-021-00556-8. Imaging of up to 60 protein markers in tissue sections using DNA-conjugated monoclonal antibodies and fluorescently labelled oligonucleotide probes.
Yang B, Treweek JB, Kulkarni RP, Deverman BE, Chen CK, Lubeck E, et al. Single-cell phenotyping within transparent intact tissue through whole-body clearing. Cell. 2014;158(4):945–58. https://doi.org/10.1016/j.cell.2014.07.017.
Weiss KR, Voigt FF, Shepherd DP, Huisken J. Tutorial: practical considerations for tissue clearing and imaging. Nat Protoc. 2021;16(6):2732–48. https://doi.org/10.1038/s41596-021-00502-8.
Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11(4):417–22. https://doi.org/10.1038/nmeth.2869.
Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, et al. Multiplexed ion beam imaging of human breast tumors. Nat Med. 2014;20(4):436–42. https://doi.org/10.1038/nm.3488.
Chang Q, Ornatsky OI, Siddiqui I, Loboda A, Baranov VI, Hedley DW. Imaging mass cytometry. Cytometry A. 2017;91(2):160–9. https://doi.org/10.1002/cyto.a.23053.
Kuett L, Catena R, Ozcan A, Pluss A, Cancer Grand Challenges IC, Schraml P, et al. Three-dimensional imaging mass cytometry for highly multiplexed molecular and cellular mapping of tissues and the tumor microenvironment. Nat Cancer. 2022;3(1):122–33. https://doi.org/10.1038/s43018-021-00301-w.
Editorial. Method of the Year 2020: spatially resolved transcriptomics. Nat Methods. 2021;18(1):1. https://doi.org/10.1038/s41592-020-01042-x.
Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353(6294):78–82. https://doi.org/10.1126/science.aaf2403.
Stickels RR, Murray E, Kumar P, Li J, Marshall JL, Di Bella DJ, et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat Biotechnol. 2021;39(3):313–9. https://doi.org/10.1038/s41587-020-0739-1.
Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 2018;361(6400) https://doi.org/10.1126/science.aat5691.
Moffitt JR, Hao J, Wang G, Chen KH, Babcock HP, Zhuang X. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc Natl Acad Sci U S A. 2016;113(39):11046–51. https://doi.org/10.1073/pnas.1612826113.
Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature. 2019;568(7751):235–9. https://doi.org/10.1038/s41586-019-1049-y.
Zhang F, Jonsson AH, Nathan A, Millard N, Curtis M, Xiao Q, et al. Deconstruction of rheumatoid arthritis synovium defines inflammatory subtypes. Nature. 2023;623(7987):616–24. https://doi.org/10.1038/s41586-023-06708-y.
Reynolds G, Vegh P, Fletcher J, Poyner EFM, Stephenson E, Goh I, et al. Developmental cell programs are co-opted in inflammatory skin disease. Science. 2021;371(6527) https://doi.org/10.1126/science.aba6500.
Morris SA. The evolving concept of cell identity in the single cell era. Development. 2019;146(12) https://doi.org/10.1242/dev.169748.
Liu Y, Wang H, Taylor M, Cook C, Martinez-Berdeja A, North JP, et al. Classification of human chronic inflammatory skin disease based on single-cell immune profiling. Sci Immunol. 2022;7(70):eabl9165. https://doi.org/10.1126/sciimmunol.abl9165.
Nakamizo S, Dutertre CA, Khalilnezhad A, Zhang XM, Lim S, Lum J, et al. Single-cell analysis of human skin identifies CD14+ type 3 dendritic cells co-producing IL1B and IL23A in psoriasis. J Exp Med. 2021;218(9) https://doi.org/10.1084/jem.20202345.
Liu J, Chang HW, Huang ZM, Nakamura M, Sekhon S, Ahn R, et al. Single-cell RNA sequencing of psoriatic skin identifies pathogenic Tc17 cell subsets and reveals distinctions between CD8(+) T cells in autoimmunity and cancer. J Allergy Clin Immunol. 2021;147(6):2370–80. https://doi.org/10.1016/j.jaci.2020.11.028.
Kasper M, Heming M, Schafflick D, Li X, Lautwein T, Zu M, Horste M, et al. Intraocular dendritic cells characterize HLA-B27-associated acute anterior uveitis. Elife. 2021:10. https://doi.org/10.7554/eLife.67396.
Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–96. https://doi.org/10.1038/s41592-019-0619-0.
Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell. 2019;177(7):1873–87 e17. https://doi.org/10.1016/j.cell.2019.05.006.
Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888–902 e21. https://doi.org/10.1016/j.cell.2019.05.031.
Qaiyum Z, Gracey E, Yao Y, Inman RD. Integrin and transcriptomic profiles identify a distinctive synovial CD8+ T cell subpopulation in spondyloarthritis. Ann Rheum Dis. 2019;78(11):1566–75. https://doi.org/10.1136/annrheumdis-2019-215349.
Weisberg SP, Ural BB, Farber DL. Tissue-specific immunity for a changing world. Cell. 2021;184(6):1517–29. https://doi.org/10.1016/j.cell.2021.01.042.
Alber S, Kumar S, Liu J, Huang ZM, Paez D, Hong J, et al. Single cell transcriptome and surface epitope analysis of ankylosing spondylitis facilitates disease classification by machine learning. Front Immunol. 2022;13:838636. https://doi.org/10.3389/fimmu.2022.838636. CITE-seq study of human PBMCs that identifies an expanded population of CD16+CD161+CD38+ NK cells in AS.
Steel KJA, Srenathan U, Ridley M, Durham LE, Wu SY, Ryan SE, et al. Polyfunctional, proinflammatory, tissue-resident memory phenotype and function of synovial interleukin-17A+CD8+ T cells in psoriatic arthritis. Arthritis Rheumatol. 2020;72(3):435–47. https://doi.org/10.1002/art.41156.
Yager N, Cole S, Lledo Lara A, Maroof A, Penkava F, Knight JC, et al. Ex vivo mass cytometry analysis reveals a profound myeloid proinflammatory signature in psoriatic arthritis synovial fluid. Ann Rheum Dis. 2021;80(12):1559–67. https://doi.org/10.1136/annrheumdis-2021-220280.
Hermann E, Yu DT, Meyer zum Büschenfelde KH, Fleischer B. HLA-B27-restricted CD8 T cells derived from synovial fluids of patients with reactive arthritis and ankylosing spondylitis. Lancet. 1993;342(8872):646–50. https://doi.org/10.1016/0140-6736(93)91760-j.
Penkava F, Velasco-Herrera MDC, Young MD, Yager N, Nwosu LN, Pratt AG, et al. Single-cell sequencing reveals clonal expansions of pro-inflammatory synovial CD8 T cells expressing tissue-homing receptors in psoriatic arthritis. Nat Commun. 2020;11(1):4767. https://doi.org/10.1038/s41467-020-18513-6.
Simone D, Penkava F, Ridley A, Sansom S, Al-Mossawi MH, Bowness P. Single cell analysis of spondyloarthritis regulatory T cells identifies distinct synovial gene expression patterns and clonal fates. Comm Biology. 2021;4(1) https://doi.org/10.1038/s42003-021-02931-3.
Deschler K, Rademacher J, Lacher SM, Huth A, Utzt M, Krebs S, et al. Antigen-specific immune reactions by expanded CD8(+) T cell clones from HLA-B*27-positive patients with spondyloarthritis. J Autoimmun. 2022;133:102901. https://doi.org/10.1016/j.jaut.2022.102901.
Yang X, Garner LI, Zvyagin IV, Paley MA, Komech EA, Jude KM, et al. Autoimmunity-associated T cell receptors recognize HLA-B*27-bound peptides. Nature. 2022;612(7941):771–7. https://doi.org/10.1038/s41586-022-05501-7. scTCR-seq analysis of CD8+ T cells from synovial fluid and aqueous humor of HLA-B27+ patients with AS and AAU. Evidence for expansion of distinct TCR clonotypes and identification of HLA-B27 restricted candidate peptides.
May E, Dulphy N, Frauendorf E, Duchmann R, Bowness P, Lopez de Castro JA, et al. Conserved TCR beta chain usage in reactive arthritis; evidence for selection by a putative HLA-B27-associated autoantigen. Tissue Antigens. 2002;60(4):299–308. https://doi.org/10.1034/j.1399-0039.2002.600404.x.
Faham M, Carlton V, Moorhead M, Zheng J, Klinger M, Pepin F, et al. Discovery of T cell receptor β motifs specific to HLA-B27-positive ankylosing spondylitis by deep repertoire sequence analysis. Arthritis Rheumatol. 2017;69(4):774–84. https://doi.org/10.1002/art.40028.
Solberg SM, Aarebrot AK, Sarkar I, Petrovic A, Sandvik LF, Bergum B, et al. Mass cytometry analysis of blood immune cells from psoriasis patients on biological therapy. Eur J Immunol. 2021;51(3):694–702. https://doi.org/10.1002/eji.202048857.
Mehta H, Mashiko S, Angsana J, Rubio M, Hsieh YM, Maari C, et al. Differential changes in inflammatory mononuclear phagocyte and T-cell profiles within psoriatic skin during treatment with guselkumab vs. secukinumab. J Invest Dermatol. 2021;141(7):1707–18 e9. https://doi.org/10.1016/j.jid.2021.01.005. Fluorescent flow cytometric analysis of skin biopsy specimens from a head-to-head randomized controlled clinical trial comparing IL-17A with IL-23p19 inhibition in psoriasis. A differential effect of the two treatment modalities on the inflammatory infiltrate is demonstrated.
Carlberg K, Korotkova M, Larsson L, Catrina AI, Stahl PL, Malmstrom V. Exploring inflammatory signatures in arthritic joint biopsies with spatial transcriptomics. Sci Rep. 2019;9(1):18975. https://doi.org/10.1038/s41598-019-55441-y.
McGonagle D, Marzo-Ortega H, O'Connor P, Gibbon W, Hawkey P, Henshaw K, Emery P. Histological assessment of the early enthesitis lesion in spondyloarthropathy. Ann Rheum Dis. 2002;61(6):534–7. https://doi.org/10.1136/ard.61.6.534.
Pachowsky ML, Raimondo MG, Xu C, Rauber S, Tascilar K, Labinsky H, et al. Concise report: a minimal-invasive method to retrieve and identify entheseal tissue from psoriatic arthritis patients. Ann Rheum Dis. 2022; https://doi.org/10.1136/annrheumdis-2021-222061. Description of a novel technique for the isolation of human enthesial cells for cellular and molecular analysis.
Cuthbert RJ, Fragkakis EM, Dunsmuir R, Li Z, Coles M, Marzo-Ortega H, et al. Brief report: group 3 innate lymphoid cells in human enthesis. Arthritis Rheumatol. 2017;69(9):1816–22. https://doi.org/10.1002/art.40150.
Cuthbert RJ, Watad A, Fragkakis EM, Dunsmuir R, Loughenbury P, Khan A, et al. Evidence that tissue resident human enthesis gammadeltaT-cells can produce IL-17A independently of IL-23R transcript expression. Ann Rheum Dis. 2019;78(11):1559–65. https://doi.org/10.1136/annrheumdis-2019-215210.
Rosine N, Rowe H, Koturan S, Yahia-Cherbal H, Leloup C, Watad A, et al. Characterization of blood mucosal-associated invariant T cells in patients with axial spondyloarthritis and of resident mucosal-associated invariant T cells from the axial entheses of non-axial spondyloarthritis control patients. Arthritis Rheumatol. 2022;74(11):1786–95. https://doi.org/10.1002/art.42090.
Appel H, Kuhne M, Spiekermann S, Ebhardt H, Grozdanovic Z, Kohler D, et al. Immunohistologic analysis of zygapophyseal joints in patients with ankylosing spondylitis. Arthritis Rheum. 2006;54(9):2845–51. https://doi.org/10.1002/art.22060.
Appel H, Loddenkemper C, Grozdanovic Z, Ebhardt H, Dreimann M, Hempfing A, et al. Correlation of histopathological findings and magnetic resonance imaging in the spine of patients with ankylosing spondylitis. Arthritis Res Ther. 2006;8(5):R143. https://doi.org/10.1186/ar2035.
Appel H, Maier R, Wu P, Scheer R, Hempfing A, Kayser R, et al. Analysis of IL-17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res Ther. 2011;13(3):R95. https://doi.org/10.1186/ar3370.
Yu T, Zhang J, Zhu W, Wang X, Bai Y, Feng B, et al. Chondrogenesis mediates progression of ankylosing spondylitis through heterotopic ossification. Bone Res. 2021;9(1):19. https://doi.org/10.1038/s41413-021-00140-6.
Egund N, Sorensen FB, Ostgard R, Loft AG, Boel LWT, Jurik AG. CT-guided transarticular biopsy of the sacroiliac joint: technique and histomorphological results. A preliminary study. Skeletal Radiol. 2020;49(3):453–60. https://doi.org/10.1007/s00256-019-03305-x.
Donlin LT, Rao DA, Wei K, Slowikowski K, McGeachy MJ, Turner JD, et al. Methods for high-dimensonal analysis of cells dissociated from cyropreserved synovial tissue. Arthritis Res Ther. 2018;20(1):139. https://doi.org/10.1186/s13075-018-1631-y.
Pisetsky DS. The basic and translational science year in review: Confucius in the era of Big Data. Semin Arthritis Rheum. 2020;50(3):373–9. https://doi.org/10.1016/j.semarthrit.2020.02.010.
Funding
J. E. was supported by NIH grant R21 AR076040-01.
Author information
Authors and Affiliations
Contributions
J. E.: Conceptualization; writing—original draft, review, and editing; and preparation of the figure
M. L.: Writing—original draft
K. W.: Writing—review and editing
M. G. A.: Writing—review and editing
Corresponding author
Ethics declarations
Ethics Approval
Not applicable
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ermann, J., Lefton, M., Wei, K. et al. Understanding Spondyloarthritis Pathogenesis: The Promise of Single-Cell Profiling. Curr Rheumatol Rep 26, 144–154 (2024). https://doi.org/10.1007/s11926-023-01132-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11926-023-01132-7