
Abstract
Metabolic bone diseases manifesting fragility fractures (such as osteoporosis) are routinely diagnosed based on bone mineral density (BMD) measurements, and the effect of various therapies also evaluated based on the same outcome. Although useful, it is well recognized that this metric does not fully account for either fracture incidence or the effect of various therapies on fracture incidence, thus, the emergence of bone quality as a contributing factor in the determination of bone strength. Infrared and Raman vibrational spectroscopic techniques are particularly well-suited for the determination of bone quality as they provide quantitative and qualitative information of the mineral and organic matrix bone components, simultaneously. Through the use of microspectroscopic techniques, this information is available in a spatially resolved manner, thus, the outcomes may be easily correlated with outcomes from techniques such as histology, histomorphometry, and nanoindentation, linking metabolic status with material properties.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Need for Evaluation of Matrix and Mineral Chemistry
Loss of bone mass, measured clinically as change in bone mineral density (BMD), is considered an important risk factor for bone fragility. However, BMD is not the sole predictor of whether an individual will experience a fracture [1, 2], and there is considerable overlap in BMD between populations that do and do not develop fractures [3, 4]. It has been demonstrated that for a given bone mass an individual’s risk to fracture increases with age [5], whereas the 1-year relative risk associated with a given change in BMD has been shown to be independent of age [6]. Consistent with these findings, numerous investigators have shown that mechanical variables directly related to fracture risk are either independent [7] or not totally accounted for bone mass itself [8–12]. Epidemiologic evidence also shows considerable overlap of bone density values between fracture and nonfracture groups suggesting that low bone quantity alone, often measured as either BMD or bone mass, is an insufficient cause of fragility fractures [13–15]. Finally, conditions such as idiopathic osteoporosis (IOP) where widely employed tools (both clinical and basic research) such as BMD, biochemical markers, micro-CT, quantitative backscattered electron imaging (qBEI), and/or finite element analysis (FE) fail to either account for, or predict the manifested bone strength, further emphasizing the need for the establishment of additional parameters that contribute to the determination of bone strength [16–19].
It is becoming evident then, that in addition to BMD, “bone quality” should also be considered when assessing bone strength and fracture risk. Bone quality is a broad term encompassing a plethora of factors such as geometry and bone mass distribution, trabecular bone microarchitecture, microdamage, increased remodeling activity, along with genetics, body size, environmental factors, and changes in bone mineral and matrix tissue properties [4, 20].
Need for Microscopic Imaging Techniques when Assessing Matrix and Mineral Chemistry
One of the obstacles to be circumvented when assessing mineral and matrix tissue properties is tissue heterogeneity at the microscopic level. In normal humans, cortical bone constitutes approximately 80 % of the human skeletal mass and trabecular bone approximately 20 % [21]. Bone surfaces may be undergoing formation or resorption, or they may be relatively inactive. Bone turnover processes, which can be visualized microscopically, occur throughout life in both cortical and trabecular bone [21]. Bone remodeling is a surface phenomenon, and in humans occurs on periosteal, endosteal, Haversian canal, and trabecular surfaces [21]. The rate of cortical bone remodeling, which may be as high as 50 % per year in the mid-shaft of the femur during the first 2 years of life, eventually declines to a rate of 2 %–5 % per year in the elderly. Rates of remodeling in trabecular bone are proportionately higher throughout life and may normally be five to ten times higher than cortical bone remodeling rates in the adult [21]. It becomes evident that: (I) bone is most metabolically active at its surfaces; (II) both mineral and matrix composition may significantly change every few microns. It is thus, important to utilize microscopic imaging techniques and account for metabolic activity/tissue age [20, 22–25]. Previously published work has reported on the relation between tissue age, bone tissue composition, and mechanical properties [26–28]. Finally, although because of the remodeling process the organic matrix and mineral chemistry are dependent on turnover rates, factors other than bone turnover may influence bone composition [29–35]. Analysis of matrix and mineral composition as a function of tissue age, thus, allows for the discrimination between bone turnover and independent effects. Utilizing techniques such as Small Angle X-ray Scattering (SAXS), quantitative backscattered electron imaging (qBEI), Raman microspectroscopy, and Fourier transform infrared microspectroscopy (FTIRM) and imaging (FTIRI), the analysis of bone mineral (poorly crystalline hydroxyapatite) at the microscopic level and the contribution of mineral crystallinity (crystallite size) and maturity (chemical composition) to bone strength is being actively pursued [20, 23–25]. Based on these analyses, an important role for the organic matrix in the determination of biomechanical properties is predicted. Specifically, the matrix is proposed to play an important role in alleviating impact damage to mineral crystallites, and to matrix/mineral interfaces, behaving like a deformable protective layer around mineral crystallites, thus, protecting them from the peak stresses caused by impact, and homogenizing stress distribution within the bone composite [36, 37]. Altered collagen properties have been associated with fragile bone, in both animals and humans [16, 18, 19, 29, 38–41]. Additionally, recent reports indicate that alterations in organic matrix composition/quality even when confined in limited anatomic areas (for example first few microns of an actively bone forming surface) may be sufficient to influence the mechanical performance of the whole bone organ [26, 29], in the absence of concomitant changes in either bone mineral quantity or quality.
Fourier Transform Infrared and Raman Spectroscopy
A Brief Overview
For 70 years, vibrational spectroscopy has been extensively applied in analytical and physical chemistry for elucidation of the structure of small molecules. The common principle underlying these two techniques is the transition between vibrational energy states of molecules; infrared transitions arise directly from absorption of energy in the infrared range, whereas Raman spectra arise from the scattering of visible or ultraviolet photons that have either gained or lost part of their energy upon interaction with the vibrating bonds. Each molecule has its own unique vibrational characteristics and therefore will result in unique IR or Raman spectral signatures. Additionally, the neighboring molecular environment influences the vibrational characteristics. The position, intensity, and width of a vibrational band can be used for monitoring a particular functional group or regions of a particular chemical species. The IR and Raman spectra of a given sample differ considerably, as some vibrations are only either IR or Raman active and hence, each technique can provide complimentary information regarding the analyzed sample [42]. Successful extension of these technologies into the biomedical arena has depended upon two factors. First (1970s‒1980s) was the development of spectra-structure correlations for proteins, nucleic acids, and lipid assemblies along with smaller molecules that are essential for the biological functions of the macromolecular entities. The second factor was the recognition that in cells and tissues, the correct spatial orientation of the structural components is essential for their optimal function, and that pathologic states may result in or may be induced by spatial disruption of the normal organization. Traditional cataloguing of spatial information has required high-resolution microscopy methods that provide visual images of biological structures. However, the approaches generally require extensive sample preparation and are lacking in molecular structure information.
Early in the history of vibrational spectroscopy, it was recognized that for successful application of IR and Raman to biomedical problems, microscopy, and imaging approaches were essential. Thus, in 1949 [43], a reflecting microscope was coupled with an IR spectrometer to produce useable spectra of nerve fibers and small crystals to demonstrate the utility of IR for identification of a specimen’s molecular features. In contrast, the weakness of ordinary Raman scattering delayed the introduction of microscopic and imaging applications of that technology until 1974 when two laboratories [44, 45] simultaneously proved the feasibility of the approach.
The extensive technical advances leading to the wider application of vibrational microscopy and imaging that have occurred in the past 25 years have been widely reviewed [46–50] and interested readers are strongly encouraged to peruse these sources. More recently, substantial technological gains have appeared for both IR and Raman, especially in the areas of improved spatial resolution and rapid data acquisition. These will be discussed briefly below.
Recent Technical Advances
Resolution Enhancement in IR Spectroscopy—Defeating the Rayleigh Criterion
Two recent instrument designs have provided improved spatial resolution for IR spectral acquisition at the AFM level (20 nm compared with the 3–10 micron values normally cited). In the first design [51], Fourier transform infrared nanospectroscopy is based on a scattering-type scanning near-field optical microscope equipped with a coherent-continuum infrared light source. This mid-infrared source emits a continuous spectrum that can be tuned between 700 − 2500 cm−1. The method can acquire IR spectra of molecules placed in the near-field of the AFM tip, with a spatial resolution of 20 nm. The backscattered radiation from the tip is spectrally analyzed with an FT-IR spectrometer.
In the second approach, a pulsed, tunable IR laser excites molecular absorption in a sample that has been mounted in ATR (attenuated total reflection) fashion, on a ZnSe prism. Samples may be prepared either by microtome sections ranging in thickness from 100 nm and 1 micron or by casting films directly from solvents. The beam from the IR laser illuminates the sample by total internal reflection. When the sample absorbs radiation, it heats up and thermally expands to excite resonant oscillations of the AFM cantilever, which are detected in a standard AFM measurement system [52]. The cantilever oscillations decay in a ringdown modality, from which amplitudes and frequencies of the oscillations may be mathematically extracted and related to IR spectral parameters. This technique has been utilized in a recently released study of bone [53] in which correlation analysis was performed across of a series of AFM-IR spectra collected outward from an osteonal center at 1 micron spatial resolution, with improvements to 200 nm resolution (or better) readily envisioned.
Tip Enhanced Raman Scattering
Tip enhanced Raman scattering (TERS) combines surface enhanced Raman spectroscopy (SERS) with AFM analysis to offer the possibility of measurements with spatial resolution in the nanometer scale. Although the basic underlying principle is rather straightforward, achieving reproducible results has proven challenging to date. The experiment incorporates a SERS AFM or STM tip from gold or silver nanoparticles to enhance the Raman signals from nearby molecules. The achievable spatial resolution is tens of nanometers. The TERS effect would be expected only in the vicinity of the tip, and would depend for its detection on the magnitude of the SERS enhancement available. The resolution enhancement comes from the size of the SERS-covered tip. The magnitude of the TERS enhancement from the relatively small number of molecules near the SERS tip must be both distinguishable from and significantly greater than the normal Raman effect from scattering molecules in the remainder of the excitation beam. Examples of TERS from single biomolecules have been reported [54, 55].
Rapid Imaging Raman Spectroscopy
Xie and coworkers, among others, have dramatically improved [56] the technique of Stimulated Raman scattering microscopy, allowing the images at particular Raman shifted frequencies to be acquired at video rates. Several applications of the technology have appeared including the imaging of drug delivery to skin [57], and detection of tumors in a mouse brain model, the latter by providing rapid intraoperative assessment of brain tissue [58].
Spatially Offset Raman Spectroscopy (SORS)
Since 2005, several laboratories [59–61] have been developing a method for noninvasive in vivo Raman measurements with specially designed fiber optic probes placed on the skin of a subject. The method is based on the fact that most materials are neither completely transparent to light nor completely block it, but that they tend to scatter it. A SORS procedure consists of at least two Raman measurements; one at the surface and one at an offset position of typically a few millimeters away. The two acquired spectra can then be subtracted using a scaled subtraction to result in two spectra representing the subsurface and surface ones. In the case of biological tissues, when the distance between the illumination and collection regions is increased, surface signals from the soft tissue become less prominent, and the collected Raman spectra contain a greater contribution from subsurface components [59, 62–68].
Parameters That are Commonly Assessed and Their Importance
Mineral to Matrix Ratio
In IR and Raman spectroscopy the area under a band is directly proportional to the concentration of the chemical species that gives rise to the specific band. As such, the ratio of the integrated areas of the v1,v3 PO4 (stretching and bending vibrations) to amide I (mainly from the peptide bond C=O stretching vibration with minor contributions from the out of phase CN stretching vibration, the CCN deformation and the NH in-plane bend) or II (out of phase combination of the NH in-plane bend and the CC and NC stretching vibration with smaller contributions from the CO in-plane bend and the CC and NC stretching vibrations) bands in FTIR, and either v1 or v2 PO4 to either amide I or amide III band in Raman, provides what is commonly reported as the mineral/matrix ratio. This is a form of bone density that, unlike other measures such as BMD by DEXA or TMC or BMD or DMB by μCT, directly measures and accounts for the amount of the organic matrix in the volume analyzed. Accounting for the organic matrix content in such considerations is important when assessing bone strength as the mechanical properties of bone tissue depend on both the mineral and the matrix (primarily type I collagen fibrils) constituents [69, 70]. It has been shown that bone exhibits a region of elastic (recoverable) deformation, followed by a region of plastic (permanent) deformation. Based on tensile testing of progressively decalcified bovine bone tissue, Burstein and Reilly postulated that the mineral provides mainly compressive, whereas the collagen, tensile strength [69]. The mineral/matrix ratio correlates with ash-weight measurements in healthy bone [71] is directly proportional to bending stiffness and failure moment, and is a superior predictor of bone-bending stiffness compared with BMD alone [72]. Mineral/matrix ratio has been shown to (1) increase with increasing tissue age in healthy premenopausal subjects [28], (2) increase immediately after menopause (but without osteoporosis) because of a reduction in the amount of organic matrix [28] compared with premenopausal healthy subjects, (3) decrease in osteoporosis [73, 74] compared with premenopausal healthy controls, and (4) be modulated by the various osteoporosis treatments [75–84].
Relative Proteoglycan Content
In bone, proteoglycans play multiple roles involving the modulation of both organic matrix mineralization and remodeling rates. Moreover, different proteoglycans are present at distinct microanatomic locations fulfilling different roles. In in vitro solution experiments, the rate of hydroxyapatite growth is regulated by the concentration of proteoglycans [85]. In osteoblastic cell culture experiments the small leucine rich (SLRP) proteoglycan decorin, modulates matrix mineralization [86]. In animal models, deficiency of biglycan, another SLRP, increases osteoblast activity and affects osteoclasts as well, resulting in an osteoporosis-like phenotype [87–89]. Interestingly, it is not only their presence or lack thereof that influences biomineralization, but also subtle posttranslational modifications in size, sulfation, and/or charge density, which are crucial for the specific role they fulfill [90, 91]. Additionally, some of these modifications are subject- and tissue age dependent [92]. Proteoglycans have also been described in perilacunar matrix around the osteocyte lacunae, and around the canaliculi [93] in compact lamellar rat and human bone, suggesting that a plausible role of these (and in particular perlecan/Hspg2 (PLN)) is to prevent mineralization and, thus, to ensure uninhibited interstitial fluid movement [94]. Proteoglycans have also been implicated in osteoclastogenesis and remodeling regulation [87, 90, 95].
A characteristic of hyaline cartilage is its high content of proteoglycan aggregates resulting in the turgid nature of the tissue, and affording the osmotic properties needed to absorb compressive loads in articular cartilage. Like bone, cartilage contains small leucine-rich repeat proteoglycans (decorin, biglycan, fibromodulin, and lumican), which contribute to the maintenance of the tissue integrity and modulate its metabolism [96].
While both FTIR and Raman spectroscopic analyses are capable of describing proteoglycan content in cartilage [22, 97–100], only Raman (spectral band ~ 1375 cm-1; CH3 symmetric deformation of glycosaminoglycan groups) can do so in mineralized tissues [18, 28, 76–79, 101] as the marker band for proteoglycans in FTIR ~ 1060 cm-1 (indicative of sugar rings) is overlapped by the v3PO4 band. To date, Raman microspectroscopic analysis cannot discriminate between different proteoglycan species, but it should be kept in mind that the Raman spectral signature of proteoglycans is due to the glycosaminoglycan (GAG) chains [102, 103], and in bone, chondroitin 4-sulfate constitutes ~ 90 % of the total GAG content and is found predominantly in biglycan and decorin [104].
Relative Lipid Content
In the literature, lipids have been reported as nucleators of collagen fiber mineralization, with a layer of lipids present just behind the first mineral deposited [105, 106]. Moreover, oxidized lipids are a substratum involved in AGEs (advanced glycation endproducts) accumulation [107]. Since increased accumulation of AGEs is associated with fracture risk [108], their distribution is important to consider. In Raman spectroscopic analysis, spectral bands ~ 1060 cm-1 (mainly phospholipids), ~1079 cm-1, ~1300 cm-1, ~1439 cm-1, and ~1745 cm-1 are characteristic of tissue lipids [109, 110]. Of particular utility for bone, is the band at ~ 1300 cm-1 (arising from methylene twisting vibrations) as it is not overlapped by collagen bands [109]. The ratio of the integrated area of this band normalized to the integrated area of the amide III band may be used to describe the relative lipid content in mineralized tissues [111].
Mineral Maturity/Crystallinity
Vibrational spectroscopic analyses, either FTIR or Raman [23, 24, 112], provide information on the chemical makeup of the poorly crystalline apatitic crystals in bone ie, the presence of impurities, and, based on comparison to X-ray line broadening analysis, on their shape and size [113, 114]. Normal bone crystallites exhibit a range of sizes, and deviations from this range have been encountered in cases of fragile bone [20, 73, 115]. In FTIR, the most commonly applied method of deriving parameters describing this bone quality metric involves resolution and quantification of the underlying peaks in the v1,v3 PO4 band by second derivative spectroscopic and curve-fitting analysis [116, 117], while in Raman spectroscopic analysis this metric is estimated from either the full width at half height of the v1 PO4 band [112, 114] or the wavelength at maxima of the same band [114]. Its contribution to bone strength may be inferred by the fact that osteoporotic bone consistently exhibits crystallites of higher maturity/crystallinity compared with healthy bone [73, 74, 116, 118–120].
Collagen Cross-Links
A distinct feature of type I collagen in mineralized tissues is its cross-linking chemistry and molecular packing structure [121], which provides the fibrillar matrices with mechanical properties such as tensile strength and viscoelasticity. The importance of collagen intermolecular cross-links to the mechanical performance of bone is very apparent in the pyridoxine deficient chick [40], as well as in lathyrism [29, 41]. To date, vibrational spectroscopic analysis is able to describe the spatial distribution of two types of the collagen cross-links, namely pyridinoline and divalent cross-links [111, 112, 122, 123]. In both FTIR and Raman analyses, this is achieved through the resolution and quantification of the amide I (amide I and II in case of FTIR) underlying peaks through second derivative spectroscopy and curve-fitting routines [123]. Second derivative spectroscopy is a popular, mathematically objective (as no initial assumptions are required as to the number and full width at half height of the anticipated underlying peaks) spectral analysis technique that allows enhanced identification of small and overlapping absorption peaks that are not resolved in the original spectrum, albeit at a loss in signal-to-noise ratio [124–126]. These peak heights and positions provide insight into the environment of the vibrating species.
The collagen cross-link parameter may well be the most important contributor to bone quality and thus, strength and/or toughness. In cases where bone resistance to fracture cannot be explained by either BMD or architecture, or predicted by finite element analysis, the ratio of pyridinoline to divalent collagen cross-links correlates with fracture incidence [16–19, 80, 127]. To further emphasize their importance in the determination of bone strength, experiments in an animal model showed that alterations in this ratio, even when confined to microanatomic locations, are sufficient to influence the mechanical properties of the whole bone organ, even in the absence of any alteration in mineral quantity and quality [29].
Other Parameters
There are a few other parameters evaluated by one or both of the vibrational spectroscopic techniques that have been correlated with fracture risk and therapies, and a few that remain to be correlated. The peak width at half-height of the pixel distribution, often referred to as “heterogeneity” [128] has been reported in FTIR imaging, X-ray diffraction, and BMDD (bone mineral density distribution) measurements, and shown to decrease with bisphosphonate treatment [82, 83, 129] and in bones from patients with fractures compared with nonfractured controls [130].
The loss of acid phosphate substitution into the HA lattice, which can be measured with synchrotron radiation [131] or by FTIR imaging [117], is inversely associated with an increase in crystallinity and hence, as indicated above, with a matrix more likely to fail under load.
Proof of Principle: Idiopathic Osteoporosis
The utility/importance of the bone quality parameters determined by vibrational spectroscopic techniques is readily seen in the case of idiopathic osteoporosis (IOP), an uncommon disorder in premenopausal women, in which fragility fractures and/or low BMD occur in otherwise healthy women with normal gonadal function [18]. A large number of iliac crest biopsies were obtained from women diagnosed with IOP and compared with sex- and age-matched control by several techniques including histology/histomorphometry, micro-CT, central QCT, BMD by DXA, p-qCT, qBEI, and vibrational spectroscopic techniques [18, 132–138]. Interestingly, the IOP cohort included women with abnormally low BMD and no fragility fractures, as well as women with normal BMD and fragility fractures. Although all of the techniques were able to discriminate between healthy controls and IOP subjects, only vibrational spectroscopic techniques, and in particular the pyridinoline/divalent collagen cross-link ratio, were able to discriminate IOP subjects based on whether they had suffered fragility fractures or not [18], verifying the results of previous preliminary studies involving smaller numbers of IOP patients [16, 19]. Moreover, these results [16, 18, 19] were reproduced independently in a different cohort of human iliac crest biopsies [17].
Examples
Figures 1 and 2 are examples of FTIR and Raman analysis of bone tissue.

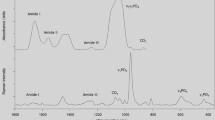
FTIR images (both x and y axes represent distance in microns) of an osteon adjacent to the edge of the section in a 6-year-old baboon. The color images all come from the same specimen. A, Optical image of a typical osteon surrounding a Haversian canal (*). In the first color image the center of the Haversian canal is shown by an *. B‒F, FTIRI images. B, Mineral to matrix ratio increases going from the newly forming osteoid surrounding the canal to the interstitial bone. C, Crystallinity also increases as the tissue age increases going from the center of the canal outwards. D, Acid phosphate substitution is greatest in the newest mineral and decreases with increasing age. E, Carbonate to mineral ratio decreases as the tissue ages going from the center of the canal to the edge. F, Collagen cross-link ratio increases as the tissue ages. Positive values in the center of the Haversian canal are due to the division of a small number by 0.

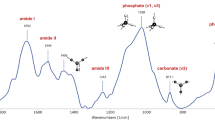
Raman imaging analysis of an osteon in the cortical bone of an iliac crest biopsy from a 43-year-old healthy female subject. A, Optical image obtained with the × 20 objective and the measured osteon shown in the shaded area; B, calculated ratio v2PO4/amide III (mineral/matrix), PGs/amide III (relative proteoglycan content), lipids/amide III (relative lipids content), and PYR/amide I (relative pyridinoline collagen cross-link content) from the osteon region (×20 objective, 35 × 35 Raman spectra, 165 × 190 μm, step size 5 μm).
Potential Pitfalls
While both FTIR and Raman analysis are very informative, they are associated with potential pitfalls that the user has to be aware of.
Because of the inherent polarization of its laser source, some of the Raman bands are not only dependent on the functional group present and its content in the tissue analyzed, but also on tissue organization/orientation. For example, the most commonly reported mineral/matrix ratio is based on the v1PO4 and amide I bands, yet it has been shown that this ratio is greatly dependent on tissue organization, unlike the ratio calculated based on the v2PO4 and amide III bands [139].
Both in FTIR and Raman spectroscopy, the bands are composite ones, consisting of underlying peaks, each specific to a unique chemical environment. As a result, useful information may be obtained through resolution of these bands into their underlying peaks, and quantification through deconvolution routines, or as noted above, second derivative spectroscopy. Currently, there are two general methods to achieve that (1) Empirical (arbitrarily chosen number and position of the underlying peaks). Although it is the fastest method as one does not have to account for spectral contribution from embedding material (such as PMMA) and water vapor, or the time consuming selection of underlying peaks based on more mathematically and spectroscopically robust methods, it only offers a mathematical solution that often has no real biological value/meaning. For example, analysis of the v1,v3 PO4 FTIR band by either empirical approach or based on more robust methodology results in significantly different number of underlying peaks, position, and interpretation of results even if performed by the same investigator [140, 141]. A recent publication based on spectroscopically robust methodology highlighted the paradox conclusions arrived at when underlying peaks are chosen arbitrarily [117]. Similarly, conflicting and biologically irrelevant conclusions may be arrived at when the amide bands are resolved to their underlying peaks empirically and arbitrarily. For example, the same investigators in one publication propose spectroscopic parameters describing collagen “maturity,” intimately related to extent of mineralization [142], while in a concurrent one analyzing human bone tissue from osteoporosis patients on long-term alendronate therapy, they report that their collagen “maturity” index changes as a function of therapy even though they compared areas of similar mineralization extent [143]; (2) Spectroscopically robust methods include deconvolution (though minimal initial parameter input by operator is required) and second derivative spectroscopy (mathematically objective as no initial parameter input is required by the operator).
Clinical Use
Although being able to offer detailed information on bone quality otherwise unattainable, FTIR and Raman techniques are not particularly well-suited to date for routine clinical use as they require excised bone tissue, something that is rather invasive and not too common in everyday clinical practice. Thus, we would suggest that these techniques be considered when pathophysiologic mechanisms need to be established, when the clinically used indices do not account for fracture incidence (eg, idiopathic osteoporosis), or when the effect of various therapies on bone material properties is investigated (especially during clinical trials). On the other hand, we follow with great interest the development of SORS Raman spectroscopic techniques [59, 61, 65–68, 144], as they hold promise for eventual everyday clinical use.
Conclusions
It is widely accepted that bone quality is an important contributor in the determination of bone strength. Infrared and Raman vibrational spectroscopic techniques are particularly well-suited methods to measure bone quality as they provide simultaneous information on the quantity and quality of both the mineral and organic matrix components, simultaneously. The outcomes that these two techniques offer are unattainable by other analytical techniques, and provide unique insights into the pathophysiology of healthy and brittle bone. A growing body of evidence suggests that these outcomes are intimately related with actual rather than predicted bone strength, thus, affording for a better, more precise, description of the contributors to bone strength, and allowing for more focused choices in the case of disease.
References
Boyce TM, Bloebaum RD. Cortical aging differences and fracture implications for the human femoral neck. Bone. 1993;14:769–78.
Marshall D, Johnell O, Wedel H. Meta-analysis of how well measures of bone mineral density predict occurrence of osteoporotic fractures. BMJ. 1996;312:1254–9.
Cummings SR. Are patients with hip fractures more osteoporotic? Review of the evidence. Am J Med. 1985;78:487–94.
McCreade RB, Goldstein AS. Biomechanics of fracture: is bone mineral density sufficient to assess risk? J Bone Miner Res. 2000;15:2305–8.
Hui S, Slemenda CW, Johnston CC. Age and bone mass as predictors of fracture in a prospective study. J Clin Invest. 1988;81:1804–9.
Siris ES, Brenneman SK, Miller PD, Barrett-Connor E, Chen YT, Sherwood LM, et al. Predictive value of low BMD for 1-year fracture outcomes is similar for postmenopausal women ages 50-64 and 65 and older: results from the National Osteoporosis Risk Assessment (NORA). J Bone Miner Res. 2004;19:1215–20.
Jepsen KJ, Schaffler MB. Bone mass does not adequately predict variations in bone fragility: a genetic approach. Trans Orthop Res Soc 47th Annual Meeting. 2001;114.
Parfitt AM. Bone remodeling and bone loss: understanding the pathophysiology of osteoporosis. Clin Obstet Gynecol. 1987;30:789–811.
Mosekilde L, Mosekilde L, Danielsen CC. Biomechanical competence of vertebral trabecular bone in relation to ash density and age in normal individuals. Bone. 1987;8(2):79–85.
McCabe F, Zhou LJ, Steele CR, Marcus R. Noninvasive assessment of ulnar bending stiffness in women. J Bone Miner Res. 1991;6:53–9.
Kanis JA, Melton LJI, Christiansen C, Johnston CJ, Haltaev N. Perspective: the diagnosis of osteoporosis. J Bone Miner Res. 1994;9:1137–42.
Kann P, Graeben S, Beyer J. Age-dependence of bone material quality shown by the measurement of frequency of resonance in the ulna. Calcif Tissue Int. 1994;54:96–100.
Schnitzler CM. Bone quality: a determinant for certain risk factors for bone fragility. Calcif Tissue Int. 1993;53:S27–31.
Ott SM. When bone mass fails to predict bone failure. Calcif Tissue Int. 1993;53(Suppl):S7–13.
Cummings SR, Black DM, Nevitt MC, Browner WS, Cauley JA, Genant HK, et al. Appendicular bone density and age predict hip fracture in women: the study of osteoporotic fractures research group. JAMA. 1990;263:665–8.
Gourion-Arsiquaud S, Faibish D, Myers E, Spevak L, Compston J, Hodsman A, et al. Use of FTIR spectroscopic imaging to identify parameters associated with fragility fracture. J Bone Miner Res. 2009;24:1565–71.
Malluche HH, Porter DS, Mawad H, Monier-Faugere MC, Pienkowski D. Low-energy fractures without low T-scores characteristic of osteoporosis: a possible bone matrix disorder. J Bone Joint Surg Am. 2013;95:e1391–6.
Misof BM, Gamsjaeger S, Cohen A, Hofstetter B, Roschger P, Stein E, et al. Bone material properties in premenopausal women with idiopathic osteoporosis. J Bone Miner Res. 2012;27:2551–61.
Paschalis EP, Shane E, Lyritis G, Skarantavos G, Mendelsohn R, Boskey AL. Bone fragility and collagen cross-links. J Bone Miner Res. 2004;19:2000–4.
Fratzl P, Gupta HS, Paschalis EP, Roschger P. Structure and mechanical quality of the collagen-mineral composite in bone. J Mater Chem. 2004;14:2115–23.
Einhorn TA. The bone organ system: form and function. In: Marcus R, Feldman D, Kelsey J, editors. Osteoporosis. New York: Academic Press Inc; 1996.
Boskey A, Pleshko Camacho N. FT-IR imaging of native and tissue-engineered bone and cartilage. Biomaterials. 2007;28:2465–78.
Paschalis EP. Fourier transform infrared analysis and bone. Osteoporos Int. 2009;20:1043–7.
Paschalis EP. Fourier transform infrared imaging of bone. Methods Mol Biol. 2012;816:517–25.
Roschger P, Paschalis EP, Fratzl P, Klaushofer K. Bone mineralization density distribution in health and disease. Bone. 2008;42:456–66.
Burket JC, Brooks DJ, MacLeay JM, Baker SP, Boskey AL, van der Meulen MC. Variations in nanomechanical properties and tissue composition within trabeculae from an ovine model of osteoporosis and treatment. Bone. 2013;52:326–36.
Donnelly E, Boskey AL, Baker SP, van der Meulen MC. Effects of tissue age on bone tissue material composition and nanomechanical properties in the rat cortex. J Biomed Mater Res A. 2010;92:1048–56.
Gamsjaeger S, Brozek W, Recker R, Klaushofer K, Paschalis EP. Transmenopausal changes in trabecular bone quality. J Bone Miner Res. 2013.
Paschalis EP, Tatakis DN, Robins S, Fratzl P, Manjubala I, Zoehrer R, et al. Lathyrism-induced alterations in collagen cross-links influence the mechanical properties of bone material without affecting the mineral. Bone. 2011;49:1232–41.
Kuroda T, Tanaka S, Saito M, Shiraki Y, Shiraki M. Plasma level of homocysteine associated with severe vertebral fracture in postmenopausal women. Calcif Tissue Int. 2013;93:269–75.
Yang J, Hu X, Zhang Q, Cao H, Wang J, Liu B. Homocysteine level and risk of fracture: a meta-analysis and systematic review. Bone. 2012;51:376–82.
El Maghraoui A, Ghozlani I, Mounach A, Rezqi A, Oumghar K, Achemlal L, et al. Homocysteine, folate, and vitamin B12 levels and vertebral fracture risk in postmenopausal women. J Clin Densitom. 2012;15:328–33.
Leboff MS, Narweker R, LaCroix A, Wu L, Jackson R, Lee J, et al. Homocysteine levels and risk of hip fracture in postmenopausal women. J Clin Endocrinol Metab. 2009;94:1207–13.
McLean RR, Jacques PF, Selhub J, Tucker KL, Samelson EJ, Broe KE, et al. Homocysteine as a predictive factor for hip fracture in older persons. N Engl J Med. 2004;350:2042–9.
van Meurs JB, Dhonukshe-Rutten RA, Pluijm SM, van der Klift M, de Jonge R, Lindemans J, et al. Homocysteine levels and the risk of osteoporotic fracture. N Engl J Med. 2004;350:2033–41.
Jager I, Fratzl P. Mineralized collagen fibrils: a mechanical model with a staggered arrangement of mineral particles. Biophys J. 2000;79:1737–46.
Gao H, Ji B, Jager IL, Arzt E, Fratzl P. Materials become insensitive to flaws at nanoscale: lessons from nature. Proc Natl Acad Sci U S A. 2003;100:5597–600.
Bailey AJ, Wotton SF, Sims TJ, Thompson PW. Post-translational modifications in the collagen of human osteoporotic femoral head. Biochem Biophys Res Comm. 1992;185:801–5.
Bailey AJ, Wotton SF, Sims TJ, Thompson PW. Biochemical changes in the collagen of human osteoporotic bone matrix. Connect Tissue Res. 1993;29:119–32.
Masse PG, Rimnac CM, Yamauchi M, Coburn PS, Rucker BR, Howell SD, et al. Pyridoxine deficiency affects biomechanical properties of chick tibial bone. Bone. 1996;18:567–74.
Oxlund H, Barckman M, Ortoft G, Andreassen TT. Reduced concentrations of collagen cross-links are associated with reduced strength of bone. Bone. 1995;17(4 Suppl):365S–71S.
Socrates G. Infrared and Raman characteristic group frequencies: tables and charts. Chichester: Wiley; 2001.
Barer R, Cole ARH, Thompson HW. Infrared spectroscopy with the reflecting microscope in physics, chemistry, and biology. Nature. 1949;163:198–201.
Delhaye M, Dhamelincourt. Laser Raman Microprobe and Microscope Proceeding Abstracts, Fourth International Conference on Raman Spectroscopy, Brunswick, Maine. 1974.
Rosasco GJ, Etz E. Investigation of the Raman Spectra of Individual Micron Sized Particles Proceeding Abstracts, Fourth International Conference on Raman Spectroscopy, Brunswick, Maine. 1974.
Raman, Infrared, and Near-Infrared Chemical Imaging. In: Sasic S, Ozaki Y editors. 2010.
Emerging Biomedical and Pharmaceutical Applications of Raman Spectroscopy. In: Morris M, Matousek P, editors. Heidelberg: Springer; 2010.
Evans CL, Xie XS. Coherent anti-stokes Raman scattering microscopy: chemical imaging for biology and medicine. Ann Rev Analytic Chem. 2008;1:883–909.
Levin IW, Bhargava R. Fourier transform infrared vibrational spectroscopic imaging: integrating microscopy and molecular recognition. Annu Rev Phys Chem. 2005;56:429–74.
Lothar Opilik L, Thomas Schmid T, Zenobi R. Modern Raman imaging: vibrational spectroscopy on the micrometer and nanometer scales. Ann Rev Analytic Chemi. 2013;6:379–98.
Huth F, Govyadinov A, Amarie SW, Nuansing W, Keilmann F, Hillenbrand R. Working principle of nano-FTIR -Nano-FTIR absorption spectroscopy of molecular fingerprints at 20 nm spatial resolution. Nano Lett. 2012;12:3973–8.
Marcott C, Lo M, Kjoller K, Domanov K, Balooch G, Luengo GS. Nanoscale infrared (IR) spectroscopy and imaging of structural lipids in human stratum corneum using an atomic force microscope to directly detect absorbed light from a tunable IR laser source. Exp Dermatol. 2013;22:419–21.
Marcott C, Lo M, Hu Q, Kjoller K, Boskey A, Noda I. Using 2D correlation analysis to enhance spectral information available from highly spatially resolved AFM-IR spectra. J Molec Struct. 2014.
Pettinger B. Single-molecule surface- and tip-enhanced Raman spectroscopy. Mol Phys. 2010;108:2039–59.
Sonntag MD, Garibay LK, Roberts JM, Seideman T, Scheidt KA, Jensen L, et al. Single-molecule tip-enhanced Raman spectroscopy. J Phys Chem. 2012;116:478–83.
Saar BG, Freudiger CW, Stanley CM, Holtom GR, Xie XS. Video-rate molecular imaging in vivo with stimulated Raman scattering. Science. 2010;330:1368–70.
Saar BG, Contreras-Rojas LR, Xie XS, Guy RH. Imaging drug delivery to skin with stimulated Raman scattering microscopy. Mol Pharm. 2011;8:969–75.
Ji M, Orringer DA, Freudiger CW, Ramkissoon S, Liu X, Lau D, et al. Rapid, label-free detection of brain tumors with stimulated Raman scattering microscopy. Sci Transl Med. 2013;5:201ra119.
Draper ER, Morris MD, Camacho NP, Matousek P, Towrie M, Parker AW. Novel assessment of bone using time-resolved transcutaneous Raman spectroscopy. J Bone Miner Res. 2005;20:1968–72.
Matousek P. Inverse spatially offset Raman spectroscopy for deep noninvasive probing of turbid media. Appl Spectrosc. 2006;60:1341–7.
Schulmerich MV, Dooley KA, Morris MD, Vanasse TM, Goldstein SA. Transcutaneous fiber optic Raman spectroscopy of bone using annular illumination and a circular array of collection fibers. J Biomed Opt. 2006;11:060502.
Ding H, Nyman JS, Sterling JA, Perrien DS, Mahadevan-Jansen A, Bi X. Development of Raman spectral markers to assess metastatic bone in breast cancer. J Biomed Opt. 2014;19:111606.
Sharma B, Ma K, Glucksberg MR, Van Duyne RP. Seeing through bone with surface-enhanced spatially offset Raman spectroscopy. J Am Chem Soc. 2013;135:17290–3.
Demers JL, Davis SC, Pogue BW, Morris MD. Multichannel diffuse optical Raman tomography for bone characterization in vivo: a phantom study. Biomed Opt Express. 2012;3:2299–305.
Okagbare PI, Begun D, Tecklenburg M, Awonusi A, Goldstein SA, Morris MD. Noninvasive Raman spectroscopy of rat tibiae: approach to in vivo assessment of bone quality. J Biomed Opt. 2012;17:90502–1.
Okagbare PI, Esmonde-White FW, Goldstein SA, Morris MD. Development of non-invasive Raman spectroscopy for in vivo evaluation of bone graft osseointegration in a rat model. Analyst. 2010;135:3142–6.
Schulmerich MV, Cole JH, Kreider JM, Esmonde-White F, Dooley KA, Goldstein SA, et al. Transcutaneous Raman spectroscopy of murine bone in vivo. Appl Spectrosc. 2009;63:286–95.
Schulmerich MV, Cole JH, Dooley KA, Morris MD, Kreider JM, Goldstein SA. Optical clearing in transcutaneous Raman spectroscopy of murine cortical bone tissue. J Biomed Opt. 2008;13:021108.
Reilly DT, Burstein AH. The mechanical properties of cortical bone. JBJS. 1974;56A:1001–22.
Martin RB, Ishida J. The relative effects of collagen fiber orientation, porosity, density, and mineralization on bone strength. J Biomech. 1989;22:419–26.
Boskey AL, Pleshko N, Doty SB, Mendelsohn R. Applications of Fourier Transform Infrared (FT-IR) Microscopy to the study of mineralization in bone and cartilage. Cells Mater. 1992;2:209–20.
Donnelly E, Chen DX, Boskey AL, Baker SP, van der Meulen MC. Contribution of mineral to bone structural behavior and tissue mechanical properties. Calcif Tissue Int. 2010;87:450–60.
Boskey AL, DiCarlo E, Paschalis E, West P, Mendelsohn R. Comparison of mineral quality and quantity in iliac crest biopsies from high- and low-turnover osteoporosis: an FT-IR microspectroscopic investigation. Osteoporos Int. 2005;16:2031–8.
Paschalis EP, Betts F, DiCarlo E, Mendelsohn R, Boskey AL. FTIR microspectroscopic analysis of human iliac crest biopsies from untreated osteoporotic bone. Calcif Tissue Int. 1997;61:487–92.
Durchschlag E, Paschalis EP, Zoehrer R, Roschger P, Fratzl P, Recker R, et al. Bone material properties in trabecular bone from human iliac crest biopsies after 3- and 5-year treatment with risedronate. J Bone Miner Res. 2006;21:1581–90.
Gamsjaeger S, Buchinger B, Zoehrer R, Phipps R, Klaushofer K, Paschalis EP. Effects of one year daily teriparatide treatment on trabecular bone material properties in postmenopausal osteoporotic women previously treated with alendronate or risedronate. Bone. 2011;49:1160–5.
Gamsjaeger S, Buchinger B, Zwettler E, Recker R, Black D, Gasser JA, et al. Bone material properties in actively bone-forming trabeculae in postmenopausal women with osteoporosis after three years of treatment with once-yearly zoledronic acid. J Bone Miner Res. 2011;26:12–8.
Gamsjaeger S, Hofstetter B, Zwettler E, Recker R, Gasser JA, Eriksen EF, et al. Effects of 3 years treatment with once-yearly zoledronic acid on the kinetics of bone matrix maturation in osteoporotic patients. Osteoporos Int. 2013;24:339–47.
Hofstetter B, Gamsjaeger S, Phipps RJ, Recker RR, Ebetino FH, Klaushofer K, et al. Effects of alendronate and risedronate on bone material properties in actively forming trabecular bone surfaces. J Bone Miner Res. 2012;27:995–1003.
Paschalis EP, Glass EV, Donley DW, Eriksen EF. Bone mineral and collagen quality in iliac crest biopsies of patients given teriparatide: new results from the fracture prevention trial. J Clin Endocrinol Metab. 2005;90:4644–9.
Roschger P, Manjubala I, Zoeger N, Meirer F, Simon R, Li C, et al. Bone material quality in transiliac bone biopsies of postmenopausal osteoporotic women after 3 years of strontium ranelate treatment. J Bone Miner Res. 2010;25:891–900.
Boskey AL, Spevak L, Weinstein RS. Spectroscopic markers of bone quality in alendronate-treated postmenopausal women. Osteoporos Int. 2009;20:793–800.
Gourion-Arsiquaud S, Allen MR, Burr DB, Vashishth D, Tang SY, Boskey AL. Bisphosphonate treatment modifies canine bone mineral and matrix properties and their heterogeneity. Bone. 2010;46:666–72.
Paschalis EP, Boskey AL, Kassem M, Eriksen EF. Effect of hormone replacement therapy on bone quality in early postmenopausal women. J Bone Miner Res. 2003;18:955–9.
Boskey AL, Spevak L, Doty SB, Rosenberg L. Effects of bone CS-proteoglycans, DS-decorin, and DS-biglycan on hydroxyapatite formation in a gelatin gel. Calcif Tissue Int. 1997;61:298–305.
Mochida Y, Duarte WR, Tanzawa H, Paschalis EP, Yamauchi M. Decorin modulates matrix mineralization in vitro. Biochem Biophys Res Commun. 2003;305:6–9.
Nielsen KL, Allen MR, Bloomfield SA, Andersen TL, Chen XD, Poulsen HS, et al. Biglycan deficiency interferes with ovariectomy-induced bone loss. J Bone Miner Res. 2003;18:2152–8.
Bi Y, Nielsen KL, Kilts TM, Yoon A, Wimer HF, Greenfield EM, et al. Biglycan deficiency increases osteoclast differentiation and activity due to defective osteoblasts. Bone. 2006;38:778–86.
Xu T, Bianco P, Fisher LW, Longenecker G, Smith E, Goldstein S, et al. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat Genet. 1998;20:78–82.
Gualeni B, de Vernejoul MC, Marty-Morieux C, De Leonardis F, Franchi M, Monti L, et al. Alteration of proteoglycan sulfation affects bone growth and remodeling. Bone. 2013;54:83–91.
Boskey AL, Maresca M, Armstrong AL, Ehrlich MG. Treatment of proteoglycan aggregates with physeal enzymes reduces their ability to inhibit hydroxyapatite proliferation in a gelatin gel. J Orthop Res. 1992;10:313–9.
Grzesik WJ, Frazier CR, Shapiro JR, Sponseller PD, Robey PG, Fedarko NS. Age-related changes in human bone proteoglycan structure. Impact of osteogenesis imperfecta. J Biol Chem. 2002;277:43638–47.
Sauren YM, Mieremet RH, Groot CG, Scherft JP. An electron microscopic study on the presence of proteoglycans in the mineralized matrix of rat and human compact lamellar bone. Anat Rec. 1992;232:36–44.
Thompson WR, Modla S, Grindel BJ, Czymmek KJ, Kirn-Safran CB, Wang L, et al. Perlecan/Hspg2 deficiency alters the peri-cellular space of the lacunocanalicular system surrounding osteocytic processes in cortical bone. J Bone Miner Res. 2011;26:618–29.
Ohtsuki T, Hatake K, Suzu S, Saito K, Motoyoshi K, Miura Y. Immunohistochemical identification of proteoglycan form of macrophage colony-stimulating factor on bone surface. Calcif Tissue Int. 1995;57:213–7.
Roughley PJ. The structure and function of cartilage proteoglycans. Eur Cell Mater. 2006;12:92–101.
Rieppo L, Rieppo J, Jurvelin JS, Saarakkala S. Fourier transform infrared spectroscopic imaging and multivariate regression for prediction of proteoglycan content of articular cartilage. PLoS One. 2012;7:e32344.
Rieppo L, Saarakkala S, Narhi T, Holopainen J, Lammi M, Helminen HJ, et al. Quantitative analysis of spatial proteoglycan content in articular cartilage with Fourier transform infrared imaging spectroscopy: critical evaluation of analysis methods and specificity of the parameters. Microsc Res Tech. 2010;73:503–12.
Saadat E, Lan H, Majumdar S, Rempel DM, King KB. Long-term cyclical in vivo loading increases cartilage proteoglycan content in a spatially specific manner: an infrared microspectroscopic imaging and polarized light microscopy study. Arthritis Res Ther. 2006;8:R147.
Yin JH, Xia Y, Ramakrishnan N. Depth-dependent anisotropy of proteoglycan in articular cartilage by fourier transform infrared imaging. Vib Spectrosc. 2011;57:338–41.
Gamsjaeger S, Srivastava AK, Wergedal JE, Zwerina J, Klaushofer K, Paschalis EP, et al. Altered bone material properties in HLA-B27 rats include reduced mineral to matrix ratio and altered collagen cross-links. J Bone Miner Res. 2014.
Bansil R, Yannas IV, Stanley HE. Raman spectroscopy: a structural probe of glycosaminoglycans. Biochim Biophys Acta. 1978;541:535–42.
Ellis R, Green E, Winlove C. Structural analysis of glycosaminoglycans and proteoglycans by means of Raman microspectrometry. Connect Tissue Res. 2009;50:29–36.
Smith AJ, Singhrao SK, Newman GR, Waddington RJ, Embery G. A biochemical and immuno-electron microscopical analysis of chondroitin sulphate-rich proteoglycans in human alveolar bone. Histochem J. 1997;29:1–9.
Boskey AL, Reddi AH. Changes in lipids during matrix: induced endochondral bone formation. Calcif Tissue Int. 1983;35:549–54.
Goldberg M, Boskey AL. Lipids and biomineralizations. Prog Histochem Cytochem. 1996;31:1–187.
Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. 2005;15:16R–28R.
Tang SY, Zeenath U, Vashishth D. Effects of non-enzymatic glycation on cancellous bone fragility. Bone. 2007;40:1144–51.
Frushour BG, Koenig JL. Raman scattering of collagen, gelatin, and elastin. Biopolymers. 1975;14:379–91.
Penel G, Delfosse C, Descamps M, Leroy G. Composition of bone and apatitic biomaterials as revealed by intravital Raman microspectroscopy. Bone. 2005;36:893–901.
Gamsjaeger S, Brozek W, Recker R, Klaushofer K, Paschalis EP. Transmenopausal changes in trabecular bone quality. J Bone Miner Res. 2014;29:608–17.
Morris MD, Mandair GS. Raman assessment of bone quality. Clin Orthop Relat Res. 2011;469:2160–9.
Gadaleta SJ, Paschalis EP, Betts F, Mendelsohn R, Boskey AL. Fourier transform infrared spectroscopy of the solution-mediated conversion of amorphous calcium phosphate to hydroxyapatite: new correlations between X-ray diffraction and infrared data. Calcif Tissue Int. 1996;58:9–16.
Kazanci M, Fratzl P, Klaushofer K, Paschalis EP. Complementary information on in vitro conversion of amorphous (precursor) calcium phosphate to hydroxyapatite from Raman microspectroscopy and wide-angle X-ray scattering. Calcif Tissue Int. 2006;79:354–9.
Fratzl P, Roschger P, Eschberger J, Abendroth B, Klaushofer K. Abnormal bone mineralization after fluoride treatment in osteoporosis: a small-angle x-ray-scattering study. J Bone Miner Res. 1994;9:1541–9.
Paschalis EP, DiCarlo E, Betts F, Sherman P, Mendelsohn R, Boskey AL. FTIR microspectroscopic analysis of human osteonal bone. Calcif Tissue Int. 1996;59:480–7.
Spevak L, Flach CR, Hunter T, Mendelsohn R, Boskey A. Fourier transform infrared spectroscopic imaging parameters describing acid phosphate substitution in biologic hydroxyapatite. Calcif Tissue Int. 2013;92:418–28.
Paschalis EP, Betts F, DiCarlo E, Mendelsohn R, Boskey AL. FTIR microspectroscopic analysis of normal human cortical and trabecular bone. Calcif Tissue Int. 1997;61:480–6.
Huang RY, Miller LM, Carlson CS, Chance MR. In situ chemistry of osteoporosis revealed by synchrotron infrared microspectroscopy. Bone. 2003;33:514–21.
Miller LM, Tibrewala J, Carlson CS. Examination of bone chemical composition in osteoporosis using fluorescence-assisted synchrotron infrared microspectroscopy. Cell Mol Biol. 2000;46:1035–44.
Yamauchi M. Collagen: the major matrix molecule in mineralized tissues. In: Anderson JJB, Garner SC, editors. Calcium and phosphorus in health and disease. New York: CRC Press; 1996. p. 127–41.
Carden A, Rajachar RM, Morris MD, Kohn DH. Ultrastructural changes accompanying the mechanical deformation of bone tissue: a Raman imaging study. Calcif Tissue Int. 2003;72:166–75.
Paschalis EP, Verdelis K, Doty SB, Boskey AL, Mendelsohn R, Yamauchi M. Spectroscopic characterization of collagen cross-links in bone. J Bone Miner Res. 2001;16:1821–8.
Dong A, Huang P, Caughey WS. Protein secondary structures in water from second-derivative amide I infrared spectra. Biochemistry. 1990;29:3303–8.
Kohler A, Bertrand D, Martens H, Hannesson K, Kirschner C, Ofstad R. Multivariate image analysis of a set of FTIR microspectroscopy images of aged bovine muscle tissue combining image and design information. Anal Bioanal Chem. 2007;389:1143–53.
Susi H, Byler DM. Protein structure by Fourier transforms infrared spectroscopy: second derivative spectra. Biochem Biophys Res Commun. 1983;115:391–7.
Blank RD, Baldini TH, Kaufman M, Bailey S, Gupta R, Yershov Y, et al. Spectroscopically determined collagen Pyr/deH-DHLNL cross-link ratio and crystallinity indices differ markedly in recombinant congenic mice with divergent calculated bone tissue strength. Connect Tissue Res. 2003;44:134–42.
Boskey AL. Bone composition: relationship to bone fragility and antiosteoporotic drug effects. Bonekey Rep. 2013;2:447.
Donnelly E, Meredith DS, Nguyen JT, Gladnick BP, Rebolledo BJ, Shaffer AD, et al. Reduced cortical bone compositional heterogeneity with bisphosphonate treatment in postmenopausal women with intertrochanteric and subtrochanteric fractures. J Bone Miner Res. 2012;27:672–8.
Gourion-Arsiquaud S, Lukashova L, Power J, Loveridge N, Reeve J, Boskey AL. Fourier transform infrared imaging of femoral neck bone: reduced heterogeneity of mineral-to-matrix and carbonate-to-phosphate and more variable crystallinity in treatment-naive fracture cases compared with fracture-free controls. J Bone Miner Res. 2013;28:150–61.
Miller LM, Vairavamurthy V, Chance MR, Mendelsohn R, Paschalis EP, Betts F, et al. In situ analysis of mineral content and crystallinity in bone using infrared micro-spectroscopy of the nu(4) PO(4)(3-) vibration. Biochim Biophys Acta. 2001;1527:11–9.
Cohen A, Dempster DW, Recker RR, Stein EM, Lappe JM, Zhou H, et al. Abnormal bone microarchitecture and evidence of osteoblast dysfunction in premenopausal women with idiopathic osteoporosis. J Clin Endocrinol Metab. 2011;96:3095–105.
Cohen A, Dempster DW, Stein EM, Nickolas TL, Zhou H, McMahon DJ, et al. Increased marrow adiposity in premenopausal women with idiopathic osteoporosis. J Clin Endocrinol Metab. 2012;97:2782–91.
Cohen A, Lang TF, McMahon DJ, Liu XS, Guo XE, Zhang C, et al. Central QCT reveals lower volumetric BMD and stiffness in premenopausal women with idiopathic osteoporosis, regardless of fracture history. J Clin Endocrinol Metab. 2012;97:4244–52.
Cohen A, Liu XS, Stein EM, McMahon DJ, Rogers HF, Lemaster J, et al. Bone microarchitecture and stiffness in premenopausal women with idiopathic osteoporosis. J Clin Endocrinol Metab. 2009;94:4351–60.
Donovan MA, Dempster D, Zhou H, McMahon DJ, Fleischer J, Shane E. Low bone formation in premenopausal women with idiopathic osteoporosis. J Clin Endocrinol Metab. 2005;90:3331–6.
Liu XS, Cohen A, Shane E, Stein E, Rogers H, Kokolus SL, et al. Individual trabeculae segmentation (ITS)-based morphological analysis of high-resolution peripheral quantitative computed tomography images detects abnormal trabecular plate and rod microarchitecture in premenopausal women with idiopathic osteoporosis. J Bone Miner Res. 2010;25:1496–505.
Rubin MR, Schussheim DH, Kulak CA, Kurland ES, Rosen CJ, Bilezikian JP, et al. Idiopathic osteoporosis in premenopausal women. Osteoporos Int. 2005;16:526–33.
Gamsjaeger S, Masic A, Roschger P, Kazanci M, Dunlop JW, Klaushofer K, et al. Cortical bone composition and orientation as a function of animal and tissue age in mice by Raman spectroscopy. Bone. 2010;47:392–9.
Rey C, Shimizu M, Collins B, Glimcher MJ. Resolution-enhanced Fourier transform infrared spectroscopy study of the environment of phosphate ion in the early deposits of a solid phase of calcium phosphate in bone and enamel and their evolution with age: 2. Investigations in the nu3PO4 domain. Calcif Tissue Int. 1991;49:383–8.
Farlay D, Panczer G, Rey C, Delmas PD, Boivin G. Mineral maturity and crystallinity index are distinct characteristics of bone mineral. J Bone Miner Metab. 2010;28:433–45.
Farlay D, Duclos ME, Gineyts E, Bertholon C, Viguet-Carrin S, Nallala J, et al. The ratio 1660/1690 cm(-1) measured by infrared microspectroscopy is not specific of enzymatic collagen cross-links in bone tissue. PLoS One. 2011;6:e28736.
Bala Y, Depalle B, Farlay D, Douillard T, Meille S, Follet H, et al. Bone micromechanical properties are compromised during long-term alendronate therapy independently of mineralization. J Bone Miner Res. 2012;27:825–34.
Schulmerich MV, Dooley KA, Vanasse TM, Goldstein SA, Morris MD. Subsurface and transcutaneous Raman spectroscopy and mapping using concentric illumination rings and collection with a circular fiber-optic array. Appl Spectrosc. 2007;61:671–8.
Compliance with Ethics Guidelines
Conflict of Interest
S. Gamsjaeger, R. Mendelsohn, A. L. Boskey, S. Gourion-Arsiquaud, K. Klaushofer, and E. P. Paschalis all declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
All studies by the authors involving animal and/or human subjects were performed after approval by the appropriate institutional review boards. When required, written informed consent was obtained from all participants.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gamsjaeger, S., Mendelsohn, R., Boskey, A.L. et al. Vibrational Spectroscopic Imaging for the Evaluation of Matrix and Mineral Chemistry. Curr Osteoporos Rep 12, 454–464 (2014). https://doi.org/10.1007/s11914-014-0238-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-014-0238-8