
Abstract
Purpose of Review
To commemorate the 50th anniversary of the groundbreaking discovery of a remarkably strong association between HLA-B*27 and ankylosing spondylitis (AS).
Recent Findings
In addition to HLA-B*27, more than 116 other recognized genetic risk variants have been identified, while epigenetic factors largely remain unexplored in this context. Among patients with AS who carry the HLA-B*27 gene, clonally expanded CD8 + T cells can be found in their bloodstream and within inflamed tissues. Moreover, the α and β chain motifs of these T-cell receptors demonstrate a distinct affinity for certain self- and microbial-derived peptides, leading to an autoimmune response that ultimately results in the onset of the disease. These distinctive peptide-binding and presentation characteristics are a hallmark of the disease-associated HLA-B*27:05 subtype but are absent in HLA-B*27:09, a subtype not associated with the disease, differing by only a single amino acid. This discovery represents a significant advancement in unraveling the 50-year-old puzzle of how HLA-B*27 contributes to the development of AS.
Summary
These findings will significantly accelerate the process of identifying peptides, both self- and microbial-derived, that instigate autoimmunity. This, in return, will pave the way for the development of more accurate and effective targeted treatments. Moreover, the discovery of improved biomarkers, in conjunction with the emerging technology of electric field molecular fingerprinting, has the potential to greatly bolster early diagnosis capabilities. A very recently published groundbreak paper underscores the remarkable effectiveness of targeting and eliminating disease-causing T cells in a HLA-B*27 patients with AS. This pivotal advancement not only signifies a paradigm shift but also bolsters the potential for preventing the disease in individuals carrying high-risk genetic variants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
This year marks the 50th anniversary of the groundbreaking discovery of a remarkably strong association between HLA-B*27 and ankylosing spondylitis (AS} [1,2,3]. In celebration of this milestone, my focus will be on AS, which represents a more homogeneous and extensively studied clinical condition, as opposed to axial spondyloarthritis (axSpA) encompassing early or milder disease forms and other SpA-related conditions [4,5,6,7].
Discovery of HLA-B*27
In 1969, Erik Thorsby of Norway made a significant breakthrough when he identified a novel human histocompatibility antigen. He accomplished this feat by developing the TH-FJH antibody by a method called “planned immunization” [8]. This specific serologic trait, initially labeled as “w27,” underwent a provisional naming evolution. Its official designation transitioned to HL-A27 at the 4th International Histocompatibility Workshop (IHW) [9]. Subsequently, at the 6th IHW, it was further designated as HLA-B27. With the advent of the DNA-based “HLA Extended Allele Nomenclature” system [10], the nomenclature for HLA loci, including the B locus, now includes an asterisk (*). Consequently, HLA-B27 is now represented as HLA-B*27, aligning with its DNA-level designation. Its prevalence varies widely among genetically unmixed (indigenous) populations and tribal groups worldwide (see Fig. 1). For consistency, throughout this review article, I will use the term HLA-B*27 and italicize it (HLA-B*27) when transitioning from the discussion of the protein molecule to the gene itself.

(Adapted from Khan MA. HLA-B27 and its subtypes in world populations. Curr Opin Rheumatol. 1995 Jul;7(4):263–9. 10.1097/00002281–199507000-00001)
The world map shows a highly variable prevalence of HLA-B*27 among the various genetically unmixed indigenous populations and tribal groups in the world. Legend: the numbers in red color (6 over North America, 5 over South America, and 8 over Australia) indicate the current average prevalence of HLA-B*27 in these regions because of European colonization. Its prevalence is noticeably very high among the various native indigenous tribes of North America in contrast to its virtual absence among a few unmixed native tribes in South America that have been available for study. The indigenous unmixed natives of Australia lack HLA-B*27. The red arrow points to Indonesia where the natives have approximately 12% prevalence of HLA-B*27 but have less AS than the Indonesians of Chinese ancestry that has only 5% prevalence. This apparent paradox results from the fact the HLA-B*27 among the native Indonesians is primarily the HLA-B*27:06 subtype that does predispose to AS, as explained in the text.
Discovery of Its Association with Ankylosing Spondylitis
The journey to this groundbreaking discovery commenced with a study undertaken by a group of British researchers at London’s Westminster Hospital [1, 2]. Notably, some members of this team have since shared firsthand accounts of their involvement in this pivotal investigation [11,12,13,14]. According to these narratives and the insights gleaned from my personal discussions, the year 1971 marked a pivotal juncture. It was during this time that David James, a hematologist who had newly established an HLA-typing laboratory and possessed a keen interest in exploring potential associations between HLA and rheumatic ailments, met with Derrick Brewerton, a rheumatologist, and inquired if there were any rheumatic diseases worth studying. Brewerton, having learned from his mentor, Professor Frank Dudley Hart, about published reports strongly suggesting a genetic predisposition to AS [15, 16], promptly suggested investigating this illness as a worthy subject.
This exchange of ideas paved the way for the journey that would lead to the groundbreaking revelation we discuss today. The investigating team drafted and submitted a study proposal, only to face rejection from their financially constrained hospital administration. Undeterred, the team proceeded to conduct HLA typing on 8 patients with AS, revealing that all possessed HLA-B*27. With the revised proposal gaining approval, their remarkable findings were eventually published in 1973 [2]. Notably, HLA-B*27 was detected in 72 out of 75 (96%) patients with classical (primary) AS, in stark contrast to a mere 3 out of 75 (4%) controls. Family studies further unveiled HLA-B*27’s presence in 31 out of 60 (51.7%) first-degree relatives (FDR) of AS patients, firmly establishing its robust genetic link to the disease. Additionally, later that same year, the team reported a strong correlation between HLA-B*27 and acute anterior uveitis (AAU) as well as reactive arthritis [17, 18].
At the same time, an independent but quite serendipitous discovery of this association was reported in 1973 by a team based at the University of California at Los Angeles and its affiliated Veterans Administration (VA) Hospital [3]. The team members had planned to investigate possible HLA association with rheumatoid arthritis (RA) for which they had a control group of healthy subjects, but they also included patient control groups comprising AS and gout patients that were abundantly available at their VA hospital. The investigators were pleasantly surprised to find that although no significant deviation from control frequencies of the then HLA specificities was noted in patients with RA and gout, HLA-B*27 was present in 35 of 40 (88%) patients with AS, as compared to 8% of the 906 normal controls [3].
In subsequent years, Erik Thorsby shared with me the information that he had tested members of his own family, revealing that some indeed possessed his newly discovered HLA antigen. Notably, one of the family members had AS, and another displayed symptoms suggestive of AS [19]. Nevertheless, as most relatives with this novel antigen enjoyed good health, he attributed these findings to serendipity. Looking back, he expressed regret for not delving deeper into this observation by investigating other AS patients. He acknowledged that had he pursued this path, he could have been a trailblazer, not only identifying a new HLA antigen but also unveiling its remarkable association with a disease [19].
Disease Heterogeneity
Studies in diverse racial/ethnic groups subsequently found differences in the strength of this association of HLA-B*27 with AS and an influence on the clinical phenotypes and endotypes of the disease [20,21,22,23,24,25]. While there are numerous similarities, HLA-B*27( +) patients with AS exhibit a greater familial disposition and a higher likelihood of associated AAU and tend to experience an earlier age of onset and age at diagnosis [26, 27•]. Furthermore, there is a slightly stronger association with HLA-B*27 among males than females, observed in both European and Chinese populations [28, 29]. Recent research underscores HLA-B*27 as an important predictor of the effectiveness of TNF inhibitors in treatment. It suggests that male gender and CRP levels are better at explaining variability in individual patient responses [30]. Acknowledging the impact of sexual dimorphism on the disease phenotype or endotype is vital for predicting outcomes and tailoring therapeutic approaches to individual patients [31].
By the mid-1980s, it became apparent that radiographic changes in the sacroiliac joints were highly prevalent in AS but not in early or atypical forms of the disease [32]. Later on, it was emphasized that the prevailing definition of AS did not encompass the broader spectrum of the disease [33], and global studies across various racial/ethnic groups have substantiated this perspective [29, 34,35,36]. Improved clinical recognition of “spondylitic disease without radiologic evidence of sacroiliitis” (now referred to as nr-axSpA) [32], which is more commonly found in females, and along with the increased use of MRI since the mid-1990s, has led to reports of the nearly equal prevalence of the extended spectrum of axSpA among both males and females [37,38,39]. A long-term Swiss study observed a gradual and progressive increase over a 36-year period (from 1980 to 2016), ultimately resulting in an equalized ratio of axSpA between the sexes [40].
HLA-B*27 Subtypes
Recent advances in molecular genotyping have unveiled a strikingly diverse landscape of the major histocompatibility complex (MHC) [41•, 42]. To address this diversity, an additional set of numbers has been introduced as suffixes to the assigned HLA names, with a preceding colon serving as a field separator. For instance, the most recent genetic variant of HLA-B*27 is named HLA-B*27:267 (https://www.ebi.ac.uk/ipd/imgt/hla). Many of them have further subvariants denoted by additional numbers after an additional colon. Among these, HLA-B*27:05:02 serves as the “parent” genotype from which other variants seem to have evolved through one or more nucleotide substitutions, altering the amino acid sequences of their encoded proteins [41•].
While many of these subtypes are exceptionally rare, differences in disease associations exist, particularly among the more common variants that have been studied [42]. HLA-B*27:05, specifically HLA-B*27:05:02, is the most widely distributed and disease-associated subtype that has been extensively studied. Other prevalent disease-associated subtypes include HLA-B*27:02 (more prevalent in Mediterranean populations) and HLA-B*27:04 (more common in Chinese, South, and Southeast Asian populations) [42]. In European populations, HLA-B*27:05 and HLA-B*27:02 confer nearly equal susceptibility to AS. However, in Chinese populations, HLA-B*27:04 exhibits a more robust association and poses a greater risk for AS compared to HLA-B*27:05 [42].
Two distinct subtypes of HLA-B*27, namely, HLA-B*27:06 (common in Southeast Asia) and HLA-B*27:09 (rare, primarily found on the Italian island of Sardinia), do not exhibit an association with classical AS [42, 43]. Intriguingly, HLA-B*27:06 differs from its closely related disease-predisposing subtype, HLA-B*27:04, by just two amino acid substitutions (His114 > Asp and Asp116 > Tyr), both located in the peptide-binding cleft [42, 43]. Similarly, HLA-B*27:09 distinguishes itself from its disease-predisposing counterpart, HLA-B*27:05, through a single amino acid substitution (Asp116 > His) in the peptide-binding cleft (as depicted in Fig. 2) [42, 43]. Thus, peptide binding seems to be the pivotal element in understanding the biology of HLA-B*27 and its role in the pathogenesis of AS [43,44,45].

(Adapted from Khan MA. Spondyloarthropathies: editorial overview. Curr Opin Rheumatol 1994; 6:351–353.” and from “Khan MA. In: Hunder GG, ed. Atlas of Rheumatology. 2005:151–181″)
A schematic ribbon drawing of the three-dimensional structure of the antigen-binding cleft of HLA-B*27. Legend: the antigen-binding cleft is made of a1 and a2 domains of the heavy chain that associates non-covalently with a light chain β2-microglobulin (not shown). A self- or foreign nonameric peptide (shown in light-blue color) is shown anchored (bound) in the antigen-binding cleft of the molecule. The view is from above, as seen from the viewpoint of a T-cell receptor. The letters N and C indicate the amino (N) and carboxy (C) terminal, respectively, of the bound peptide. The floor of the antigen-binding cleft is formed by the beta strands (broad arrows), while its margins are formed by alpha helices shown as helical ribbons. The top alpha helix and the four beta strands to the left are from the alpha-1 domain of the heavy chain, and the bottom alpha helix and the four beta strands to the right are from the alpha-2 domain. The disulfide bond is shown as two connecting spheres. Not marked are the six side pockets (assigned the letters A, B, C, D, E, and F) on the surface of the antigen-binding cleft. Pockets A and F are highly conserved deep pockets at the two terminals of the antigen-binding cleft. The residues that form the B pocket are marked by black arrowheads (at positions 7, 9, 24, 34, 45, 63, 67, and 99). The side chain of the second amino acid of the bound peptide anchors into pocket B. The non-disease-associated HLA-B*27:09 is distinguished from its closely related but disease-associated HLA-B*27:05 subtype by only one amino acid substitution at position 116. The other non-disease-associated subtype HLA-B*27:06 is distinguished from its closely related but disease-associated HLA-B*27:04 subtype by amino acid substitutions at positions 114 and 116 that are located on the floor of the peptide binding groove within the F pocket.
It is conceivable that disease-predisposing HLA-B*27 subtypes might induce pathogenicity through non-canonical mechanisms (as illustrated in Fig. 3) [46,47,48]. Both genetic and functional interactions can influence antigen presentation by altering the equilibrium between epitope generation and destruction. The accumulation of misfolded or unfolded free heavy chains, in conjunction with β2m and endoplasmic reticulum (ER) chaperones, can trigger intracellular stress, the unfolded protein response (UPR), and autophagy. This cascade results in HLA-B*27 pathogenicity through this non-canonical mechanism [46,47,48].

The interaction of genetic and epigenetic factors triggering innate and adaptive immunity resulting in the production of pro-inflammatory cytokines. Legend: accumulation of misfolded or unfolded free heavy chains along with β2m and ER chaperones can cause intracellular stress, unfolded protein response (UPR), and autophagy. Moreover, the free heavy chains may reach the cell surface and they can then form dimers and multimers that are amenable to recognition by CD4 + T cells or by NK cells. These events promote release of various pro-inflammatory cytokines. (This figure is used with permission from Carlo Perricone (Fatica M, D'Antonio A, Novelli L, Triggianese P, Conigliaro P, Greco E, et al. How Has Molecular Biology Enhanced Our Undertaking of axSpA and Its Management. Curr Rheumatol Rep. 2023;25(1):12–33. 10.1007/s11926-022–01092-4))
Additionally, HLA-B*27-free heavy chains have the capacity to form homodimers, both intracellularly and on the cell surface [47]. This can play a pathogenic role by binding to natural killer family receptors, including KIRs (killer Ig-like receptors) expressed on both natural killer cells and T cells, as well as LILRs (leukocyte Ig-like receptors) found on monocytes, B lymphocytes, and dendritic cells, which are enriched for IL-17 production [47]. However, it is worth noting that a subsequent study has challenged the role of HLA-B*27 homodimers in AS and implicated other mechanisms, such as peptide binding and antigen presentation, as pivotal in disease pathogenesis [49].
Additional Disease-Predisposing Genes
Researchers have long favored the involvement of HLA-B*27 in disease pathogenesis [50,51,52]. In 1992, a significant breakthrough occurred when a three-dimensional structure of HLA-B*27 was resolved at 2.1 A resolution, offering insights into the tight binding of peptide in its antigen-binding cleft [53]. Recent genome-wide association studies (GWAS) have identified 116 single-nucleotide polymorphisms (SNPs) associated with an increased risk of AS [28, 55,56,57, 58•, 59, 60]. Only 28% of the total genetic risk is attributed to these SNPs, with just over 20% stemming from the MHC, primarily the HLA-B*27 gene, and 7.4% from non-MHC risk loci, including ERAP1, ERAP2, NPEPPS, IL23R, TNFRSF1A, TYK2, ILGR, IL27, IL12B, ANTXR2, PTGER4, KIF21B, STAT3, CARD9, and MEVF [54,55,56,57, 58•, 59, 60].
Interestingly, these non-MHC risk loci have a disproportionate impact on gene expression and epigenetic markers in the gastrointestinal tract [55, 56]. Genetic changes can alter protein production, while epigenetic changes control gene activation, effectively switching genes “on” and “off.” Ongoing research seeks to unveil how the interplay between genetic and epigenetic factors leads to immune-mediated events and the release of pro-inflammatory cytokines, contributing to the disease [61, 62, 63••] ERAP1 and ERAP2 play a significant role, often interacting with susceptibility to MHC class-I alleles in a phenomenon known as epistasis, thereby influencing the peptidome [64,65,66,67,68]. Notably, after HLA-B*27, ERAP1 emerges as the second most influential gene in increasing the risk of AS [54,55,56,57,57, 58•] Genetic polymorphisms in the ERAP genes can affect their enzymatic function in the ER, potentially resulting in the production of oligomeric peptides with low affinity for binding to disease-associated HLA-B*27 subtypes (and HLA-B*40) within the ER-derived cytoplasmic vehicles, subsequently presented on the cell surface via the protein loading complex (PLC) mechanism [65,66,67,68]. ERAP1 and ERAP2 variants that cause loss of function or expression are associated with reduced disease risk.
ERAP’s role in infectious diseases has garnered significant attention in recent research [69, 70]. A particularly noteworthy investigation delved into the genetic impact of the Yersinia pestis pandemic (plague or famously known as the “Black Death”), which struck in the mid-fourteenth century, claiming the lives of 30 to 50% of the population across Europe, the Middle East, and Asia [71]. This study identified 245 genetic variants that exhibited changes in frequency before and after the plague, focusing on London, England’s inhabitants. ERAP2 stood out prominently in these changes. It featured two alleles, differing by a single genetic letter, determining whether the gene produces a full-size or truncated peptide. Intriguingly, individuals inheriting two copies of the allele produced full-size peptide were twice as likely to survive the plague compared to those with the allele produced truncated peptides. Furthermore, within just three to four subsequent generations, a 10% increase in the protective allele’s frequency was observed in the London population [71]. It is intriguing to note that the selective advantages of ERAP2’s protective alleles in the past intersect with alleles now associated with increased susceptibility to autoimmune diseases, such as Crohn’s disease and AS. A subsequent report has pointed out that there is insufficient evidence for natureal selection associated with the Black Death [72]. However, it is worth mentioning that there are several ERAP2-dependent cellular peptides with striking similarities to some from arthritogenic bacteria, including one HLA-B*27:05 ligand fully conserved in a protein from Campylobacter jejuni [73].
There are some additional MHC antigens, besides HLA-B*27 and HLA-B*40 (a split of HLA-Bw60), that may also be operative in genetic predisposition to AS. They include HLA-A*2, HLA-A*29, HLA-B*38 and HLA-B*39 splits of HLA-B16, HLA-B*47, HLA-B*49, HLA-B*51, HLA-B*52, HLA-C*15, HLA-DRB1*01:03, and HLA-DQB1*04 [74,75,76,77]. On the other hand, HLA-B*07, HLA-B*57, HLA-DRB1*15:01, HLA-DQB1*02:01, and HLA-DQB1*06:02 show a negative association, suggesting that they may have some protective effect [74].
HLA-B*27, Gut Microbiome, and Arthritogenic Peptide Hypothesis
HLA-B*27 in healthy subjects influences the composition of the gut microbiome because bacterial dysbiosis has been demonstrated in both healthy HLA-B*27( +) subjects and AS patients, as compared to controls [78], and is influenced by both disease activity and HLA-B*27 status [79]. It has been proposed that alteration of the gut microbiome and intracellular microbial handling of putative arthritogenic organisms by HLA-B*27 can result in an aberrant or impaired immune response in conjunction with an up-regulated production of pro-inflammatory cytokines [58•, 78,79,80,81,82].
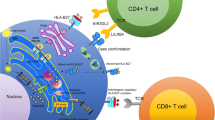
X-ray crystallography has shown that HLA-B*27:05 binds pVIPR (a well-defined self-peptide from the vasoactive intestinal peptide receptor type 1) in two distinct conformations, whereas non-disease-associated HLA-B*27:09 presents the same peptide in only one of these two conformations [83, 84]. Considering the subtle structural alterations that impact peptide binding and presentation, it has been proposed that disease-predisposing HLA-B*27 subtypes may present currently unidentified non-self-derived peptide(s) that could lead to the activation of CD8 + cytotoxic T-cell clonotypes, which, during thymus ontogenesis, were not eliminated (as depicted in Fig. 4) [43, 44]. The presence of these T cells could potentially play a role in the development of an autoimmune inflammatory process leading to AS in later life in HLA-B*27( +) individuals in the presence of other disease-predisposing genetic and epigenetic factors.

Generation of autoreactive CD8 + T cells and their role in disease. Legend: considering the subtle structural alterations that impact peptide binding and presentation, it has been proposed that disease-predisposing HLA-B*27 subtypes may present currently unidentified non-self-derived peptide(s). This presentation could lead to the activation of CD8 + cytotoxic T-cell clonotypes, which, during thymus ontogenesis, were not eliminated. The presence of these T cells in early life could potentially play a role in the development of an autoimmune inflammatory process leading to AS in individuals born with HLA-B*27 and other disease-predisposing genetic factors in later life. TEC = thymus endothelial cells, APC = antigen-presenting cells. (From: Khan MA, Mathieu A, Sorrentino R, Akkoc N. The pathogenetic role of HLA-B27 and its subtypes. Autoimmun Rev. 2007;6(3):183–9. 10.1016/j.autrev.2006.11.003)
There is ample published evidence for the presence of clonally expanded CD8 + T lymphocytes in the blood and synovial fluid of AS patients but only in a small proportion of healthy HLA B*27( +) subjects [58•, 85,86,87,88,89]. The T-cell receptor (TCR) expresses disease-associated β-chain variable region-complementary-determining region 3β (BV9–CDR3β) motif. However, there was no description of the corresponding TCRα chains. Yang et al. [90••] have used single-cell RNA sequencing to isolate orphan TCRs expressing the AS-associated BV9–CDR3β-TRBJ2.3 motif from the blood and synovial fluid in HLA-B*27( +) patients with AS and from the blood and eye (aqueous humor) of those with AAU. They showed that TCRβ chains consistently paired with TRAV21( +) TCRα chains. Additionally, bulk TCR sequencing of these biosamples showed that when compared with blood, there was at least a tenfold enrichment of the disease-associated β-chain motif in synovial fluid or in ocular fluid. This suggests that T cells expressing the disease-associated TCRβ with TCRα pairing have undergone clonal expansion in the inflamed tissues.
Yang et al. [90••] then used single-cell sequencing of the TCRs of these clonally expanded CD8 + T cells and expressed them as recombinant TCRs to screen for potential HLA-B*27:05-restricted peptides that engage these disease-associated TCRs. They used HLA-B*27:05 yeast display peptide libraries to identify shared self-peptides and microbial peptides that engage the TCRs of these patients. Subsequently, they searched the human proteome as well as the proteomes from the five potentially arthritogenic bacteria (Chlamydia, Salmonella, Shigella, Yersinia, and Klebsiella species) to identify the proteins from which such peptides may have originated. By using these approaches, the investigators were able to narrow down to a very short list of human and microbial proteins from the millions of possibilities. The structural analysis of these proteins revealed that a shared binding motif present in both self-antigens and microbial antigens engages the BV9–CDR3β TCRs. They also observed that these patients showed striking similarities in the structure of their TCRs, including those of the eye-specific clonal expansion in an HLA-B*27( +) patient with isolated AAU without AS [90••]. These distinctive peptide-binding and presentation characteristics are a hallmark of the disease-associated HLA-B*27:05 subtype but are absent in HLA-B*27:09, a subtype not associated with the disease, differing by only a single amino acid. This discovery represents a significant advancement in unraveling the 50-year-old puzzle of how HLA-B*27 contributes to the development of AS.
These recent findings strongly support that AS is driven by the presentation of antigenic peptides by HLA-B*27 to the adaptive immune system and that there may be multiple microbial triggers with shared structural features, thus broadening the concept of the arthritogenic peptide. Likewise, several self-peptides might be involved in cross-reactivity, some derived from the entheses or joints and others from the eye (iris or ciliary body). Possibility exists that this inflammatory response may later become independent of the initial trigger. Autoimmunity in AS patients could also be caused by neoantigens formed due to post-translational modifications of proteins that break immune tolerance, according to Zhai et al. [91•]. This break in tolerance to the self-proteome appears to be critical for autoimmune response in AS, just as citrulline-modified peptides are a critical source of neoantigens in RA, another autoimmune disease [92]. These discoveries will accelerate efforts to identify the involved antigens, discover more effective targeted treatments, find better tools for very early diagnosis, and identify people who are at high risk and discover preventive measures for them.
HLA-B*27 and Other Biomarkers as Aids to Diagnosis
Diagnosing AS in most patients often relies on a combination of clinical evaluation and imaging, although an unnecessary delay of several years is sometimes encountered, more so among women. The presence of HLA-B*27, in an appropriate clinical context, can enhance clinical suspicion for difficult-to-diagnose cases, such as early or atypical presentations, juvenile onset, or undifferentiated forms of SpA [93]. However, it is important to note that HLA-B*27 is not a definitive confirmation of the disease and cannot serve as a standalone screening test. This is because AS can affect individuals without this genetic marker, and conversely, over 95% of those who carry this gene in the general population remain unaffected by the condition. Moreover, the strength of disease association can differ among the various forms of SpA and depends on the racial/ethnic background of the patient [20,21,22,23, 29, 42]. For example, only about 50% of African American patients with classical (primary) AS unassociated with psoriasis or Crohn’s disease possess HLA-B*27 as compared to approximately 90% of patients of Scandinavian, Chinese, and Korean ancestry [20, 21, 28, 42]. It is worth noting that the HLA-B*27 test, being a genetic marker, does not require additional repetition unless specific technical or laboratory discrepancies arise.
Ophthalmologists commonly request HLA-B*27 test for patients exhibiting anterior uveitis [94]. The Standardization of Uveitis Nomenclature (SUN) Working Group has proposed classification criteria for SpA- and HLA-B*27-associated anterior uveitis [95]. Key criteria include (1) episodes of acute or recurrent acute unilateral or alternating unilateral anterior uveitis in conjunction with either SpA or a positive HLA-B*27 test result or (2) persistent anterior uveitis characterized by a history of the classic course along with either SpA or HLA-B*27 or (3) anterior uveitis occurring simultaneously with both SpA and HLA-B*27 [95].
Special emphasis should be placed on swift referral and early diagnosis [96]. Early-stage axSpA has recently been defined as a duration of ≤ 2 years with axial symptoms [97]. Numerous potential biomarkers have been explored to facilitate early diagnosis. Acute phase reactants, such as erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), may not be elevated in up to 50% axSpA patients with clinically active disease. Genetic and “omics” profiling (transcriptomic, proteomic, and metabolomic technologies, as well as microbiome analysis) have been investigated for early diagnosis and to enhance the prediction of treatment responses and long-term outcomes [98,99,100,101,102,103,104,105,106]. A recent development in the field is the introduction of an innovative in vitro diagnostic analytical technique known as electric field molecular fingerprinting (EMF), which can detect biomarker changes in blood samples and other bioliquids [107••, 108•]. This breakthrough will enable personalized molecular fractionation (“fingerprinting”) for the very early detection of the disease.
Polygenic Risk Score
Polygenic risk scores (PRSs) are indeed a valuable tool in precision medicine, as they consider the combined influence of multiple genetic risk variants in polygenic diseases like AS and related SpA [109, 110]. It can be an effective tool for precision medicine wherein an individual’s genetics is used as an aid to diagnose disease. Its usefulness, as with any diagnostic test, depends on the prior likelihood of the disease in an individual versus that in the general population.
In the case of diagnosing AS, PRS has shown promise. It has an impressive area under the curve (AUC) of 0.924 in receiver operator characteristics (ROC) analysis among Europeans. This outperforms individual tests such as HLA-B*27 (AUC = 0.885), MRI (AUC = 0.869), or CRP (AUC = 0.700) [110] (see Fig. 5). It is worth noting that the performance of PRS can vary based on the ethnicity of the subjects. For instance, among people of East Asian ethnicity, the AUC for PRS is even higher at 0.948, compared to 0.901 for HLA-B*27 testing alone [110]. Validated optimal- and population-specific PRSs have the potential for broad-scale clinical use as they are well suited for AS, a disease with a high heritability and a relatively low prevalence.

Polygenic risk score (PRS) has higher discriminatory capacity for AS than HLA-B*27. Legend: the area under the curve (AUC) in receiver operator characteristics (ROC) analysis among Europeans is 0.924 is better than for the HLA-B*27 test alone (AUC = 0.869). It is also better than MRI alone (AUC = 0.885) and CRP alone (AUC = 0.700). *The AUC among East Asians, which is 0.948, is not shown. (This figure is modified and used with permission of Zhixiu Li (Reference. Li Z, Wu X, Leo PJ, et al. Polygenic Risk Scores have high diagnostic capacity in ankylosing spondylitis. Ann Rheum Dis. 2021 Sep; 80(9):1168–1174. Correction: 2021 Nov; 80(11): e187)
PRSs hold significant promise to help in early diagnosis, enhance clinical certainty for difficult-to-diagnose cases, improve the current disease classification, explain differences in response to treatment, and help in identifying AAU patients who may eventually develop AS [111, 112]. PRS can also be used to explore the interaction between genetic and epigenetic factors, but the epigenetic characterization is more complicated because it is affected by non-heritable variables including age, sex, diet, medications, and smoking status [61,62,63••]. The recent advances in our understanding of the polygenic architecture of AS/axSpA can lead to the development of precision medicine to personalize disease management and hopefully prevention [113, 114•].
Lifetime Disease Risk
HLA-B*27 carries a markedly increased risk for AS, with more than 120 odds ratios, and it remains to this day the strongest genetic association with a polygenic disease [28, 56, 57, 58•, 59, 60]. Individuals homozygous for the HLA-B*27 gene have approximately double the lifetime risk for AS when compared to the heterozygous subjects [115, 116]. However, homozygosity does not influence disease manifestations and functional disability of the patients [117].
Family studies of HLA-B*27( +) patients with AS have highlighted a markedly increased familial occurrence of the disease [118, 119••, 120•]. A prospective family study of Swiss AS probands and their FDR has reported that, after 35 years of follow-up, 27.1% of HLA-B*27( +) developed axSpA versus 1.6% of HLA-B*27( −) FDR [119••, 120•]. Since all the FDR in the study were older than 45 years of age by the study conclusion (with a mean age of 58.4 years), it is improbable that further disease occurrence will take place among this group. The study also confirmed that affected mothers were more likely to pass on the disease to their offspring compared to affected fathers [119••].
The occurrence of AAU among FDR was identified as a potential indicator for prompt screening for axSpA [120•]. In contrast, chronic inflammatory back pain (IBP) alone was found to be an unreliable predictor for the subsequent development of axSpA in these families [120•]. Instead, a more reliable predictor was the combined presence of chronic inflammatory pain or discomfort in three regions of the axial skeleton: the lumbar spine and gluteal region, the thoracic spine, and the anterior chest wall, which was a better predictor of disease occurrence. This is referred to as the “3-Region Index” that defines “chronic IBP in the axial (spinal) region” with a sensitivity of 83.1% and specificity of 87.2%, with a positive predictive value of 6.4 [120•].
Concluding Remarks
Genetic and epigenetic factors, microbiome dysbiosis, and entheseal micro-damage influence disease induction and progression [28, 54,55,56,57, 58•, 59,60,61,62, 63••, 121,122,123, 124•]. HLA-B*27-presented microbial peptides seem to act as a trigger for autoimmunity by activating CD8 + T cells that cross-react with self-peptides and result in AS [90••]. The prospect of targeting and eliminating the disease- causing T cells presents the potential for both curing affected individuals and preventing the onset of the disease in individuals harboring high-risk genetic variants. Identificationof these exogenous and endogenous antigens could help to identify cellular biomarkers for early and improved diagnosis and discover more effective targeted treatments. Better serum biomarkers are being identified to facilitate disease diagnosis and monitoring [98,99,100,101,102,103,104,105,106].
The use of TNF inhibitors and other biologics and, most recently, the oral treatment with Janus kinase (JAK) inhibitors has improved treatment outcomes [125,126,127,128,129,130]. However, only up to 50% of patients achieve good response, and most of them require lifelong medication with consequent potential adverse effects. There is a need to strategize and individualize optimal treatment initiation at the earliest stage of the disease.
The search continues for novel therapeutic targets by searching for additional susceptibility genes to identify new drug targets [125]. The association of IL23R with AS was the first clear evidence that the IL-23/IL-17 pathway was involved in the disease, and it stimulated the studies that led to the approval of the first IL-17 inhibitor—secukinumab—for the treatment of AS and related forms of SpA [58•, 59, 60]. Thus, even a small contribution of a risk locus may have substantial functional importance and could help discover highly effective novel therapies. Therapeutic potential of mesenchymal stem cells is also being investigated [131, 132•]
The identification of enhanced biomarkers, coupled with the emergent application of electric field molecular “fingerprinting,” holds the potential to greatly enhance early diagnosis capabilities [98,99,100,101,102,103,104,105,106, 107••, 108•]. Availability of ultra-low-dose CT, MRI-based synthetic CT, and the use of artificial intelligence–based “deep learning” algorithms are expected to facilitate early diagnosis by detecting inflammatory and structural lesions in patients with suspected sacroiliitis [133,134,135,136].
Despite the availability of a growing array of more effec-tive therapies spanning over five decades, the HLAB* 27( +) patients with AS continue to experience a shortened lifes-pan [137, 138••]. It is noteworthy to emphasize that the presence of the HLA-B*27 gene in the general population does not result in a reduction of overall lifespan [138••]. An insightful paper titled “Fifty years after the discovery of the association of HLA-B*27 with ankylosing spondylitis” has recently been published, offering valuable additional insights into this topic [139•].
A very recently published groundbreaking paper has demonstrated the remarkable effectiveness of targeted depletion of TRBV9+ T cell in an HLA-B*27 patients with AS. This pivotal advancement not only signifies a paradigm shift but also bolster the potiential for preventing the desease in individuals high-risk genetic varriants [140••]
In conclusion, I would like to share a fascinating insight from my ongoing prospective study, spanning four generations within my own family. This journey began with my paternal grandfather’s diagnosis of AS, and it has revealed that 9 family members have subsequently developed AS or the broader spectrum of SpA (a finding yet to be published). Remarkably, one of these individuals lacks the HLA-B*27 gene. This count of affected family members may grow, as only four individuals in the fourth generation have crossed the age of 45 and can now consider themselves beyond the typical risk threshold. It is worth mentioning that in 1973, my elder son was born, inheriting from me his HLA-B*27 gene, just days before reports linking this gene to AS emerged. Coincidentally, in the same year, I embarked on my academic career as a rheumatologist. It is truly intriguing to reflect on how medical knowledge and personal experiences have intersected throughout my academic journey.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Caffrey MF, James DC. Human lymphocyte antigen association in ankylosing spondylitis. Nature. 1973;242(5393):121. https://doi.org/10.1038/242121a0.
Brewerton DA, Hart FD, Nicholls A, Caffrey M, James DC, Sturrock RD. Ankylosing spondylitis and HL-A 27. Lancet. 1973;1(7809):904–7. https://doi.org/10.1016/s0140-6736(73)91360-3.
Schlosstein L, Terasaki PI, Bluestone R, Pearson CM. High association of an HL-A antigen, W27, with ankylosing spondylitis. N Engl J Med. 1973;288(14):704–6. https://doi.org/10.1056/NEJM197304052881403.
Rudwaleit M, Khan MA, Sieper J. The challenge of diagnosis and classification in early ankylosing spondylitis: do we need new criteria? Arthritis Rheum. 2005;52(4):1000–8. https://doi.org/10.1002/art.20990.
Akkoc N, Khan MA. Radiographic axial spondyloarthritis versus ankylosing spondylitis. Clin Exp Rheumatol. 2016;34(1 Suppl 95):S7.
Khan MA, van der Linden S. Axial spondyloarthritis: a better name for an old disease: a step toward uniform reporting. ACR Open Rheumatol. 2019;1(5):336–9. https://doi.org/10.1002/acr2.11044.
Robinson PC, van der Linden S, Khan MA, et al. Axial spondyloarthritis: concept, construct, classification, and implications for therapy. Nat Rev Rheumatol. 2021;17:109–18. https://doi.org/10.1038/s41584-020-00552-4.
Thorsby E. HL-A antigens and genes. I. A study of unrelated Norwegians. Vox Sang. 1969;17(2):81–92. https://doi.org/10.1111/j.1423-0410.1969.tb00377.x.
Allen E, Amos DB, Batchelor R. Joint report of 4th International Histocompatibility Workshop. In: Terasaki PI, editor. Histocompatibility Testing Copenhagen 1970;17–47.: Munksgaard; 1970.
Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE. IPD-IMGT/HLA database. Nucleic Acids Res. 2020;48(D1):D948–55. https://doi.org/10.1093/nar/gkz950.
Brewerton DA. Discovery: HLA and disease. Curr Opin Rheumatol. 2003;15(4):369–73. https://doi.org/10.1097/00002281-200307000-00001.
James DC. HLA-B27 in clinical medicine–historical reflections on the discovery of the disease association. Br J Rheumatol. 1983;22(4 Suppl 2):20–4. https://doi.org/10.1093/rheumatology/xxii.suppl_2.20.
Sturrock RD. Human leukocyte antigen B27. Clin Med (Lond). 2010;10(2):162–3. https://doi.org/10.7861/clinmedicine.10-2-162.
Spencer DG, Sturrock RD, Buchanan WW. Ankylosing spondylitis: yesterday and today. Med Hist. 1980;24(1):60–9. https://doi.org/10.1017/s0025727300039788.
Stecher RM, Hauser H. Ankylosing spondylitis; report of occurrence in 2 brothers. Am J Roentgenol Radium Ther. 1946;56(5):601–6.
Stecher RM, Hersh AH. Familial occurrence of ankylosing spondylitis. Br J Phys Med. 1955;18(8):176–83.
Brewerton DA, Caffrey M, Nicholls A, Walters D, James DC. Acute anterior uveitis and HL-A 27. Lancet. 1973;302(7836):994–6. https://doi.org/10.1016/s0140-6736(73)91090-8.
Brewerton DA, Caffrey M, Nicholls A, Walters D, Oates JK, James DC. Reiter’s disease and HL-A 27. Lancet. 1973;302(7836):996–8. https://doi.org/10.1016/s0140-6736(73)91091-x.
Khan MA. HLA-B27 and its pathogenic role. J Clin Rheumatol. 2008;14(1):50–2. https://doi.org/10.1097/RHU.0b013e3181637a38.
Khan MA, Kushner I, Braun WE. Comparison of clinical features in HLA-B27 positive and negative patients with ankylosing spondylitis. Arthritis Rheum. 1977;20(4):909–12. https://doi.org/10.1002/art.1780200401.
Khan MA, Braun WE, Kushner I, Grecek DE, Muir WA, Steinberg AG. HLA B27 in ankylosing spondylitis: differences in frequency and relative risk in American Blacks and Caucasians. J Rheumatol Suppl. 1977;3:39–43.
Khan MA, Kushner I, Braun WE. Genetic heterogeneity in primary ankylosing spondylitis. J Rheumatol. 1980;7(3):383–6.
Feldtkeller E, Khan A, van der Heijde D, van der Linden S, Braun J. Age at disease onset and diagnosis delay in HLA-B27 negative vs. positive patients with ankylosing spondylitis. Rheumatol Int. 2003;23(2):61–6. https://doi.org/10.1007/s00296-002-0237-4.
Akkoc N, Yarkan H, Kenar G, Khan MA. Ankylosing spondylitis: HLA-B*27-positive versus HLA-B*27-negative disease. Curr Rheumatol Rep. 2017;19(5):26. https://doi.org/10.1007/s11926-017-0654-8.
Cui Z, Hou G, Meng X, Feng H, He B, Tian Y. Bidirectional causal associations between inflammatory bowel disease and ankylosing spondylitis: a two-sample Mendelian randomization analysis. Front Genet. 2020;19(11):587876. https://doi.org/10.3389/fgene.2020.587876.
Arévalo M, Gratacós Masmitjà J, Moreno M, Calvet J, Orellana C, Ruiz D, et al; REGISPONSER group. Influence of HLA-B27 on the ankylosing spondylitis phenotype: results from the REGISPONSER database. Arthritis Res Ther. 2018;20(1):221. https://doi.org/10.1186/s13075-018-1724-7. Erratum in: Arthritis Res Ther. 2020 Jun 11;22(1):140.
• Li Z, van der Linden SM, Khan MA, Baumberger H, Zandwijk HV, Khan MK, Villiger PM, Brown MA. Heterogeneity of axial spondyloarthritis: genetics, sex, and structural damage matter. RMD Open. 2022;8(1):e002302. https://doi.org/10.1136/rmdopen-2022-002302. (This study provides evidence for a high degree of heterogeneity of primary AS and recommends that stratified or adjusted analysis of effectiveness studies be performed, taking genetics, sex, and radiographic damage (sacroiliitis) into account.)
Brown MA, Xu H, Li Z. Genetics and the axial spondyloarthritis spectrum. Rheumatology (Oxford). 2020;59(Suppl4):iv58–66. https://doi.org/10.1093/rheumatology/keaa464.
Zhang S, Peng L, Li Q, Zhao J, Xu D, Zhao J, et al. Spectrum of spondyloarthritis among Chinese populations. Curr Rheumatol Rep. 2022;24(8):247–58. https://doi.org/10.1007/s11926-022-01079-1.
Fröhlich F, Micheroli R, Hebeisen M, Kissling S, Bürki K, Exer P, et al. HLA-B27 as a predictor of effectiveness of treatment with TNF inhibitors in axial spondyloarthritis: data from the Swiss Clinical Quality Management Registry. Clin Rheumatol. 2023;42(5):1267–74. https://doi.org/10.1007/s10067-022-06490-8.
Stovall R, van der Horst-Bruinsma IE, Liu SH, Rusman T, Gensler LS. Sexual dimorphism in the prevalence, manifestation and outcomes of axial spondyloarthritis. Nat Rev Rheumatol. 2022;18(11):657–69. https://doi.org/10.1038/s41584-022-00833-0.
Khan MA, van der Linden SM, Kushner I, Valkenburg HA, Cats A. Spondylitic disease without radiologic evidence of sacroiliitis in relatives of HLA-B27 positive ankylosing spondylitis patients. Arthritis Rheum. 1985;28(1):40–3. https://doi.org/10.1002/art.1780280107.
Khan MA, van der Linden SM. A wider spectrum of spondyloarthropathies. Semin Arthritis Rheum. 1990;20(2):107–13. https://doi.org/10.1016/0049-0172(90)90023-9.
Malaviya AN, Kalyani A, Rawat R, Gogia SB. Comparison of patients with ankylosing spondylitis (AS) and non-radiographic axial spondyloarthritis (nr-axSpA) from a single rheumatology clinic in New Delhi. Int J Rheum Dis. 2015;18(7):736–41. https://doi.org/10.1111/1756-185X.12579.
Bittar M, Yong WC, Magrey M, Khan MA. Worldwide differences in clinical phenotype of axial spondyloarthritis. Curr Rheumatol Rep. 2021;23(10):76. https://doi.org/10.1007/s11926-021-01043-5.
Burgos-Varga R, Wei JC, Rahman MU, Akkoc N, Haq SA, Hammoudeh M, et al. The prevalence and clinical characteristics of nonradiographic axial spondyloarthritis among patients with inflammatory back pain in rheumatology practices: a multinational, multicenter study. Arthritis Res Ther. 2016;18(1):132. https://doi.org/10.1186/s13075-016-1027-9.
Haroon NN, Paterson JM, Li P, Haroon N. Increasing proportion of female patients with ankylosing spondylitis: a population-based study of trends in the incidence and prevalence of AS. BMJ Open. 2014;4(12):e006634. https://doi.org/10.1136/bmjopen-2014-006634.
Tournadre A, Pereira B, Lhoste A, Dubost JJ, Ristori JM, Claudepierre P, et al. Differences between women and men with recent-onset axial spondyloarthritis: results from a prospective multicenter French cohort. Arthritis Care Res (Hoboken). 2013;65(9):1482–9. https://doi.org/10.1002/acr.22001.
Rusman T, van Vollenhoven RF, van der Horst-Bruinsma IE. Gender differences in axial spondyloarthritis: women are not so lucky. Curr Rheumatol Rep. 2018;20(6):35. https://doi.org/10.1007/s11926-018-0744-2.
Baumberger H, Khan M. SAT0417|Gradual progressive change to equal prevalence of ankylosing spondylitis among males and females in Switzerland: data from the Swiss ankylosing spondylitis society (SVMB). Ann Rheumatic Dis. 2017;76(Suppl 2):929-. https://doi.org/10.1136/annrheumdis-2017-eular.3961.
• Barker DJ, Maccari G, Georgiou X, Cooper MA, Flicek P, Robinson J, et al. The IPD-IMGT/HLA database. Nucleic Acids Res. 2023;51(D1):D1053–60. https://doi.org/10.1093/nar/gkac1011. (The IPD-IMGT/HLA database provides data for genes located within the human MHC region.)
Khan MA. An update on the genetic polymorphism of HLA-B*27 with 213 alleles encompassing 160 subtypes (and still counting). Curr Rheumatol Rep. 2017;19(2):9. https://doi.org/10.1007/s11926-017-0640-1.
Khan MA, Mathieu A, Sorrentino R, Akkoc N. The pathogenetic role of HLA-B27 and its subtypes. Autoimmun Rev. 2007;6(3):183–9. https://doi.org/10.1016/j.autrev.2006.11.003.
Sorrentino R, Bockmann RA, Fiorillo MT. HLA-B27 and antigen presentation: at the crossroads between immune defense and autoimmunity. Mol Immunol. 2014;57(1):22–7. https://doi.org/10.1016/j.molimm.2013.06.017.
Tedeschi V, Alba J, Paladini F, Paroli M, Cauli A, Mathieu A, et al. Unusual placement of an EBV epitope into the groove of the ankylosing spondylitis-associated HLA-B27 allele allows CD8+ T cell activation. Cells. 2019;8(6):572. https://doi.org/10.3390/cells8060572.
Jah N, Jobart-Malfait A, Ermoza K, Noteuil A, Chiocchia G, Breban M, et al. HLA-B27 subtypes predisposing to ankylosing spondylitis accumulate in an endoplasmic reticulum-derived compartment apart from the peptide-loading complex. Arthritis Rheumatol. 2020;72(9):1534–46. https://doi.org/10.1002/art.41281.
Bowness P. HLA-B27. Annu Rev Immunol. 2015;33(1):29–48. https://doi.org/10.1146/annurev-immunol-032414-112110.
Taurog JD, Chhabra A, Colbert RA. Ankylosing spondylitis and axial spondyloarthritis. N Engl J Med. 2016;374(26):2563–74. https://doi.org/10.1056/NEJMra1406182.
Lim Kam Sian TCC, Indumathy S, Halim H, Greule A, Cryle MJ, Bowness P, et al. Allelic association with ankylosing spondylitis fails to correlate with human leukocyte antigen B27 homodimer formation. J Biol Chem. 2019;294(52):20185–95. https://doi.org/10.1074/jbc.RA119.010257.
Hammer RE, Maika SD, Richardson JA, Tang JP, Taurog JD. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 1990;63(5):1099–112. https://doi.org/10.1016/0092-8674(90)90512-d.
Benjamin R, Parham P. Guilt by association: HLA-B27 and ankylosing spondylitis. Immunol Today. 1990;11(4):137–42. https://doi.org/10.1016/0167-5699(90)90051-a.
Feltkamp TE, Khan MA, Lopez de Castro JA. The pathogenetic role of HLA-B27. Immunol Today. 1996;17(1):5–7. https://doi.org/10.1016/0167-5699(96)80559-7.
Madden DR, Gorga JC, Strominger JL, Wiley DC. The three-dimensional structure of HLA-B27 at 2.1 A resolution suggests a general mechanism for tight peptide binding to MHC. Cell. 1992;70(6):1035–48. https://doi.org/10.1016/0092-8674(92)90252-8.
Welcome Trust Case Control Consortium; Australo-Anglo-American Spondylitis Consortium (TASC); Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39(11):1329–37. https://doi.org/10.1038/ng.2007.17.
International Genetics of Ankylosing Spondylitis Consortium (IGAS); Cortes A, Hadler J, Pointon JP, Robinson PC, Karaderi T, Leo P, et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet. 2013;45(7):730–8. https://doi.org/10.1038/ng.2667
Ellinghaus D, Jostins L, Spain SL, Cortes A, Bethune J, Han B, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet. 2016;48(5):510–8. https://doi.org/10.1038/ng.3528.
Li Z, Akar S, Yarkan H, Lee SK, Çetin P, Can G, et al. Genome-wide association study in Turkish and Iranian populations identify rare familial Mediterranean fever gene (MEFV) polymorphisms associated with ankylosing spondylitis. PLoS Genet. 2019;15(4):e1008038. https://doi.org/10.1371/journal.pgen.1008038.
• Garrido-Mesa J, Brown MA. T cell repertoire profiling and the mechanism by which HLA-B27 causes ankylosing spondylitis. Curr Rheumatol Rep. 2022;24(12):398–410. https://doi.org/10.1007/s11926-022-01090-6. (A good review explaining that AS is driven by the presentation of antigenic peptides to the adaptive immune system by HLA-B*27.)
Wordsworth BP, Cohen CJ, Davidson C, Vecellio M. Perspectives on the genetic associations of ankylosing spondylitis. Front Immunol. 2021;12:603726. https://doi.org/10.3389/fimmu.2021.603726.
Fatica M, D’Antonio A, Novelli L, Triggianese P, Conigliaro P, Greco E, et al. How has molecular biology enhanced our undertaking of axSpA and its management. Curr Rheumatol Rep. 2023;25(1):12–33. https://doi.org/10.1007/s11926-022-01092-4.
Liao HT, Tsai CY, Lai CC, Hsieh SC, Sun YS, Li KJ, et al. The potential role of genetics, environmental factors, and gut dysbiosis in the aberrant non-coding RNA expression to mediate inflammation and osteoclastogenic/osteogenic differentiation in ankylosing spondylitis. Front Cell Dev Biol. 2022;20(9):748063. https://doi.org/10.3389/fcell.2021.748063.
ENCODE Project Consortium; Snyder MP, Gingeras TR, Moore JE, Weng Z, Gerstein MB, Ren B, Perspectives on ENCODE. Nature. 2020;583(7818):693–698. https://doi.org/10.1038/s41586-020-2449-8. Epub 2020 Jul 29. Erratum in: Nature. 2022 May;605(7909):E4.
•• Brown AC, Cohen CJ, Mielczarek O, Migliorini G, Costantino F, Allcock A, et al. Comprehensive epigenomic profiling reveals the extent of disease-specific chromatin states and informs target discovery in ankylosing spondylitis. Cell Genom. 2023;3(6):100306. https://doi.org/10.1016/j.xgen.2023.100306. (These investigators have provided comprehensive transcriptomic and epigenomic mapping in AS.)
York IA, Chang SC, Saric T, Keys JA, Favreau JM, Goldberg AL, et al. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat Immunol. 2002;3(12):1177–84. https://doi.org/10.1038/ni860.
Lopez de Castro JA, Alvarez-Navarro C, Brito A, Guasp P, Martin-Esteban A, Sanz-Bravo A. Molecular and pathogenic effects of endoplasmic reticulum aminopeptidases ERAP1 and ERAP2 in MHC-I-associated inflammatory disorders: towards a unifying view. Mol Immunol. 2016;77:193–204. https://doi.org/10.1016/j.molimm.2016.08.005.
Tsui FW, Haroon N, Reveille JD, Rahman P, Chiu B, Tsui HW, Inman RD. Association of an ERAP1 ERAP2 haplotype with familial ankylosing spondylitis. Ann Rheum Dis. 2010;69(4):733–6. https://doi.org/10.1136/ard.2008.103804.
Evans DM, Spencer CC, Pointon JJ, Su Z, Harvey D, Kochan G, et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet. 2011;43(8):761–7. https://doi.org/10.1038/ng.873.
Robinson PC, Costello ME, Leo P, Bradbury LA, Hollis K, Cortes A, et al. ERAP2 is associated with ankylosing spondylitis in HLA-B27-positive and HLA-B27-negative patients. Ann Rheum Dis. 2015;74(8):1627–9. https://doi.org/10.1136/annrheumdis-2015-207416.
Saulle I, Vicentini C, Clerici M, Biasin M. An overview on ERAP roles in infectious diseases. Cells. 2020;9(3). https://doi.org/10.3390/cells9030720
Yao Y, Liu N, Zhou Z, Shi L. Influence of ERAP1 and ERAP2 gene polymorphisms on disease susceptibility in different populations. Hum Immunol. 2019;80(5):325–34. https://doi.org/10.1016/j.humimm.2019.02.011.
Klunk J, Vilgalys TP, Demeure CE, Cheng X, Shiratori M, Madej J, et al. Evolution of immune genes is associated with the Black Death. Nature. 2022;611(7935):312–9. https://doi.org/10.1038/s41586-022-05349-x.
Barton AR, Santander CG, Skoglund P, Moltke I, Reich D, Mathieson I. Insufficient evidence for natural selection associated with the Black Death. bioRxiv. 2023;15:2023.03.14.532615. https://doi.org/10.1101/2023.03.14.532615.
Lorente E, Fontela MG, Barnea E, Martín-Galiano AJ, Mir C, Galocha B, et al. Modulation of natural HLA-B*27:05 ligandome by ankylosing spondylitis-associated endoplasmic reticulum aminopeptidase 2 (ERAP2). Mol Cell Proteomics. 2020;19(6):994–1004. https://doi.org/10.1074/mcp.RA120.002014.
Reveille JD, Zhou X, Lee M, Weisman MH, Yi L, Gensler LS, et al. HLA class I and II alleles in susceptibility to ankylosing spondylitis. Ann Rheum Dis. 2019;78(1):66–73. https://doi.org/10.1136/annrheumdis-2018-213779.
Robinson WP, van der Linden SM, Khan MA, Rentsch HU, Cats A, Russell A, et al. HLA-Bw60 increases susceptibility to ankylosing spondylitis in HLA-B27+ patients. Arthritis Rheum. 1989;32(9):1135–41. https://doi.org/10.1002/anr.1780320912.
Khan MA, Kushner I, Braun WE. B27-negative HLA-BW16 in ankylosing spondylitis. Lancet. 1978;1(8078):1370–1. https://doi.org/10.1016/s0140-6736(78)92455-8.
Khan MA, Kushner I, Braun WE. Association of HLA-A2 with uveitis in HLA-B27 positive patients with ankylosing spondylitis. J Rheumatol. 1981;8(2):295–8.
Asquith M, Sternes PR, Costello ME, Karstens L, Diamond S, Martin TM, et al. HLA alleles associated with risk of ankylosing spondylitis and rheumatoid arthritis influence the gut microbiome. Arthritis Rheumatol. 2019;71(10):1642–50. https://doi.org/10.1002/art.40917.
Berland M, Meslier V, Berreira Ibraim S, Le Chatelier E, Pons N, Maziers N, et al. Both disease activity and HLA-B27 status are associated with gut microbiome dysbiosis in spondyloarthritis patients. Arthritis Rheumatol. 2023;75(1):41–52. https://doi.org/10.1002/art.42289.
So J, Tam LS. Gut microbiome and its interaction with immune system in spondyloarthritis. Microorganisms. 2020;8(11). https://doi.org/10.3390/microorganisms8111727
Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. 2012;18(7):1069–76. https://doi.org/10.1038/nm.2817.
Gracey E, Yao Y, Green B, Qaiyum Z, Baglaenko Y, Lin A, et al. Sexual dimorphism in the Th17 signature of ankylosing spondylitis. Arthritis Rheumatol. 2016;68(3):679–89. https://doi.org/10.1002/art.39464.
Loll B, Fabian H, Huser H, Hee CS, Ziegler A, Uchanska-Ziegler B, et al. Increased conformational flexibility of HLA-B*27 subtypes associated with ankylosing spondylitis. Arthritis Rheumatol. 2016;68(5):1172–82. https://doi.org/10.1002/art.39567.
Loll B, Rückert C, Uchanska-Ziegler B, Ziegler A. Conformational plasticity of HLA-B27 molecules correlates inversely with efficiency of negative T cell selection. Front Immunol. 2020;11:179. https://doi.org/10.3389/fimmu.2020.00179.
Faham M, Carlton V, Moorhead M, Zheng J, Klinger M, Pepin F, et al. Discovery of T cell receptor β motifs specific to HLA-B27-positive ankylosing spondylitis by deep repertoire sequence analysis. Arthritis Rheumatol. 2017;69(4):774–84. https://doi.org/10.1002/art.40028.
Huang H, Sikora MJ, Islam S, Chowdhury RR, Chien YH, Scriba TJ, et al. Select sequencing of clonally expanded CD8(+) T cells reveals limits to clonal expansion. Proc Natl Acad Sci U S A. 2019;116(18):8995–9001. https://doi.org/10.1073/pnas.1902649116.
Zheng M, Zhang X, Zhou Y, Tang J, Han Q, Zhang Y, et al. TCR repertoire and CDR3 motif analyses depict the role of αβ T cells in ankylosing spondylitis. EBioMedicine. 2019;47:414–26. https://doi.org/10.1016/j.ebiom.2019.07.032.
Hanson AL, Nel HJ, Bradbury L, Phipps J, Thomas R, Lê Cao KA, et al. Altered repertoire diversity and disease-associated clonal expansions revealed by T cell receptor immunosequencing in ankylosing spondylitis patients. Arthritis Rheumatol. 2020;72(8):1289–302. https://doi.org/10.1002/art.41252.
Komech EA, Koltakova AD, Barinova AA, Minervina AA, Salnikova MA, Shmidt EI, et al. TCR repertoire profiling revealed antigen-driven CD8+ T cell clonal groups shared in synovial fluid of patients with spondyloarthritis. Front Immunol. 2022;13:973243. https://doi.org/10.3389/fimmu.2022.973243.
•• Yang X, Garner LI, Zvyagin IV, Paley MA, Komech EA, Jude KM, et al. Autoimmunity-associated T cell receptors recognize HLA-B*27-bound peptides. Nature. 2022;612(7941):771–7. (HLA-B*27:05-presented microbial peptides act as a trigger for autoimmunity by activating CD8+ T cells that cross-react with self-peptides and result in AS, virtually solving the 50-year-old puzzle of how HLA-B*27 leads to disease.)
• Zhai Y, Chen L, Zhao Q, Zheng ZH, Chen ZN, Bian H, et al. Cysteine carboxyethylation generates neoantigens to induce HLA-restricted autoimmunity. Science. 2023;379(6637):eabg2482. https://doi.org/10.1126/science.abg2482. (This study reports that autoimmunity in patients with AS results from neoantigens formed by post-translational modification of proteins that break immune tolerance.)
Law SC, Street S, Yu CH, Capini C, Ramnoruth S, Nel HJ, et al. T-cell autoreactivity to citrullinated autoantigenic peptides in rheumatoid arthritis patients carrying HLA-DRB1 shared epitope alleles. Arthritis Res Ther. 2012;14(3):R118. https://doi.org/10.1186/ar3848.
Khan MA, Khan MK. Diagnostic value of HLA-B27 testing ankylosing spondylitis and Reiter’s syndrome. Ann Intern Med. 1982;96(1):70–6. https://doi.org/10.7326/0003-4819-96-1-70.
Khan MA, Haroon M, Rosenbaum JT. Acute anterior uveitis and spondyloarthritis: more than meets the eye. Curr Rheumatol Rep. 2015;17(9):59. https://doi.org/10.1007/s11926-015-0536-x.
Standardization of Uveitis Nomenclature (SUN) Working Group.Classification criteria for spondyloarthritis/HLA-B27-associated anterior uveitis. Am J Ophthalmol. 2021;228:117–25. https://doi.org/10.1016/j.ajo.2021.03.049.
Ozgocmen S, Khan MA. Current concept of spondyloarthritis: special emphasis on early referral and diagnosis. Curr Rheumatol Rep. 2012;14(5):409–14. https://doi.org/10.1007/s11926-012-0274-2.
Navarro-Compán V, Benavent D, Capelusnik D, van der Heijde D, Landewé RB, Poddubnyy D, et al. ASAS consensus definition of early axial spondyloarthritis. Ann Rheum Dis. 2023. https://doi.org/10.1136/ard-2023-224232.
Brown MA, Li Z, Cao KL. Biomarker development for axial spondyloarthritis. Nat Rev Rheumatol. 2020;16(8):448–63. https://doi.org/10.1038/s41584-020-0450-0.
Prajzlerova K, Grobelna K, Pavelka K, Senolt L, Filkova M. An update on biomarkers in axial spondyloarthritis. Autoimmun Rev. 2016;15(6):501–9. https://doi.org/10.1016/j.autrev.2016.02.002.
Maksymowych WP. Biomarkers for diagnosis of axial spondyloarthritis, disease activity, prognosis, and prediction of response to therapy. Front Immunol. 2019;10:305. https://doi.org/10.3389/fimmu.2019.00305.
Liu S, Ji W, Lu J, Tang X, Guo Y, Ji M, et al. Discovery of potential serum protein biomarkers in ankylosing spondylitis using tandem mass tag-based quantitative proteomics. J Proteome Res. 2020;19(2):864–72. https://doi.org/10.1021/acs.jproteome.9b00676.
Li H, Wang L, Zhu J, Xiao J, Yang H, Hai H, et al. Diagnostic serum biomarkers associated with ankylosing spondylitis. Clin Exp Med. 2023;23(5):1729–39. https://doi.org/10.1007/s10238-022-00958-2.
Jarlborg M, Courvoisier DS, Lamacchia C, Martinez Prat L, Mahler M, Bentow C, et al. Serum calprotectin: a promising biomarker in rheumatoid arthritis and axial spondyloarthritis. Arthritis Res Ther. 2020;22(1):105. https://doi.org/10.1186/s13075-020-02190-3.
De Craemer AS, Witte T, Lobaton Ortega T, Hoorens A, De Vos M, Cuvelier C, et al. Anti-CD74 IgA antibodies show diagnostic potential for axial spondyloarthritis but are not associated with microscopic gut inflammation. Rheumatology (Oxford). 2023;62(2):984–90. https://doi.org/10.1093/rheumatology/keac384.
Li D, Cao R, Dong W, Cheng M, Pan X, Hu Z, et al. Identification of potential biomarkers for ankylosing spondylitis based on bioinformatics analysis. BMC Musculoskelet Disord. 2023;24(1):413. https://doi.org/10.1186/s12891-023-06550-3.
Hwang M, Assassi S, Zheng J, Castillo J, Chavez R, Vanarsa K, et al. Quantitative proteomic screening uncovers candidate diagnostic and monitoring serum biomarkers of ankylosing spondylitis. Arthritis Res Ther. 2023;25(1):57. https://doi.org/10.1186/s13075-023-03044-4.
•• Pupeza I, Huber M, Trubetskov M, Schweinberger W, Hussain SA, Hofer C, et al. Field-resolved infrared spectroscopy of biological systems. Nature. 2020;577(7788):52–9. https://doi.org/10.1038/s41586-019-1850-7. (The senior author of this paper—Ferenc Krauz—has just been awarded the 2023 Nobel Prize in Physics for his role in the discovery of this new technology.)
• Eissa T, Kepesidis KV, Zigman M, Huber M. Limits and prospects of molecular fingerprinting for phenotyping biological systems revealed through in silico modeling. Anal Chem. 2023;95(16):6523–32. https://doi.org/10.1021/acs.analchem.2c04711. (The authors describe the potential benefits of their newly developed technologies in health diagnostics.)
Brown MA, Aletaha D. Genetic risk scores in inflammatory arthritis: a new era? Nat Rev Rheumatol. 2020;16(10):545–6. https://doi.org/10.1038/s41584-020-0473-6.
Li Z, Wu X, Leo PJ, De Guzman E, Akkoc N, Breban M, et al. Polygenic risk scores have high diagnostic capacity in ankylosing spondylitis. Ann Rheum Dis. 2021;80(9):1168–74. https://doi.org/10.1136/annrheumdis-2020-219446.
Huang XF, Li Z, De Guzman E, Robinson P, Gensler L, Ward MM, et al. Genomewide association study of acute anterior uveitis identifies new susceptibility loci. Invest Ophthalmol Vis Sci. 2020;61(6):3. https://doi.org/10.1167/iovs.61.6.3.
Robinson PC, Claushuis TA, Cortes A, Martin TM, Evans DM, Leo P, et al. Genetic dissection of acute anterior uveitis reveals similarities and differences in associations observed with ankylosing spondylitis. Arthritis Rheumatol. 2015;67(1):140–51. https://doi.org/10.1002/art.38873.
Ramaswami R, Bayer R, Galea S. Precision medicine from a public health perspective. Annu Rev Public Health. 2018;39:153–68. https://doi.org/10.1146/annurev-publhealth-040617-014158.
• Allard-Chamard H, Li Q, Rahman P. Emerging concepts in precision medicine in axial spondyloarthritis. Curr Rheumatol Rep. 2023;25(10):204–12. https://doi.org/10.1007/s11926-023-01113-w. (A good review on precision medicine in axSpA.)
Khan MA, Kushner I, Braun WE, Zachary AA, Steinberg AG. HLA-B27 homozygosity in ankylosing spondylitis: relationship to risk and severity. Tissue Antigens. 1978;11(5):434–8. https://doi.org/10.1111/j.1399-0039.1978.tb01280.x.
Jaakkola E, Herzberg I, Laiho K, Barnardo MC, Pointon JJ, Kauppi M, et al. Finnish HLA studies confirm the increased risk conferred by HLA-B27 homozygosity in ankylosing spondylitis. Ann Rheum Dis. 2006;65(6):775–80. https://doi.org/10.1136/ard.2005.041103.
Kim TJ, Na KS, Lee HJ, Lee B, Kim TH. HLA-B27 homozygosity has no influence on clinical manifestations and functional disability in ankylosing spondylitis. Clin Exp Rheumatol. 2009;27(4):574–9.
Morin M, Hellgren K, Frisell T. Familial aggregation and heritability of ankylosing spondylitis - a Swedish nested case-control study. Rheumatology (Oxford). 2020;59(7):1695–702. https://doi.org/10.1093/rheumatology/kez519.
•• van der Linden SM, Khan MA, Li Z, Baumberger H, Zandwijk HV, Khan MK, et al. Recurrence of axial spondyloarthritis among first-degree relatives in a prospective 35-year-follow-up family study. RMD Open. 2022;8(2). https://doi.org/10.1136/rmdopen-2022-002208. (This prospective family study of 125 Swiss AS probands and their 360 first-degree relatives found that axSpA developed in 27.1% of HLA-B27(+) FDR after 35 years of follow-up.)
• van der Linden SM, Khan MA, Li Z, Baumberger H, Zandwijk HV, Khan K, et al. Factors predicting axial spondyloarthritis among first-degree relatives of probands with ankylosing spondylitis: a family study spanning 35 years. Ann Rheum Dis. 2022;81(6):831–7. https://doi.org/10.1136/annrheumdis-2021-222083. (A “3-Region Index” that defines “chronic inflammatory axial spinal pain” is a much better predictor of the occurrence of axSpA than chronic inflammatory back pain.)
Chen S, Li Z, Chen D, Cui H, Wang J, Li Z, Li X, Zheng Z, Zhan Z, Liu H. Piezo1-mediated mechanotransduction promotes entheseal pathological new bone formation in ankylosing spondylitis. Ann Rheum Dis. 2023;82(4):533–45. https://doi.org/10.1136/ard-2022-223428.
Mauro D, Thomas R, Guggino G, Lories R, Brown MA, Ciccia F. Ankylosing spondylitis: an autoimmune or autoinflammatory disease? Nat Rev Rheumatol. 2021;17(7):387–404. https://doi.org/10.1038/s41584-021-00625-y.
Watad A, Cuthbert RJ, Amital H, McGonagle D. Enthesitis: much more than focal insertion point inflammation. Curr Rheumatol Rep. 2018;20(7):41. https://doi.org/10.1007/s11926-018-0751-3.
• Nam B, Jo S, Bang SY, Park Y, Shin JH, Park YS, et al. Clinical and genetic factors associated with radiographic damage in patients with ankylosing spondylitis. Ann Rheum Dis. 2023;82(4):527–32. https://doi.org/10.1136/ard-2022-222796. (This is a GWAS study to identify clinical and genetic factors associated with severe radiographic damage in AS.)
Nancy Z, Yan L, Hui S, Paul B, Liye C. From the genetics of ankylosing spondylitis to new biology and drug target discovery. Front Immunol. 2021;17(12):624632. https://doi.org/10.3389/fimmu.2021.624632.
Ward MM, Deodhar A, Gensler LS, Dubreuil M, Yu D, Khan MA, et al. 2019 Update of the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network Recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthritis Rheumatol. 2019;71(10):1599–613. https://doi.org/10.1002/art.41042.
Ramiro S, Nikiphorou E, Sepriano A, Ortolan A, Webers C, Baraliakos X, et al. ASAS-EULAR recommendations for the management of axial spondyloarthritis: 2022 update. Ann Rheum Dis. 2023;82(1):19–34. https://doi.org/10.1136/ard-2022-223296.
Akkoc N, Khan MA. JAK inhibitors for axial spondyloarthritis: what does the future hold? Curr Rheumatol Rep. 2021;23(6):34. https://doi.org/10.1007/s11926-021-01001-1.
Baraliakos X, van der Heijde D, Sieper J, Inman RD, Kameda H, Li Y, et al. Efficacy and safety of upadacitinib in patients with ankylosing spondylitis refractory to biologic therapy: 1-year results from the open-label extension of a phase III study. Arthritis Res Ther. 2023;25(1):172. https://doi.org/10.1186/s13075-023-03128-1.
Danve A, Deodhar A. Treatment of axial spondyloarthritis: an update. Nat Rev Rheumatol. 2022;18(4):205–16. https://doi.org/10.1038/s41584-022-00761-z.
Armaka M, Apostolaki M, Jacques P, Kontoyiannis DL, Elewaut D, Kollias G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J Exp Med. 2008;205(2):331–7. https://doi.org/10.1084/jem.20070906.
• Tavasolian F, Inman RD. Biology and therapeutic potential of mesenchymal stem cell extracellular vesicles in axial spondyloarthritis. Commun Biol. 2023;6(1):413. https://doi.org/10.1038/s42003-023-04743-z. (A current review on the therapeutic potential of mesenchymal stem cells in axSpA.)
Jans LBO, Chen M, Elewaut D, Van den Bosch F, Carron P, Jacques P, et al. MRI-based synthetic CT in the detection of structural lesions in patients with suspected sacroiliitis: comparison with MRI. Radiology. 2021;298(2):343–9. https://doi.org/10.1148/radiol.2020201537.
Fritz J, Kijowski R, Recht MP. Artificial intelligence in musculoskeletal imaging: a perspective on value propositions, clinical use, and obstacles. Skeletal Radiol. 2022;51(2):239–43. https://doi.org/10.1007/s00256-021-03802-y.
Diekhoff T, Hermann KGA, Lambert RG. Future of low-dose computed tomography and dual-energy computed tomography in axial spondyloarthritis. Curr Rheumatol Rep. 2022;24(6):198–205. https://doi.org/10.1007/s11926-022-01075-5.
Van Den Berghe T, Babin D, Chen M, Callens M, Brack D, Maes H, et al. Neural network algorithm for detection of erosions and ankylosis on CT of the sacroiliac joints: multicentre development and validation of diagnostic accuracy. Eur Radiol. 2023. https://doi.org/10.1007/s00330-023-09704-y.
Khan MA, Khan MK, Kushner I. Survival among patients with ankylosing spondylitis: a life-table analysis. J Rheumatol. 1981;8(1):86–90.
•• Li Z, Khan MK, van der Linden S, Villiger PM, Baumberger H, van Zandwijk H, Khan MA, Brown MA. HLA-B*27, axial spondyloarthritis, and survival. Ann Rheum Dis. 2023; Sept 7. https://doi.org/10.1136/ard-2023-224434. (HLA-B*27(+) patients with AS have reduced life expectancy. But HLA-B*27(+) individuals in the general population have a normal lifespan.)
• Braun J, Sieper J. Fifty years after the discovery of the association of HLA B27 with ankylosing spondylitis. RMD Open. 2023;9(3):e003102. https://doi.org/10.1136/rmdopen-2023-003102. (A nice and concise review on this subject.)
•• Britanova OV, Lupyr KR, Staroveros DB, Shagina IA, Somov DV, Klimenko A, et al. Targeted depletion of TRBV+ T cell as immunotheraphy in a ptient with ankylosing spondylitis. Nat Med. 2023. https://doi.org/10.1038/s41591-023-02613-z. The first report of dramatic efficacy of targeted depletion of TRBV9+ T cells as immunotherapy in an HLA-B*27(+) patient with ankylosing spondylitis has just been published.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
The author confirms compliance with ethical standards, and there was no need for informed consent. This article does not contain any studies with human or animal subjects performed by the author. Moreover, there were no human rights issues.
Conflict of Interest
The author declares no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Khan, M.A. HLA-B*27 and Ankylosing Spondylitis: 50 Years of Insights and Discoveries. Curr Rheumatol Rep 25, 327–340 (2023). https://doi.org/10.1007/s11926-023-01118-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11926-023-01118-5