
Abstract
Purpose of Review
Systemic sclerosis (SSc) is a heterogeneous autoimmune disease which has defined three hallmarks: Small vessel vasculopathy, production of autoantibodies and fibroblast dysfunction. The exact aetiology of the disease remains unknown, due to the complex nature of the cellular signalling pathways involved. However, there is strong and consistent evidence that the innate system, in particular toll-like receptor signalling, is contributing to the progression and perhaps onset of systemic sclerosis. In light of this evidence, this review examines the role of innate immunity in systemic sclerosis and where appropriate suggests avenues for therapeutic modulation in SSc.
Recent Findings
Multiple lines of evidence suggest that Toll-like receptors (TLRs) are dysregulated and emerging evidence suggests that many endogenous ligands are also elevated in the disease leading to ‘sterile inflammation’ and ultimately the induction of fibrosis. Currently, no effective therapy exists and exploiting the innate immune system perturbation may be one possible avenue.
Summary
Innate immune dysregulation is key in SSc pathogenesis and may represent a novel target.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Systemic sclerosis (SSc), also referred to as scleroderma, is an autoimmune connective tissue disease that causes fibrosis of the skin and internal organs [1]. Mortality in SSc patients is typically due to interstitial lung disease and pulmonary arterial hypertension [2, 3]. The exact aetiology of SSc is unknown; however, vascular injury and dysfunction of the immune system are typically associated with the disease. It is suggested that tissue damage to the endothelium is the initial triggering factor in SSc and that this damage leads to fibrosis via inflammation and immune cell activation. Numerous autoantibodies have been detected in SSc patients [4,5,6], with anti-nuclear antibodies being particularly prevalent [7]. Additionally, there has been observed epigenetic changes to multiple genes in SSc patients [8].
The critical role of the innate immune system in SSc will be discussed in this review. The innate immune system is often viewed as the body’s rapid response against pathogens and chemical or mechanical damage. This is achieved through the recognition of damage-, microbe- and pathogen-associated molecular patterns (known as DAMPs, MAMPs and PAMPs, respectively). Markers of activation of the innate immune system are present in both early and late stage SSc patients; this suggests a role for the innate immune system in both onset and progression of SSc; making research into this area a key focus for developing novel therapeutics.
Molecular patterns can have foreign or native origins and will interact with pattern recognition receptors (PRRs), chief among them are Toll-like receptors (TLRs), which upon activation with their ligand will trigger an inflammatory response [9]. As a result of this, in the initial stages of systemic sclerosis, there is a migration of mononuclear cells into the dermis which has been associated with increased collagen synthesis in nearby fibroblasts [10]. The initial triggering of TLRs and the innate immune system may precede fibrosis and lead to the persistent fibrotic state through a variety of mechanisms.
Toll-Like Receptors in Systemic Sclerosis
TLRs are evolutionary conversed receptors, which play a critical role in the innate immune system. TLRs recognise PAMPs and their subsequent signalling cascade can induce many cellular changes, Fig. 1a. However, TLRs can also act in response to DAMPs released from endogenous cells, Table 1, often under unfavourable conditions. In general, released DAMPs serve as an alarm ‘signal’ for cells, which results in an inflammatory response. The precise mechanisms regulating the release of DAMPs and the environment that favours this release are not clear but generally include stress or damage.

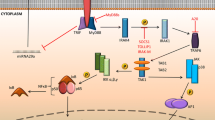
The role of innate immunity in systemic sclerosis. a Once activated by a ligand TLR induce a signalling cascade, involving MyD88 and TRIF, the downstream consequences of this activation result in the activation of various transcription factors. In systemic sclerosis TLR signalling can have multiple cellular effect including increase in ECM protein and immune cell activation. b IL-6 secretion by dermal fibroblasts after incubation with SAA, pre-treatment with IKK 2 (an NF-κB inhibitor) or DMSO vehicle control. IL-6 secretion was measured by ELISA. c Dermal fibroblasts were co-transfected an NF-κB luciferase reporter construct and a ‘dominant negative’ IκBα plasmid, cells treated after 24 h with SAA or HKLM (a natural TLR 2 agonist). Asterisk indicates significant differences assessed by ANOVA. d The image shows a skin section stained with H&E, the dark purple shows monocytes around the vasculature in SSc tissue skin. There is evidence that these cells are typically found between thickened collagen fibres and in the perivascular regions in SSc. Asterisk indicates accumulation of monocytes around the perivascular space. Abbreviations: Toll-like Receptors (TLR), TRIF-related adapt molecule (TRAM), TIR-domain-containing adaptor protein-inducing IFNβ (TRIF), extra cellular matrix (ECM), interluekin 6 (IL6), serum amyloid A (SAA), nuclear factorκB (NF-κB), dimethyl sulfoxide (DMSO), heat-killed Listeria monocytogenes (HKLM), hematoxylin and eosin (H&E) and systemic sclerosis (SSc)
The role TLRs play in many autoimmune diseases is well-documented [11,12,13,14]. To date, 13 TLRs have been described with 10 in humans, each responding to distinct ligands. TLRs are expressed both on the cell membrane and intracellularly in endocytic vesicles; this makes each set of TLRs uniquely placed to respond to the appropriate ligands. Interestingly, the molecular make-up of the DAMPs and PAMPs share no similarity and are a hugely diverse set of molecules despite this all trigger inflammation through activation of NF-KB. It is thought in SSc, DAMPs are released from damaged epithelial cells caused by disruption of the vasculature, and that aberrant TLR signalling may contribute to both the onset and progression of SSc [15]. Multiple TLR ligands have been described in SSc, consisting of both endogenous and microbial origin. One hypothesis is that the release of mediators from initial damage to the vasculature is the major stimulus for inflammation and wound healing and that a compromised resolution of this response underpins the fibrosis seen in the disease.
Although the inhibition of TLR signalling via interference with DAMPs may seem like a promising prospect, novel DAMPs and associated pathways are still being discovered. One such novel DAMP/TLR signalling pathway is that involving serum amyloid A (SAA)/TLR2 [13]. It has long been known that SAA is associated with immune disorders including rheumatoid arthritis and pulmonary fibrosis [16] and is elevated in SSc [15]. It was found that SAA induced interleukin 6 (IL-6) in dermal fibroblasts, whilst treatment with a neutralising antibody to TLR2 attenuated induction of IL-6 [13]. However, cells deleted for TLR4 did not show any activation demonstrating that TLR4 plays no role in SAA-mediated inflammation. Interestingly, it was found SAA induction of IL-6 was nuclear factor-кB (NF-кB) dependant [13]. Figure 1b shows a marked decrease in IL-6 after treatment with an IKK-2 inhibitor; the dependence of SAA on NF-кB was further confirmed with a double-negative mutant of NF-кB which saw a decrease in associated luciferase activity, Fig. 1c [13]. SSA-mediated induction of IL-6 may provide a mechanism into increased collagen 1 level in SSc, as IL-6 has been shown to directly induce collagen secretion in fibroblasts [17]. Thus, if SSA could be decreased, this could potentially lead to a decrease in the deposition of collagen, via a reduction of IL-6.
Studies have found an increase in DAMPs, such as hyaluronic acid (HA) and high-mobility group box-1 (HMGB-1) associated with TLR-4, in the serum of patients with SSc [18]. Takahashi et al. 2015 showed stimulation of TLR4 via HA and HMGB-1 resulted in fibroblast activation, whilst mice with a point mutation in the TLR4 gene showed attenuated skin sclerosis when challenged with bleomycin [19], also HMGB-1 has been found to activate hepatic stellate cells [20]. Again, showing reduction of DAMPs has the potential to be utilised as a therapeutic strategy in patients with SSc.
Skin biopsies analysed from SSc patients have shown a significant increase in TLR4 compared with healthy controls [18]. Strikingly, the same study also found enhanced Smad signalling with downregulation of BAMBI, a TGF-β antagonist, and suppression of the anti-fibrotic miR-29, which usually moderates both the duration and intensity of fibrotic responses by targeting the collagen messenger RNA (mRNA) 3’UTRs [18]. The study suggests a continuous feed-forward loop involving TLR4 is, in part, responsible for the persistence of fibrosis in SSc; thus, inhibition of TLR 4 is presented as novel therapeutic strategy in the progression, but not the onset, of fibrosis in SSc [18]. Furthermore, it has been shown that TLR4 is overexpressed in lesional skin from SSc patients along with the TLR4 co-receptor MD-2 and CD14[21]. Interestingly, in vivo administration of the TLR4 ligand to mice (LPS) resulted in immune activation and increased expression of the pro-fibrotic molecule TGF-β1, which was abrogated in TLR4-deficient mice [21].
A recent study demonstrated elevated levels of fibronectin EDA in both tissue skin samples and serum derived from patients with SSc. Fibronectin EDA is an alternatively spliced form of fibronectin, which is elevated in tissue damage. It was found that incubation with fibronectin EDA leads to upregulation of various extra cellular matrix (ECM) molecules, alpha SMA (α-SMA) expression and an increase in contractility in cells [22]. Remarkably, this was reduced in mice lacking fibronectin EDA or TLR4 expression demonstrating fibronectin EDA dependence on TLR4. To further confirm this, TLR 4 was blocked using pharmaceuticals; this blockade leads to a reduction in fibrosis [22]. Fibronectin EDA is hugely upregulated after tissue damage; this holds the possibly to explain a positive feed-forward loop. In the suggested feed-forward loop, the initial damage leads to increased fibronectin EDA which then activates TLR4 to decrease miR29a and increased collagen expression in a Smad-dependant pathway. We have also shown downregulation of miR29a in fibrosis via lipopolysaccharide (LPS) [23]. Previous reports have also demonstrated that animals lacking the fibronectin EDA domain gene have reduced ability to heal skin wounds [24], and fibronectin EDA is also associated with cardiac fibrosis through TLR signalling [25].
Tenascin C is an extracellular matrix glycoprotein, which is transiently expressed upon tissue injury. As with fibronectin EDA, it has recently been demonstrated that tenascin C is elevated in not only skin biopsies from SSc patients, but also the serum [26••]. The study cleverly used three-dimensional skin models to show that treatment with tenascin C increased collagen and α-SMA mRNA and that tenascin C can induce a wide pro-fibrotic response [26••]. The study later goes on to show that the pro-fibrotic response induced by Tenascin C is TLR4-dependant [26••]. This is not only important in the management of SSc but also may hold potential in reducing SSc-associated complications, as tenascin C is also increased in SSc-associated pulmonary fibrosis [27]. When taken in combination, these papers suggest that TLR4 activation by endogenous ligands is critical in the progression of fibrosis and therefore targeting TLR4 either by reduction of DAMPs or TLR 4 antagonism could be of therapeutic benefit. Indeed the TLR agonist S100A8 is elevated in SSc serum and has been suggested to serve as a biomarker [28].
The differentiation of fibroblasts to myofibroblasts is pivotal in SSc, due to the increased level of extracellular matrix proteins. TLR9 has been shown to increase and co-localise with myofibroblasts in SSc skin biopsies, over 60% of myofibroblasts were TLR 9 positive [29]. Moreover, the study used two murine models of SSc to show the accumulation of TLR9-positive fibroblasts around lesion tissue [29]. The activation of TLR 9, via CpG, in this study, lead to an increase in activated TGF-β, which when blocked by neutralising antibodies eliminated the pro-fibrotic effects of CpG [29]. TGF-β1 is known to increase collagen and promote fibroblast to myofibroblast transdifferentiation.
Interestingly, a separate study found that TGF-β increased mRNA TLR9 levels in lung fibroblasts [30]. If TGF-β1 is shown to increase TLR9 in dermal fibroblasts, targeting of TGF-β1 could be of benefit via two mechanisms of action, by lowering both TGFβ itself and TLR9 mRNA. A study using the drug bortzomib has already shown to decrease in the autocrine release of TGF-β [31]. Interestingly, the study used the bleomycin murine model and found attenuation of both lung and skin fibrosis after treatment with bortzomib [31].
Although the direct targeting of DAMPs and TLRs has been discussed in depth, it must also be taken into account that downstream molecules such as interferon regulatory factor 5 (IRF5) and Fos-related antigen 2 (Fra 2) could also play a role in SSc pathogenesis, alongside complex epigenetic regulation. Basal levels of, the endosomal TLR, TLR8 have been found to be 2.1-fold lower in monocytes from patients with SSc [32•]. Despite this, after stimulation with single-stranded RNA (ssRNA) and a histone methyltransferase inhibitor, monocytes displayed increased levels of Fra-2 and tissue inhibitor of metalloproteases 1 (TIMP 1) [32•]. ssRNA is commonly found in viruses suggesting that a virus could be responsible for the induction of a pro-fibrotic state in SSc. The relationship between TIMP 1 and matrix metalloproteinases (MMPs) is critical in ECM accumulation, increased levels of TIMPs inhibit MMPs from degrading ECM proteins, thus the net effects are an increase in ECM, and we could show that this was MyD88-dependant. Murine models of SSc, such as bleomycin, have shown that induction of fibrosis is mediated through IRF5, and that the signalling axis involving TLR 4 and IRF-5 is directly involved in regulating B cells [33]. Interestingly, IRF 5 knockout mice do not show immune abnormalities such as increased levels of IL-6 when challenged with bleomycin [33]. Also polyI:C, the endosomal TLR3 ligand, activates monocytes in SSc to cause fibrosis [34]. Furthermore, type I interferon has been shown to upregulate TLR3 expression in dermal fibroblasts and that this interferon mediated upregulation of TLR3 leads to enhanced IL-6 and MCP-1 levels and ultimately enhanced recruitment of macrophages into the tissue [35].
The targeting of TLRs and their signalling cascades is viewed as an attractive therapeutic intervention in a disease with little available therapy. However, despite heavy scrutiny across several autoimmune diseases, the role of TLRs in the pathogenesis of SSc is complex and not yet fully understood. Recently, several factors ranging from novel signalling cascades [13], Heme oxgenase-1 levels [36], rare TLR2 polymorphisms [37•] and epigenetic changes [32•] have been shown to affect the progression of the disease. Whilst scrutiny of TLRs and their associated pathways will remain, rightly, at the forefront of research into SSc, the natural protective role of TLRs must be taken into account before progress can be made into the clinic.
Monocyte/Macrophage Involvement in SSc
Post-injury, precursor cells such as monocytes are recruited from the bone marrow to the site of injury. Recruited cells, along with resident tissue macrophages, cause inflammation and clear cellular debris. If the inflammation and tissue damage fail to resolve, fibrosis may arise [38]. In SSc, there is infiltration of mononuclear cells, including macrophages [39] and monocytes [40], Fig. 1d. These cells were found to be situated in between thickened collagen fibres and in the perivascular and periappendageal regions [40]. It is speculated that here, the macrophages secrete high amounts of pro-fibrotic molecules and thus mediate fibrosis in situ.
In SSc patients, there is a statistically significant increase in the number of monocytes in the peripheral blood. Interestingly, monocytes from SSc patients have lower rate of apoptosis than observed in healthy individuals [41] suggesting that they are somehow resistant to apoptosis.
When stimulated by IL-4 or IL-13, resident macrophages in the skin can adopt a phenotype known as M2, associated with vascularization and wound repair. This M2 subtype of macrophage can produce TGβ-1 [40], a pro-fibrotic molecule that stimulates production of type I collagen in fibroblasts [42]. M2 macrophages are typically considered to be anti-inflammatory [40] but have been associated with certain fibrotic diseases, including SSc [43]; likely due to their production of TGF-β and other inflammatory molecules [44]. We have demonstrated high levels of macrophages in the skin and co-localised with the molecule TIMP-1 [32•]; here, they are releasing TIMP-1 and the net effect is the deposition of ECM via inhibiting MMPs. Also, though the macrophages are releasing TIMP-1 that directly activated the normal fibroblast to a myofibroblast via an unknown mechanism. However, deletion of the transcription factor Fra-2 ameliorates the effect. What is also interesting is that Fra-2 levels are increased by ssRNA, and if we inhibit histone methyltransferase activity with Dznep, this enhances Fra2-mediated TIMP-1 levels [32•]; thus, epigenetic mechanisms are at play also [45]. It is likely that the macrophage population is not as binary as M1 and M2 and that this is a plastic system that is modified rather quickly likely through epigenetic mechanisms [46]. We have also found previously a dysregulated level of microRNA-135b and that this targets STAT6, thus alterations in STAT6 may mediate enhanced IL-4 signaling involved in M2 generation [47].
If the macrophage population could be skewed towards to an M1 type, with the use of pharmaceuticals, this may help slow collagen production and reduce fibrosis. However, as the M1 subtype is pro-inflammatory, this may not be a viable option.
Monocyte chemoattractant protein-1 (MCP-1), also known as CCL2, is an important mediator of inflammation and fibrosis, particularly in SSc [10]. Expression of CCL2 has been found to be upregulated in fibroblasts in SSc skin at both the mRNA and protein level [48]. Multiple ligands have been identified dysregulated in SSc and these target TLRs to cause downstream signalling and blocking; these may be therapeutic, but this may reduce the innate immune response. Another therapy may be blocking the chemokines that are released to drive the influx of monocytes into the skin. Chemokines are elevated in the disease, and recently, new chemokine antagonists have been developed.
Complement in SSc
The complement system, comprised of approximately 30 circulating and membrane proteins, is a complex proteolytic cascade; it acts as one of the central components of innate immunity and plays a pivotal role in the initial defence against infection and in the elimination of dead/modified self cells [49, 50]. The activation of complement is controlled by multiple mechanisms forming three pathways: the classical pathway is activated by C1 interacting with antibody bound to pathogens [51]; the lectin pathway is activated by mannose binding lectin interacting with sugars on the cell surface and alternative pathway activation occurs continuously at low levels in the plasma [52], allowing for quick response to initial pathogen exposure and amplification of both the classical and lectin pathways [53].
Several studies have suggested a role for complement in SSc; however, few define how the complement cascade is activated in SSc [54,55,56,57]. In 2010, a study of SSc patients showed increased levels of factor H, a complement regulator, which had impaired function [58]. This was shown again in a later study, along with a decrease in regulator CD46 expression in vasculature of SSc patients [59]. This suggests a potential reason for the increase in complement terminal complexes/ fragments seen in previous research. The development of a mini factor H has been shown to reduce abnormal C3 deposition in a factor H deficient mouse model [60]; once fully developed, this may become a therapeutic option for SSc patients with impaired factor H function.
In 2014, it was shown that whilst the lectin pathway of complement was not a cause of SSc, it served to exacerbate symptoms following ischemia and reperfusion [61]. Treatment of the lectin pathway has been shown to be protective against myocardial reperfusion injury [62] and against renal damage caused by reperfusion [63]. A future therapeutic option in SSc may therefore be to target the lectin pathway and protect against further complications.
Mast Cells in SSc
Mast cells are densely granulated cells of the innate immune system with the primary purpose of combatting parasitic infection. To do this, mast cells will degranulate, releasing the contents of the vesicles within their cytosol into the surrounding environment. These vesicles contain various cytokines and pro-inflammatory molecules including histamine and TGF-β. Mast cells have been implicated in cardiac fibrosis, renal fibrosis and pulmonary fibrosis [64]. In SSc, the density of dermal mast cells in the fingers is reflective of the severity of sclerosis, correlating with the patient’s modified Rodnan skin score. The density of dermal mast cells in the fingers was also found to be higher in patients who were positive for anti-topoisomerase I antibody [65].
Inflammasomes in SSc
The inflammasome is a multi-molecular complex that is activated by diverse ligands that activates caspase-1 into its active protease; this leads to the release of interleukin-1β and IL-18 from the cell. Activation of nod-like receptors (NLR) ultimately results in the activation of the inflammasome. NLRP3 is one of the most studied inflammasome components and has a variety of molecular triggers. The inflammasome NLRP3 has been investigated in SSc. Mice with deleted NLRP3 were resistant to fibrosis when challenged with bleomycin; however, the endogenous ligand associated with the activation of NLR in SSc remains unknown[66]. Indeed, a small nucleotide polymorphism (SNP) has been demonstrated in SSc patients [67], whether this is non-synonymous has yet to be determined.
Recent evidence has also suggested a role for the inflammasome in myofibroblast generation by mitochondrial NLRP3 via enhanced Smad signalling and genetic reduction of NLRP3 is protective in experimentally induced liver fibrosis [68, 69]. The fact that few ligands have been described for the inflammasome does not discount that there may be many ligands released upon cellular stress.
Conclusion
Transition of research into the clinic in SSc remains an issue; this in part is due to the complex pathways involved in its pathogenesis. A wealth of evidence is now emerging that the innate immune system is critical in driving and initiating the disease. Chief among these are the TLRs and their ligands. Targeting these therapeutically may be a useful option. Alternatively, targeting the downstream effector molecules could also be useful or the epigenetic modifications that also occur. Multiple epigenetic modifications in response to TLR ligation occur, and these could be targeted with currently used clinical drugs. Although progress has been made in the mechanisms involved in sustaining fibrosis, the initiating events in SSc remain somewhat of a mystery.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Valentini G, Black C. Systemic sclerosis. Best Pract Res Clin Rheumatol. 2002;16(5):807–16.
Yasuoka H. Recent treatments of interstitial lung disease with systemic sclerosis. Clin Med Insights Circ Respir Pulm Med. 2015;9 Suppl 1:97–110. doi:10.4137/CCRPM.S23315.
Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest. 2013;144(4):1346–56. doi:10.1378/chest.12-2396.
Patterson KA, Roberts-Thomson PJ, Lester S, Tan JA, Hakendorf P, Rischmueller M, et al. Interpretation of an extended autoantibody profile in a well-characterized Australian systemic sclerosis (Scleroderma) cohort using principal components analysis. Arthritis Rheumatol. 2015;67(12):3234–44. doi:10.1002/art.39316.
Chang WS, Schollum J, White DH, Solanki KK. A cross-sectional study of autoantibody profiles in the Waikato systemic sclerosis cohort. New Zealand Clin Rheumatol. 2015;34(11):1921–7. doi:10.1007/s10067-015-2981-3.
Ochoa E, Martin JE, Assasi S, Beretta L, Carreira P, Guillen A, et al. Confirmation of CCR6 as a risk factor for anti-topoisomerase I antibodies in systemic sclerosis. Clin Exp Rheumatol. 2015;33(4 Suppl 91):S31–5.
Mierau R, Moinzadeh P, Riemekasten G, Melchers I, Meurer M, Reichenberger F, et al. Frequency of disease-associated and other nuclear autoantibodies in patients of the German Network for Systemic Scleroderma: correlation with characteristic clinical features. Arthritis Res Ther. 2011;13(5):R172. doi:10.1186/ar3495.
Dowson C, O’Reilly S. DNA methylation in fibrosis. Eur J Cell Biol. 2016;95(9):323–30. doi:10.1016/j.ejcb.2016.06.003.
Martin SF. Adaptation in the innate immune system and heterologous innate immunity. Cell Mol Life Sci. 2014;71(21):4115–30. doi:10.1007/s00018-014-1676-2.
Yamamoto T. Autoimmune mechanisms of scleroderma and a role of oxidative stress. Self Nonself. 2011;2(1):4–10. doi:10.4161/self.2.1.14058.
Roelofs M, Joosten L, Abdollahi-Roodsaz S, Van Lieshout A, Sprong T, Van Den Hoogen F, et al. The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum. 2005;52(8):2313–22.
Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202(8):1131–9.
O’Reilly S, Cant R, Ciechomska M, Finnigan J, Oakley F, Hambleton S, et al. Serum amyloid A induces interleukin-6 in dermal fibroblasts via Toll-like receptor 2, interleukin-1 receptor-associated kinase 4 and nuclear factor-κB. Immunology. 2014;143(3):331–40.
Duffy L, O’Reilly SC. Toll-like receptors in the pathogenesis of autoimmune diseases: recent and emerging translational developments. ImmunoTargets Therapy. 2016;5:69.
Fullard N, O’Reilly S, editors. Role of innate immune system in systemic sclerosis. Seminars in immunopathology; 2015: Springer.
Lakota K, Carns M, Podlusky S, Mrak-Poljsak K, Hinchcliff M, Lee J, et al. Serum amyloid A is a marker for pulmonary involvement in systemic sclerosis. PLoS One. 2015;10(1), e0110820.
O’Reilly S, Ciechomska M, Cant R, van Laar JM. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-β (TGF-β) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J Biol Chem. 2014;289(14):9952–60.
Bhattacharyya S, Kelley K, Melichian DS, Tamaki Z, Fang F, Su Y, et al. Toll-like receptor 4 signaling augments transforming growth factor-β responses: a novel mechanism for maintaining and amplifying fibrosis in scleroderma. Am J Pathol. 2013;182(1):192–205.
Takahashi T, Asano Y, Ichimura Y, Toyama T, Taniguchi T, Noda S, et al. Amelioration of tissue fibrosis by toll-like receptor 4 knockout in murine models of systemic sclerosis. Arthritis Rheumatol. 2015;67(1):254–65.
Zhang Z, Lin C, Peng L, Ouyang Y, Cao Y, Wang J, et al. High mobility group box 1 activates Toll like receptor 4 signaling in hepatic stellate cells. Life Sci. 2012;91(5):207–12.
Stifano G, Affandi AJ, Mathes AL, Rice LM, Nakerakanti S, Nazari B, et al. Chronic Toll-like receptor 4 stimulation in skin induces inflammation, macrophage activation, transforming growth factor beta signature gene expression, and fibrosis. Arthritis Res Ther. 2014;16(4):1.
Bhattacharyya S, Tamaki Z, Wang W, Hinchcliff M, Hoover P, Getsios S, et al. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci Transl Med. 2014;6(232):232ra50–ra50.
Ciechomska M, O’Reilly S, Suwara M, Bogunia-Kubik K, van Laar JM. MiR-29a reduces TIMP-1 production by dermal fibroblasts via targeting TGF-β activated kinase 1 binding protein 1, implications for systemic sclerosis. PLoS One. 2014;9(12), e115596.
Muro et al. Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan Journal of Cell Biology 2003 162: 149–160.ᅟ
Booth AJ, Wood SC, Cornett AM, Dreffs AA, Lu G, Muro AF, et al. Recipient-derived EDA fibronectin promotes cardiac allograft fibrosis. J Pathol. 2012;226(4):609–18.
•• Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X et al. Tenascin-C drives persistence of organ fibrosis. Nature communications. 2016;7. Shows the possible feed-forward loop with a fragment only expressed after tissue damage.
Brissett M, Veraldi KL, Pilewski JM, Medsger TA, Feghali-Bostwick CA. Localized expression of tenascin in systemic sclerosis–associated pulmonary fibrosis and its regulation by insulin-like growth factor binding protein 3. Arthritis Rheum. 2012;64(1):272–80.
Van Bon L, Cossu M, Loof A, Gohar F, Wittkowski H, Vonk M, et al. Proteomic analysis of plasma identifies the Toll-like receptor agonists S100A8/A9 as a novel possible marker for systemic sclerosis phenotype. Ann Rheum Dis. 2014;73(8):1585–9.
Fang F, Marangoni RG, Zhou X, Yang Y, Ye B, Shangguang A, et al. TLR9 signaling is augmented in systemic sclerosis and elicits TGF-β-dependent fibroblast activation. Arthritis Rheumatol. 2016.
Kirillov V, Siler JT, Ramadass M, Ge L, Davis J, Grant G, et al. Sustained activation of toll-like receptor 9 induces an invasive phenotype in lung fibroblasts: possible implications in idiopathic pulmonary fibrosis. Am J Pathol. 2015;185(4):943–57.
Mutlu GM, Budinger GS, Wu M, Lam AP, Zirk A, Rivera S, et al. Proteasomal inhibition after injury prevents fibrosis by modulating TGF-β1 signalling. Thorax. 2012;67(2):139–46.
• Ciechomska M, O’Reilly S, Przyborski S, Oakley F, Bogunia-Kubik K, van Laar JM. Histone demethylation and toll-like receptor 8–dependent cross-talk in monocytes promotes transdifferentiation of fibroblasts in systemic sclerosis via Fra-2. Arthritis Rheumatol. 2016;68(6):1493–504. This is an important paper that describes the activation of monocytes by ssRNA and an alteration of lysine trimethylation on the histones that can aggravate fibrosis via upregulation of Fra2 and TIMP-1 expression.
Saigusa R, Asano Y, Taniguchi T, Yamashita T, Ichimura Y, Takahashi T, et al. Multifaceted contribution of the TLR4-activated IRF5 transcription factor in systemic sclerosis. Proc Natl Acad Sci. 2015;112(49):15136–41.
Farina GA, York MR, Di Marzio M, Collins CA, Meller S, Homey B, et al. Poly (I: C) drives type I IFN-and TGFβ-mediated inflammation and dermal fibrosis simulating altered gene expression in systemic sclerosis. J Investig Dermatol. 2010;130(11):2583–93.
Agarwal SK, Wu M, Livingston CK, Parks DH, Mayes MD, Arnett FC, et al. Toll-like receptor 3 upregulation by type I interferon in healthy and scleroderma dermal fibroblasts. Arthritis Res Ther. 2011;13(1):1.
van Bon L, Cossu M, Scharstuhl A, Pennings BWC, Vonk MC, Vreman HJ, et al. Low heme oxygenase-1 levels in patients with systemic sclerosis are associated with an altered Toll-like receptor response: another role for CXCL4? Rheumatology. 2016;55(11):2066–73. doi:10.1093/rheumatology/kew251.
• Broen J, Bossini-Castillo L, Van Bon L, Vonk MC, Knaapen H, Beretta L, et al. A rare polymorphism in the gene for Toll-like receptor 2 is associated with systemic sclerosis phenotype and increases the production of inflammatory mediators. Arthritis Rheum. 2012;64(1):264–71. This paper was the first to describe the rare SNP in TLR2 associated with a particular phenotype in SSc.
Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44(3):450–62. doi:10.1016/j.immuni.2016.02.015.
Fleischmajer R, Perlish JS, Reeves JR. Cellular infiltrates in scleroderma skin. Arthritis Rheum. 1977;20(4):975–84.
Higashi-Kuwata N, Jinnin M, Makino T, Fukushima S, Inoue Y, Muchemwa FC, et al. Characterization of monocyte/macrophage subsets in the skin and peripheral blood derived from patients with systemic sclerosis. Arthritis Res Ther. 2010;12(4):R128. doi:10.1186/ar3066.
Lopez-Cacho JM, Gallardo S, Posada M, Aguerri M, Calzada D, Mayayo T, et al. Association of immunological cell profiles with specific clinical phenotypes of scleroderma disease. Biomed Res Int. 2014;2014:148293. doi:10.1155/2014/148293.
Ghosh AK, Mori Y, Dowling E, Varga J. Trichostatin A blocks TGF-beta-induced collagen gene expression in skin fibroblasts: involvement of Sp1. Biochem Biophys Res Commun. 2007;354(2):420–6. doi:10.1016/j.bbrc.2006.12.204.
Higashi-Kuwata N, Makino T, Inoue Y, Takeya M, Ihn H. Alternatively activated macrophages (M2 macrophages) in the skin of patient with localized scleroderma. Exp Dermatol. 2009;18(8):727–9. doi:10.1111/j.1600-0625.2008.00828.x.
Raes G, Beschin A, Ghassabeh GH, De Baetselier P. Alternatively activated macrophages in protozoan infections. Curr Opin Immunol. 2007;19(4):454–9. doi:10.1016/j.coi.2007.05.007.
O’Reilly S. Epigenetics in fibrosis. Mol Asp Med. 2016.
Ciechomska M, van Laar J, O’Reilly S. Emerging role of epigenetics in systemic sclerosis pathogenesis. Genes Immun. 2014;15(7):433–9.
O’Reilly S, Ciechomska M, Fullard N, Przyborski S, van Laar JM. IL-13 mediates collagen deposition via STAT6 and microRNA-135b: a role for epigenetics. Sci Rep. 2016;6.
Distler O, Pap T, Kowal-Bielecka O, Meyringer R, Guiducci S, Landthaler M, et al. Overexpression of monocyte chemoattractant protein 1 in systemic sclerosis: role of platelet-derived growth factor and effects on monocyte chemotaxis and collagen synthesis. Arthritis Rheum. 2001;44(11):2665–78.
Harris CL, Heurich M, Rodriguez de Cordoba S, Morgan BP. The complotype: dictating risk for inflammation and infection. Trends Immunol. 2012;33(10):513–21. doi:10.1016/j.it.2012.06.001.
Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci. 2013;126(Pt 13):2903–13. doi:10.1242/jcs.124388.
Lachmann PJ, Hughes-Jones NC. Initiation of complement activation. Springer Semin Immunopathol. 1984;7(2–3):143–62.
Turner MW. The role of mannose-binding lectin in health and disease. Mol Immunol. 2003;40(7):423–9.
Harris CL, Pettigrew DM, Lea SM, Morgan BP. Decay-accelerating factor must bind both components of the complement alternative pathway C3 convertase to mediate efficient decay. J Immunol. 2007;178(1):352–9.
Senaldi G, Vergani D, McWhirter A, Black CM. Complement activation in progressive systemic sclerosis. Lancet. 1987;1(8542):1143–4.
Benbassat C, Schlesinger M, Luderschmidt C, Valentini G, Tirri G, Shoenfeld Y. The complement system and systemic sclerosis. Immunol Res. 1993;12(3):312–6.
Senaldi G, Lupoli S, Vergani D, Black CM. Activation of the complement system in systemic sclerosis. Relationship to clinical severity. Arthritis Rheum. 1989;32(10):1262–7.
Sprott H, Muller-Ladner U, Distler O, Gay RE, Barnum SR, Landthaler M, et al. Detection of activated complement complex C5b-9 and complement receptor C5a in skin biopsies of patients with systemic sclerosis (scleroderma). J Rheumatol. 2000;27(2):402–4.
Scambi C, La Verde V, De Franceschi L, Barausse G, Poli F, Benedetti F, et al. Comparative proteomic analysis of serum from patients with systemic sclerosis and sclerodermatous GVHD. Evidence of defective function of factor H. PLoS One. 2010;5(8), e12162. doi:10.1371/journal.pone.0012162.
Scambi C, Ugolini S, Jokiranta TS, De Franceschi L, Bortolami O, La Verde V, et al. The local complement activation on vascular bed of patients with systemic sclerosis: a hypothesis-generating study. PLoS One. 2015;10(2), e0114856. doi:10.1371/journal.pone.0114856.
Nichols EM, Barbour TD, Pappworth IY, Wong EK, Palmer JM, Sheerin NS, et al. An extended mini-complement factor H molecule ameliorates experimental C3 glomerulopathy. Kidney Int. 2015;88(6):1314–22. doi:10.1038/ki.2015.233.
Osthoff M, Ngian GS, Dean MM, Nikpour M, Stevens W, Proudman S, et al. Potential role of the lectin pathway of complement in the pathogenesis and disease manifestations of systemic sclerosis: a case–control and cohort study. Arthritis Res Ther. 2014;16(6):480. doi:10.1186/s13075-014-0480-6.
Jordan JE, Montalto MC, Stahl GL. Inhibition of mannose-binding lectin reduces postischemic myocardial reperfusion injury. Circulation. 2001;104(12):1413–8.
Castellano G, Melchiorre R, Loverre A, Ditonno P, Montinaro V, Rossini M, et al. Therapeutic targeting of classical and lectin pathways of complement protects from ischemia-reperfusion-induced renal damage. Am J Pathol. 2010;176(4):1648–59. doi:10.2353/ajpath.2010.090276.
Hugle T. Beyond allergy: the role of mast cells in fibrosis. Swiss Med Wkly. 2014;144:w13999. doi:10.4414/smw.2014.13999.
Yukawa S, Yamaoka K, Sawamukai N, Shimajiri S, Kubo S, Miyagawa I, et al. Dermal mast cell density in fingers reflects severity of skin sclerosis in systemic sclerosis. Mod Rheumatol. 2013;23(6):1151–7. doi:10.1007/s10165-012-0813-8.
Artlett CM, Sassi-Gaha S, Rieger JL, Boesteanu AC, Feghali-Bostwick CA, Katsikis PD. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum. 2011;63(11):3563–74.
Dieude P, Guedj M, Wipff J, Ruiz B, Riemekasten G, Airo P et al. NLRP1 influences the systemic sclerosis phenotype: a new clue for the contribution of innate immunity in systemic sclerosis-related fibrosing alveolitis pathogenesis. Ann Rheum Dis. 2010:annrheumdis131243.
Bracey NA, Gershkovich B, Chun J, Vilaysane A, Meijndert HC, Wright JR, et al. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J Biol Chem. 2014;289(28):19571–84.
Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59(3):898–910.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Scleroderma
Rights and permissions
About this article

Cite this article
Dowson, C., Simpson, N., Duffy, L. et al. Innate Immunity in Systemic Sclerosis. Curr Rheumatol Rep 19, 2 (2017). https://doi.org/10.1007/s11926-017-0630-3
Published:
DOI: https://doi.org/10.1007/s11926-017-0630-3