
Abstract
Purpose of Review
Porphyrias constitute a group of rare metabolic disorders that result in a deficiency of the heme biosynthetic pathway and lead to the accumulation of metabolic intermediaries. Patients with porphyria can experience recurrent neurovisceral attacks which are characterized by neuropathic abdominal pain and acute gastrointestinal symptoms, including nausea, vomiting, and constipation. Depending on the type of porphyria, patients can present with cutaneous manifestations, such as severe skin photosensitivity, chronic hemolysis, or evidence of neurologic dysfunction, including alterations in consciousness, neurovascular involvement, seizures, transient sensor-motor symptoms, polyneuropathy, and behavioral abnormalities.
Recent Findings
More recently, cases of posterior reversible encephalopathy syndrome, cerebral vasoconstriction, and acute flaccid paralysis have also been described. While the exact pathogenic mechanisms linking the accumulation of abnormal heme biosynthetic intermediaries to neurologic manifestations have not been completely elucidated, it has been proposed that these manifestations are more common than previously thought and can result in permanent neurologic injury.
Summary
This article reviews the basic principles of heme synthesis as well as the pathogenic mechanism of disease, presentation, and treatment of acute hepatic porphyrias with emphasis on those with neurologic manifestations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Porphyrias encompass a group of inherited or acquired metabolic disorders in which there is an abnormal synthesis of heme. There are seven types of porphyrias caused by particular enzymatic deficiencies in the metabolism of protoporphyrins resulting in the accumulation of toxic metabolic intermediaries. In 2008, a new X-linked dominant subtype, X-Linked protoporphyria, resulting from a gain-of-function mutation was described [1]. Every cell in the body synthesizes heme which is used in respiratory and oxidation–reduction biochemical processes. However, heme biosynthesis is particularly active in erythropoietic cells where it is used for hemoglobin synthesis and hepatocytes where it is necessary for the synthesis of hemoproteins and cytochromes. Thus, based on the tissue affected, porphyrias can be categorized as hepatic or erythropoietic. X-Linked protoporphyria, congenital erythropoietic porphyria, and erythropoietic protoporphyria are erythropoietic subtypes that present with cutaneous manifestations. There are five hepatic porphyrias. The most common, porphyria cutanea tarda, can be acquired and is characterized by skin photosensitivity. The remaining hepatic porphyrias, namely acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP), and δ-aminolevulinic acid (ALA) dehydratase deficiency porphyria (ADP), have periods of clinical latency which are interrupted by life-threatening neurovisceral attacks (Table 1) [2, 3]. Classic neurologic manifestations include posterior reversible encephalopathy syndrome, neuropathy, seizures, transient sensory-motor deficits, cognitive decline, neurobehavioral abnormalities, and psychiatric disorders. In this article, the authors provide a review of the presentation, classification, pathogenic mechanisms of disease, and treatment of the acute hepatic porphyrias with emphasis on the neurologic manifestations.
Heme Biosynthetic Pathway
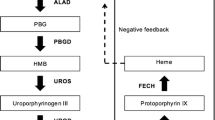
Heme is an iron-containing prosthetic group that is found in different proteins and enzymes, such as hemoglobin, myoglobin, catalase, nitric oxide synthase (NOS), and cytochromes. The most common type of heme, called heme b, combines with globin chains to form hemoglobin, which is required for the production of red blood cells. By binding oxygen reversibly via the positively charged Fe2+, heme allows its transport in red blood cells from the lungs to the rest of the body. From the structural standpoint, heme is a porphyrin ring complexed with Fe2+ and protoporphyrin IX. Heme is both absorbed in the intestine and synthesized endogenously. The first step in heme biosynthesis is under the control of the ALA synthase, a regulated mitochondrial enzyme that catalyzes the condensation of glycine and succinyl-CoA to form ALA. There are two forms of ALA synthase (ALAS); ALAS1 is ubiquitously expressed, but primarily in the liver, while ALA2 is erythroid-specific and largely found in the bone marrow. ALA is transported to the cytosol where it is metabolized to porphobilinogen (PBG) by the PBG synthase (aka ALA dehydratase). The PBG deaminase (aka hydroxymethylbilane synthase) transforms PBG into hydroxymethylbilane, which is subsequently metabolized to uroporphyrinogen III by the uroporphyrinogen III synthase and to coproporphyrinogen III by the uroporphyrinogen decarboxylase. Coproporphyrinogen III is transported back to the mitochondria, where it is sequentially oxidized by the coproporphyrinogen oxidase and the protoporphyrinogen oxidase to protoporphyrin IX. The synthesis of heme is completed by the ferrochelatase which inserts Fe2+ into the protoporphyrin ring [3].
The expression of most of the enzymes involved in heme synthesis is under the control of erythroid-specific regulatory DNA elements, which recruit transcription factors and coregulators. At low oxygen levels, the kidneys produce erythropoietin, increasing the bone marrow production of red blood cells and heme. In reticulocytes, heme stimulates the synthesis of globin chains necessary for the maturation of red blood cells. In addition, heme biosynthesis is controlled by the availability of iron. In the liver, heme is required for cytochrome P450. Thus, cytochrome P450 inducers stimulate the synthesis of heme. Since free heme is hepatotoxic, its production is tightly regulated. To this end, ALAS1 is subject to a negative feedback that involves free heme and its oxidized form hemin, which contains protoporphyrin IX and Fe3+ instead of Fe2+[4•].
Figure 1 depicts the heme biosynthetic pathway and the different types of defects described in porphyrias, and highlights those with cutaneous and neurologic manifestations. With the exception of porphyria cutanea tarda, which is typically acquired, and the X-Linked protoporphyria, which results in a gain-of-function mutation that leads to the accumulation of protoporphyrin despite normal ferrochelatase activity, all the porphyrias are autosomal disorders that result in a partial deficiency of enzymes involved in the biosynthesis of heme. As a group, porphyrias are characterized by genetic heterogeneity, variable expression, and incomplete penetrance, with more than a thousand mutations being described in genes that encode for enzymes involved in heme biosynthesis. Attempts to establish genotype–phenotype correlations have been made with little success to date. It should be noted that significant genetic variations exist based on ethnic background, which may result in different susceptibility to triggers and clinical presentations.

Heme biosynthetic pathway. The porphyrias in red are characterized by neurologic dysfunction as opposed to those in green which have cutaneous manifestations represented, most commonly, by skin photosensitivity. ALA synthase 1 is the rate-limiting enzyme in the production of heme in the liver. The activity of this enzyme is controlled via negative-feedback regulation by the intracellular heme pool. ALA, δ-aminolevulinic acid; Gly, glycine
Compounds that induce cytochrome P450 enzymes in the liver, including alcohol, phenytoin, phenobarbital, and endogenous steroids, upregulate ALAS1. Similarly, aromatic compounds, such as those contained in cigarette smoke, are also cytochrome inducers and upregulate ALAS. In comparison, starvation and infections induce heme oxygenase which decreases heme levels resulting in the activation of ALAS. Conversely, glucose is an ALAS inhibitor.
General Manifestations
A variety of environmental stressors can precipitate porphyric exacerbations, including drugs (Table 2), hormonal changes, fasting, alcohol, surgery, and infections. From the clinical standpoint, patients can be categorized into three main presentations: (1) exclusive cutaneous manifestations are commonly seen in erythropoietic porphyrias; (2) exclusive neurovisceral symptoms are seen in ADP and AIP; and (3) neurovisceral symptoms with or without cutaneous manifestations occur in HCP and VP [3]. The cutaneous symptoms in HCP and VP may be independent from the acute porphyric flares. Patients may present with blisters and skin sensitivity, particularly on the hands, with hyperpigmentation and scaring which represent the sequelae of repeated skin eruptions.
During acute attacks, patients may note discoloration of the urine due to the increased excretion of porphyrins, particularly after exposure to sunlight. Frequently encountered manifestations of acute neurovisceral attacks are neuropathic abdominal pain, which resembles an acute abdomen and can be associated with gastrointestinal symptoms such as nausea, vomiting, constipation, and paralytic ileus [2]. Thus, the diagnosis of porphyria is commonly delayed and preceded by often unnecessary gastrointestinal surgical procedures, including appendectomies or cholecystectomies. Due to the severe pain and/or dysautonomia, patients can present with findings suggestive of sympathetic overactivity such as tachycardia and hypertension [5]. Less commonly, patients can report urinary retention, tremors, and hyperhidrosis. Electrolyte abnormalities such as hyponatremia and hypomagnesemia, resulting from hypothalamic involvement or excessive loss in feces or urine, can also be seen during acute flares. Neuropsychiatric manifestations are common in porphyria; delirium and confusion are often self-limited and can occur in as many as 50% of the patients with acute attacks. Other psychiatric manifestations include anxiety, depression, and psychotic symptoms [6]. A subset of patients can also experience neurologic symptoms during these acute attacks including alterations in consciousness, seizures, axonal and/or autonomic neuropathy, and transient sensory-deficits. In small cohorts, posterior reversible encephalopathy syndrome (PRES) and reversible cerebral vasoconstriction syndrome (RCVS) have also been described [7, 8]. Given the rarity of porphyria, and the even less frequent neurologic manifestations, there are no large studies from which to draw clear conclusions on, but there exists an ever growing body of evidence linking acute hepatic porphyria and involvement of both the peripheral (PNS) and central nervous (CNS) systems [6]. The neurologic manifestations of porphyria are described in the following sections.
Neurologic Manifestations
Posterior Reversible Encephalopathy Syndrome and Vascular Complications
One of the most common documented neurologic manifestations of acute porphyria, cited in numerous case reports, is PRES. PRES is a clinical-radiographic diagnosis most often seen in the setting of elevated blood pressure or with the use of some medications. While the exact underlying mechanism is incompletely understood, the prevailing theory is that there is loss of autoregulation in the CNS vasculature leading to vasogenic edema, which can be seen on MRI. Typical symptoms of PRES include alterations in cognition, headache, seizures, and visual disturbances [9].
Many case reports and series, over the past few decades, document this in various types of porphyria during acute attacks including AIP, VP, and HCP. Many articles speculate that elevated ALA levels may cause endothelial dysfunction and thus inhibit autoregulation [8, 10,11,12]. PRES has been observed both in adult and pediatric cases and there are case reports that it can be recurring with further acute porphyric flares [8, 11,12,13].
While PRES itself appears to be well documented, there are also additional rarer cases that have exhibited other vascular manifestations including vasoconstriction and ischemic stroke [8]. PRES is often considered on a spectrum with RCVS, which is certainly associated with ischemia [14]. In 2012, there was at least one case reported with HCP in a young woman with evidence of cerebral vasospasm and frank stroke on MRI [15].
Seizures
While there are reports of clinical seizures during acute attacks of porphyria, especially in AIP, but also in HCP and VP, there appears to be few captured on EEG upon literature review when compared with radiographic changes seen in PRES on MRI. Further complicating the picture, PRES is known to be associated with EEG changes and seizures, including non-convulsive seizures [16]. There are a few cases that do reveal PRES and EEG changes in the form of lateralized periodic discharges in the setting of porphyria flare with AIP [7]. However, many cases with EEG data reveal diffuse slowing consistent with encephalopathy, which is non-specific and this is often in the setting of other processes including PRES or electrolyte abnormalities [10].
However, there does exist reports of seizures without evidence of characteristic PRES changes on MRI. These cases include patients that have experienced non-convulsive status and epilepsia partialis continua, but it should be noted that MRI data in these cases appears to be missing or CT was used, which could miss more subtle findings consistent with PRES [17, 18]. Treatment for seizures will be discussed later in this article, as it presents a unique challenge since so many antiseizure drugs are processed by the liver’s cytochrome P450 system, which is greatly affected by porphyria as discussed above.
Alterations in Consciousness
Alterations in consciousness on a spectrum from mild cognitive deficits to severe encephalopathy are among the most common neurologic abnormalities described in ADP, AIP, HCP, and VP [19]. What is not clear is the mechanism by which this occurs. Many studies have proposed that this occurs as a consequence of other neurologic processes such as those described previously, including PRES and seizures, which are well known to cause cerebral dysfunction and cognitive changes that are typically reversible [20].
However, there are other derangements which might contribute to mental status changes. A well-known example is hyponatremia during an acute porphyria attack. This is thought to be a result of a syndrome of inappropriate antidiuretic hormone secretion or excessive losses [19]. Electrolyte derangement, such as this, is well known to cause changes in mentation, decreased alertness, and seizures [20].
Furthermore, another common symptom of acute porphyria, hypertension, is also known to cause changes in cognition, as seen in patients without porphyria diagnosed with hypertensive encephalopathy [21]. Given these multiple insults, including electrolyte disturbance, autonomic disturbance, PRES, and seizures, it is not entirely clear if the particular heme metabolites are also contributing to altered consciousness directly as well. If a patient presents with mental status changes during an acute attack, it is likely multifactorial in etiology and it would be difficult to assess if simply elevated heme metabolite levels could be solely responsible.
Neuropathy
Another well-described area of neurologic symptoms in patients with porphyria is in regard to the peripheral nervous system. There is evidence of transient sensory and motor disturbance in the PNS as well as involvement of the autonomic nervous system. This again is seen mostly in AIP cases, as it is the most common hepatic porphyria, but as with seizures, VP and HCP case can be seen.
The most concerning manifestation in the PNS associated with acute hepatic porphyric attack, which has been well documented, is motor weakness. There are case reports and series that demonstrate an axonal polyradiculopathy that can range from mild symptoms in selected peripheral nerves to a much more profound and diffuse weakness appearing similar to Guillain-Barré syndrome including respiratory failure in patients with AIP. Axonal damage is evident more typically in proximal muscles, particularly in the upper extremities, with muscle fibrillations and decreased muscle fiber recruitment [22]. These cases of weakness can mimic the progression of Guillain-Barré syndrome and may even present with recurrent quadriparesis [23, 24••]. It should be noted that many of the case reports note a lack of cytoalbuminologic dissociation and the more classic ascending paralysis expected with Guillain-Barré syndrome [24••]. Though non-specific, conduction studies typically confirm the findings of axonal neuropathy without evidence of conduction block or severe slowing. The differential diagnosis in these cases should include lead toxicity, which can also affect heme biosynthesis, lead to the accumulation of porphyrins, and present with gastrointestinal symptoms. On biopsy studies, there is axonal degeneration with or without evidence of segmental demyelination.
Cranial nerve involvement has also been documented, albeit, not as robustly. There have been recent case reports of multiple cranial nerves being affected in one patient and even the absence of some brainstem reflexes [25, 26]. The most commonly affected cranial nerves are the facial and the vagus nerves, though cases of trigeminal, hypoglossal, accessory, and oculomotor involvement have also been described. When present, cranial neuropathies generally develop after involvement of the limbs or trunk.
In 2012, a small case series presented patients with what was described as dysautonomia, not only tachycardia and hypertension, but also unusual sweating and diarrhea. In addition, there is evidence of demyelination and axonal degeneration of the vagus nerve and decreased density of sympathetic ganglia cells has been observed in autopsy studies. These findings suggest that the hallmark abdominal symptoms of acute porphyrias, including the early abdominal pain, constipation, diarrhea, nausea, and vomiting, may be a manifestation of splanchnic autonomic pathology. Similarly, vagal nerve damage, such as seen with cranial nerves, could explain some of the patient’s vital sign irregularities and fluctuations [27].
A published retrospective study from Sweden of 107 patients found that, indeed, about 33% of patients had a motor neuropathy manifested by weakness. Perhaps more surprisingly, 65% of symptomatic patients reported sensory symptoms including both pain and numbness largely in the bilateral lower extremities. It should be noted that sensory symptoms in porphyria may follow either a “bathing-trunk” or “glove and stocking” distribution. While sensory disturbance was known before, it was previously thought that motor symptoms were far less common than sensory neuropathy. Furthermore, this study also reported that autonomic disturbance was by far the most common manifestation in about 96% of the patients. Of note, 5.6% reported chronic deficits including persisting sensory neuropathy, motor weakness, or non-specific neurobehavioral symptoms such as insomnia, fatigue, and irritability; all of these patients with chronic deficits had recurrent attacks documented [28••].
Pathogenesis of Neurologic Dysfunction
Different mechanisms have been proposed to explain the neurologic dysfunction seen in neurovisceral porphyrias. Based on evidence obtained in preclinical models, it has been suggested that the elevated ALA levels that occur during acute attacks have a direct cytotoxic effect on different neural cell lines. Mechanistically, ALA undergoes autoxidation and upregulates the production of oxygen free radicals resulting in lipid peroxidation and mitochondrial dysfunction [29•]. Heme is necessary for the production of cytochromes, which are essential for the mitochondrial production of ATP. Thus, it is plausible that ALA-mediated oxidative damage and heme depletion lead to the mitochondrial swelling and loss of transmembral potential observed in in vitro models [30]. Additional mechanisms of neurologic dysfunction include oxidative stress–associated demyelination and abnormal axonal transport secondary to mitochondrial dysfunction. This is in addition to the inhibition of myelinogenesis and increased susceptibility of glial cells to oxidative stress observed in response to ALA in vitro models. Biochemically, ALA has structural similarities with GABA and glutamate. In addition, there is experimental evidence that indicates that ALA acts as an agonist of presynaptic GABAA receptors and inhibits glutamate uptake [31, 32]. Furthermore, the intracerebral injection of ALA in rats induces convulsions which can be inhibited by glutamate receptor antagonists. Together, these findings suggest that, in addition to oxidative stress, ALA can interfere with the normal functioning of endogenous neurotransmitters. Alternatively, ALA can inhibit the Na+/K+ ATPase in human brain tissue which affects axonal membrane potential and promotes depolarization [33]. Interestingly, depolarization has been reported in association with acute peripheral neuropathy [34].
ALA can cross the blood–brain barrier. However, the influx of ALA from the periphery to the CNS is quite slow and it has been postulated that the choroid plexus could clear whatever ALA might permeate to the CNS [29•]. Thus, alternative mechanisms of CNS injury are currently under investigation. In addition to its role in mitochondrial dysfunction, heme depletion has also been implicated in other pathogenic mechanisms of disease in neurovisceral porphyrias. PRES, for example, has been associated with endothelial dysfunction and a reduction in the production of the endogenous vasodilator nitric oxide. Similarly, a wealth of evidence obtained in subarachnoid hemorrhage has associated dysregulation of nitric oxide synthesis to vasospasm. Nitric oxide is produced by the NOS, a hemoprotein that requires heme. Interestingly, in a case of AIP with PRES, the treatment with heme arginate improved NO levels [8]. Also, the expression of NOS is particularly elevated in neuronal subpopulations of the myenteric plexus [35]. Thus, it is possible that heme depletion, by affecting the bioavailability of NO, could cause PRES, cerebral vasoconstriction, and/or the dysautonomic gastrointestinal symptoms seen during acute attacks.
Treatment
The treatment of acute porphyric attacks has for a long time relied on heme infusions, removal of triggers such as medications and, in the case of low carbohydrate diets, increasing caloric intake [29•]. Intravenous glucose can abort mild attacks. In addition, treatment with erythropoietin can reduce ALA, PBG, and porphyrin levels and reduce the severity of attacks. The safety of medications for the treatment of patients with porphyria can be found at the European Drug Database for Acute Porphyrias of the American Porphyria Foundation drug database [36••, 37••].
For the treatment of patients with epilepsy or for those in need of antiseizure medications during acute porphyria, it is recommended to avoid agents that are highly metabolized by the liver including carbamazepine, phenytoin, and valproic acid (Table 2). A recent review of anticonvulsants in the pediatric population suggested that newer agents, particularly those that avoid hepatic metabolism, should be prioritized such as levetiracetam, topiramate, gabapentin, and lacosamide [38].
In 2019, the FDA approved givosiran for the treatment of acute hepatic porphyrias. This is an ALA synthase small interfering RNA that inhibits the synthesis of heme. It has shown that it can reduce the number of acute porphyria episodes in patients with recurrent attacks [39]. Overall, the mainstay of treatment of the neurologic symptoms of acute porphyria is to avoid attacks in the first place and, should one occur, to treat it aggressively to avoid long-term complications.
Patients are at risk of new attacks and should receive information regarding exacerbating factors and the importance of adopting healthy habits, including smoking cessation, abstinence from alcohol, and eating a well-balanced diet. Early treatment of infections and metabolic abnormalities as well as long-term monitoring of iron levels and kidney function is recommended. Suppression of ovulation with gonadotropin-releasing hormone agonist may be considered for women who experience recurrent attacks confined to the luteal phase of the menstrual cycle [40].
Prognosis
With appropriate treatment and supportive care, patients typically recover from acute attacks. However, it has been shown that some patients may experience persistent hypertension, liver dysfunction, and chronic renal insufficiency. In addition, there is an increased prevalence of psychiatric disorders among individuals with porphyria. These include anxiety, depression, adjustment disorder, behavioral changes, and substance use disorder. The recovery of the polyneuropathy is usually slow but complete, though this is influenced by the degree of axonal involvement. Approximately 1% of the attacks are fatal and cases of sudden death, presumably due to cardiac arrhythmia, may occur during acute flares [41].
Conclusion
Acute hepatic porphyrias are rare genetic disorders of heme synthesis that can cause largely gastro-neurologic symptoms and dysfunction thought to be related to a buildup of toxic metabolites. As the disorders are fairly rare, they are challenging to study and much of the information published is from case reports and series published from all over the world. Generally, there is consensus that these acute porphyrias are associated with mental status changes, seizures, PRES, dysautonomia, and motor and sensory neuropathies. The exact underlying mechanism is not entirely clear and many of these manifestations likely interplay, although rarer still, with the potential for permanent neurologic damage. If at all possible, it is best to avoid any known triggers and to otherwise treat attacks quickly and aggressively to mitigate any long-term consequences for the patients. More study is still needed for these elusive disorders but their early recognition, diagnosis, and treatment are critical.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Yasuda M, Chen B, Desnick RJ. Recent advances on porphyria genetics: inheritance, penetrance & molecular heterogeneity, including new modifying/causative genes. Mol Genet Metab. 2019;128:320–31.
Puy H, Gouya L, Deybach J. Porphyrias. Lancet. 2010;375:924–37.
Szlendak U, Bykowska K, Lipniacka A. Clinical, biochemical and molecular characteristics of the main types of porphyria. Adv Clin Exp Med. 2016;25:361–8.
Phillips JD. Heme biosynthesis and the porphyrias. Mol Genet Metab. 2019;128:164–77 (• This publication provides a detailed description of the biochemistry of heme.).
Pischik E, Kazakov V, Kauppinen R. Is screening for urinary porphobilinogen useful among patients with acute polyneuropathy or encephalopathy? J Neurol. 2008;255:974–9.
Suh Y, Gandhi J, Seyam O, Jiang W, Joshi G, Smith NL, Ali KS. Neurological and neuropsychiatric manifestations of porphyria. Int J Neurosci. 2019;129:1226–33.
Silveira DC, Bashir M, Daniel J, Lucena MH, Bonpietro F. Acute intermittent porphyria presenting with posterior reversible encephalopathy syndrome and lateralized periodic discharges plus fast activity on EEG. Epilepsy Behav Case Rep. 2016;6:58–60.
Takata T, Kume K, Kokudo Y, Ikeda K, Kamada M, Touge T, Deguchi K, Masaki T. Acute intermittent porphyria presenting with posterior reversible encephalopathy syndrome, accompanied by prolonged vasoconstriction. Intern Med. 2017;56:713–7.
Anderson R, Patel V, Sheikh-Bahaei N, Liu CSJ, Rajamohan AG, Shiroishi MS, Kim PE, Go JL, Lerner A, Acharya J. Posterior reversible encephalopathy syndrome (PRES): pathophysiology and neuro-imaging. Front Neurol. 2020;11:463.
Zheng X, Liu X, Wang Y, Zhao R, Qu L, Pei H, Tuo M, Zhang Y, Song Y, Ji X, et al. Acute intermittent porphyria presenting with seizures and posterior reversible encephalopathy syndrome: two case reports and a literature review. Medicine (Baltimore). 2018;97: e11665.
Zhao B, Wei Q, Wang Y, Chen Y, Shang H. Posterior reversible encephalopathy syndrome in acute intermittent porphyria. Pediatr Neurol. 2014;51:457–60.
Rasheed F, Mehdi QS, Bhatti S, Khan MMA. Posterior reversible encephalopathy syndrome in a patient with variegate porphyria: a case report. Cureus . 2018;10.
Hagemann G, Ugur T, Witte OW, Fitzek C. Recurrent posterior reversible encephalopathy syndrome (PRES). J Hum Hypertens. 2004;18:287–9.
Pilato F, Distefano M, Calandrelli R. Posterior reversible encephalopathy syndrome and reversible cerebral vasoconstriction syndrome: clinical and radiological considerations. Front Neurol. 2020;11:34.
Mullin S, Platts A, Randhawa K, Watts P. Cerebral vasospasm and anterior circulation stroke secondary to an exacerbation of hereditary corproporphyria. Pract Neurol. 2012;12:384–7.
Bastide L, Legros B, Rampal N, Gilmore EJ, Hirsch LJ, Gaspard N. Clinical correlates of periodic discharges and nonconvulsive seizures in posterior reversible encephalopathy syndrome (PRES). Neurocrit Care. 2018;29:481–90.
Dawit S, Bhatt SK, Das DM, Pines AR, Shiue HJ, Goodman BP, Drazkowski JF, Sirven JI. Nonconvulsive status epilepticus secondary to acute porphyria crisis. Epilepsy Behav Case Rep. 2019;11:43–6.
Tran TPY, Leduc K, Savard M, Dupré N, Rivest D, Nguyen DK. Acute porphyria presenting as epilepsia partialis continua. Case Rep Neurol. 2013;5:116–24.
Bonkovsky HL, Dixon N, Rudnick S. Pathogenesis and clinical features of the acute hepatic porphyrias (AHPs). Mol Genet Metab. 2019;128:213–8.
Jaramillo-Calle DA, Solano JM, Rabinstein AA, Bonkovsky HL. Porphyria-induced posterior reversible encephalopathy syndrome and central nervous system dysfunction. Mol Genet Metab. 2019;128:242–53.
Price RS, Kasner SE. Hypertension and hypertensive encephalopathy. Handb Clin Neurol. 2014;119:161–7.
Alqwaifly M, Bril V, Dodig D. Acute intermittent porphyria: a report of 3 cases with neuropathy. CRN. 2019;11:32–6.
Mutluay B, Köksal A, Çelık RGG, Bülbül HH. A case of acute intermittent porphyria mimicking Guillain-Barré syndrome. Noro Psikiyatr Ars. 2019;56:311–2.
Rad N, Beydoun SR. Porphyria-induced recurrent quadriplegia misdiagnosed as Guillain-Barré syndrome. US Neurol. 2020;16:66.
Barraza G, Serranova T, Herrero C, Casanova-Mollá J, To-Figueras J, Herranz J, Valls-Solé J. Brainstem dysfunction in variegate porphyria. Muscle Nerve. 2012;46:426–33.
Spiritos Z, Salvador S, Mosquera D, Wilder J. Acute intermittent porphyria: current perspectives and case presentation. TCRM. 2019;15:1443–51.
Nabin A, Thapa LJ, Paudel R, Rana PVS. Acute intermittent porphyria with SIADH and fluctuating dysautonomia. Kathmandu Univ Med J. 2012;10:96–9.
Baumann K, Kauppinen R. Long-term follow-up of acute porphyria in female patients: update of clinical outcome and life expectancy. Mol Genet Metab Rep. 2022;30: 100842 (•• Large database of porphyria cases with long-term follow-up providing insight into frequency of complications.).
Ricci A, Di Pierro E, Marcacci M, Ventura P. Mechanisms of neuronal damage in acute hepatic porphyrias. Diagnostics (Basel). 2021;11:2205 (• Detailed description of the pathogenic mechanisms associated with neuronal damage in porphyrias.).
Hermes-Lima M, Castilho RF, Valle VG, Bechara EJ, Vercesi AE. Calcium-dependent mitochondrial oxidative damage promoted by 5-aminolevulinic acid. Biochim Biophys Acta. 1992;1180:201–6.
Emanuelli T, Pagel FW, Porciúncula LO, Souza DO. Effects of 5-aminolevulinic acid on the glutamatergic neurotransmission. Neurochem Int. 2003;42:115–21.
Müller WE, Snyder SH. delta-Aminolevulinic acid: influences on synaptic GABA receptor binding may explain CNS symptoms of porphyria. Ann Neurol. 1977;2:340–2.
Russell VA, Lamm MC, Taljaard JJ. Inhibition of Na+, K+-ATPase activity by delta-aminolevulinic acid. Neurochem Res. 1983;8:1407–15.
Lin CS-, Krishnan AV, Lee M, Zagami AS, You H, Yang C, Bostock H, Kiernan MC. Nerve function and dysfunction in acute intermittent porphyria. Brain . 2008;131:2510–2519.
Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–70.
•• The drug database for acute porphyria. Available at: http://www.drugs-porphyria.org/. Accessed April 11, 2022. (Updated database containing safety of drugs in porphyria.)
•• American Porphyria Foundation Drug Database. Available at: https://porphyriafoundation.org/drugdatabase/. Accessed Apr 11, 2022. (Updated database containing safety of drugs in porphyria.)
Vidaurre J, Gedela S, Yarosz S. Antiepileptic drugs and liver disease. Pediatr Neurol. 2017;77:23–36.
Syed YY. Givosiran: a review in acute hepatic porphyria. Drugs. 2021;81:841–8.
Stein P, Badminton M, Barth J, Rees D, Stewart MF. Best practice guidelines on clinical management of acute attacks of porphyria and their complications. Ann Clin Biochem. 2013;50:217–23.
Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. N Engl J Med. 2017;377:862–72.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Kyle Wylie and Fernando D. Testai each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neurology of Systemic Diseases
Rights and permissions
About this article

Cite this article
Wylie, K., Testai, F.D. Neurological Manifestations of Acute Porphyrias. Curr Neurol Neurosci Rep 22, 355–362 (2022). https://doi.org/10.1007/s11910-022-01205-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11910-022-01205-7