
Abstract
Aldosterone binds to the mineralocorticoid receptor and has an important regulatory role in body fluid and electrolyte balance. It also influences a variety of different cell functions such as oxidative stress, inflammation and organ fibrosis. The important role of the tissue-specific mineralocorticoid receptors in cardiovascular and renal injury has been shown in knockout animals and in clinical studies Mineralocorticoid receptor antagonists seem to exert their beneficial effects via anti-oxidative, anti-inflammatory and anti-fibrotic effects. Spironolactone and eplerenone were the first steroidal mineralocorticoid receptor antagonist. The established steroidal mineralocorticoid receptor antagonists show important therapeutic effects but are hampered by a variety of side effects, most importantly clinically significant hyperkaliemia. Selective non-steroidal mineralocorticoid receptor antagonists have been recently developed and demonstrate effectiveness in early clinical trials. Finereroneholds promise for the future application of this new mineralocorticoid receptor antagonist class in patients with chronic kidney disease since it has shown a significant reduction in UACR combined with a safety profile similar to that in the placebo group. However, further long-term studies investigating relevant clinical end points like reduction in cardiovascular or renal event rate are warranted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease is the leading cause of mortality and morbidity in patients with chronic kidney disease, and this is especially true for patients with diabetic nephropathy. However, despite the world-wide importance of this global health challenge, there are only few evidence-based treatments for reducing cardiovascular events and ameliorate organ function in these patients. The clinical evaluation of several new therapeutic candidates for diabetic nephropathy therapy, such as bardoxolone, aliskiren, and darbepoetin alfa, has failed to limit both the progression to end-stage renal disease and cardiovascular morbidity/mortality in long-term studies. The failure of novel drug candidates to delay progression to end-stage renal disease and limit or abrogate cardiovascular morbidity and mortality raises interest in the improvement of existing therapeutic strategies. One of the most promising strategies to reduce cardiovascular risk in patients with chronic kidney disease and end-stage renal disease are mineralocorticoid receptor antagonist-based treatment regimens. In this short review, we will summarize the latest development in this area and outline future prospects.
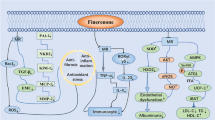
Aldosterone is secreted in the glomerular zone of the adrenal cortex in response to hyperkaliemia or sodium depletion as the endpoint of activation of the renin–angiotensin system and contributes importantly to regulation of blood pressure [1••]. The hormone classically acts in the distal tubule and collecting duct of the nephron where it increases water and sodium reabsorption thereby leading to extracellular fluid expansion. Aldosterone binds to the mineralocorticoid receptor, a ligand-dependent transcription factor belonging to the nuclear receptor family [2–4]. A multitide of studies over the last decade have convincingly shown that in addition to the regulatory role in body fluid and electrolyte balance, aldosterone influences a variety of different cell functions. It seems that mineralocorticoid receptors play a major role in oxidative stress, inflammation and, last but not least, organ fibrosis.
These novel functions of mineralocorticoid receptors, besides the classic role in ion and water transport, have been detected also in other cell types, especially in cardiac and vascular tissues. In addition, local production of aldosterone may occur in peripheral tissue [5••, 6]. Experimental evidence indicates that prolonged exposure to inappropriately elevated concentrations of aldosterone causes cardiac and renal damage independent of blood pressure levels [1••, 3]. Aldosterone concentrations and mineralocorticoid-receptor signaling are associated with an enhanced risk of cardiovascular injury and the incidence of sudden cardiac death. The mineralocorticoid-dependent mechanisms are complex and involve several molecular and cellular systems (Table 1).
Molecular and Cellular Effects of Mineralocorticoid Receptor Activation
An increase in oxidative stress by aldosterone and mineralocorticoid receptor activation has been demonstrated in a large number of cell types, such as cardiomyocytes, endothelial cells, vascular smooth muscle cells, adipocytes, macrophages, and collecting duct cells. Aldosterone increases the expression of the NADPH oxidase subunits Nox 2 and Nox4 as well as p47phox, p67phox, and rac1 [7–9], thereby leading to an increase in Nox activity with enhanced production of superoxides. In addition, aldosterone may induce mitochondrial dysfunction, thus contributing further to oxidative stress. The consequences of oxidative stress are multiple, including DNA damage and protein carbonylation. The mineralocorticoid-induced oxidative stress leads also to uncoupling of the NO synthase with a decreased availability of NO. Besides the deleterious effects of vasorelaxation, oxidative stress may directly activate the nuclear factor-κB pathway leading to inflammation [10, 11••].
Low-grade inflammation is a hallmark of cardiovascular and renal disease. Patients with hypertension, cardiovascular, and renal disease often present with a chronic vascular inflammation. In some patients, inflammatory biomarkers such as C-reactive protein or inflammatory cytokines (MCP-1) and their receptors are increased. These patients have a poor clinical outcome and prognosis [12••]. Indeed, several experimental studies have shown that chronic activation of the mineralocorticoid receptor enhances and stimulates a variety of different inflammatory mechanisms. Macrophages, dendritic cells, and T lymphocytes have been described as target cells for mineralocorticoids. The activation of the mineralocorticoid receptor enhances the expression of interleukin (IL)-6 and tumor necrosis factor-α (TNF-α) and nuclear factor κB activation in both immune and nonimmune cells [13]. Schiffrin and coworkers have recently demonstrated that the infiltration of the arterial wall and perivascular space by immune cells is a relatively early event after activation of the mineralocorticoid receptor [14]. This pro-inflammatory effect of aldosterone can be abolished in monocyte/macrophage deficient animals [15]. Interestingly, oxidative stress and endothelial dysfunction are also reduced in these animals indicating that the effects of aldosterone on immune cells is centrally important in this “trio infernal” of oxidative stress, microinflammation and fibrosis. The important role of the mineralocorticoid receptor in macrophage activation has been demonstrated in several studies with macrophage-specific mineralocorticoid receptor KO mice [16]. Deletion of the mineralocorticoid receptor protected against cardiac fibrosis after angiotensin II challenge or thoracic aortic constriction [17, 18]. Aldosterone may directly influence a switch in macrophage polarization and the differentiation of monocytes into an inflammatory phenotype [19]. Besides monocytes, it seems that T-cells are also involved in aldosterone-induced microinflammation. Aldosterone increases IL-6 and transforming growth factor β expression in dendritic cells and influences the cell-mediated Th17 polarization of T lymphocytes [20•, 21]. Aldosterone may be involved in autoimmune renal damage via these mechanisms. It has been shown that aldosterone stimulates Th17-mediated immunity [20•]. Schiffrin and coworkers have shown that T regulatory cells prevent aldosterone-induced vascular injury in mice [22]. The pro-inflammatory effects of aldosterone are not only present in vascular but also in adipose tissue, where it stimulates secretion of proinflammatory adipokines and production of TNFα, MCP1, PAI-1, and IL-6, an effect prevented by pharmacological mineralocorticoid receptor antagonism ( [23]; Table 2).
Whether aldosterone potentiates the cellular effects of DAMPs (danger-associated molecular pattern molecules, such as tenascine-C/fibronectin, heat shock proteins, IL-1α, osteopontin, uric acid, etc.) or whether aldosterone plays a role in the generation of these molecules is an important issue [24••, 25]. In support of the latter hypothesis are findings that several of these factors, such as tenascine-C, osteopontin, galectin-3, collagen, fibronectin, are influenced by activation of the mineralocorticoid receptor.
All these studies support the hypothesis that aldosterone and its nuclear receptor modulate both innate and adaptive immunity.
In summary, there is a large body of evidence showing that inhibition of the mineralocorticoid receptor can decrease microinflammation in animal models and in patients [20•, 21] supporting the hypothesis that the beneficial effects of mineralocorticoid antagonists in cardiovascular and renal diseases could in part rely on their anti-inflammatory properties.
The third molecular mechanism whereby aldosterone promotes organ damage is its role in fibrosis [26]. The first studies have been carried out in myocardial tissue and have clearly shown that chronic mineralocorticoid receptor activation is associated with fibrosis, extracellular matrix remodeling and cell growth and survival [27, 28]. It was already mentioned that mineralocorticoid receptors influence several profibrotic factors, such as NGAL, CT1, Gal-3, and osteopontin. In randomized clinical trials, the beneficial effects of mineralocorticoid antagonists in heart failure and cardiovascular disease were associated with a reduction of fibrosis markers [29, 30]. Activation of the mineralocorticoid receptor enhances cardiovascular and renal tubulointerstitial fibrosis [31]. This anti-fibrotic effect of mineralocorticoid antagonists has also been shown in other tissues: spironolactone limits peritoneal fibrosis in animal models [32–34], reduces liver fibrosis [35], and even influences skin fibrosis [36]. The onset of fibrosis seems to involve several different cell types dependent on the specific organ system including fibroblasts but also cardiomyocytes, vascular smooth muscle cells, endothelial cells, tubular cells, as well as inflammatory cells that secondarily stimulate extracellular matrix production.
In summary, mineralocorticoid receptor antagonists seem to exert their beneficial effects via anti-oxidative, anti-inflammatory and anti-fibrotic effects. Whether this triad is due to specific effects of the mineralocorticoid receptor on the respective mechanism or whether one underlying molecular pathway is responsible for the induction of all three remains to be demonstrated. Aldosterone stimulates the expression of endothelin 1 (ET1), transforming growth factor β, and plasminogen activator inhibitor [37]. Importantly, the interaction between the mineralocorticoid receptor and angiotensin II (AngII) receptor AT1R signaling cascades plays a key role in aldosterone-induced organ damage. AngII can activate the mineralocorticoid receptor pathway indirectly via the EGFR pathway [38] while expression of AT1R is obligatory for aldosterone activation in vascular smooth muscle cells [39]. The functional importance of this interaction between the two pathways shows the prevention of AngII-induced cardiac and vascular remodeling by spironolacton [7, 40].
Mineralocorticoid Receptor and Renal Diseases
After the initial decsription of renal disease in hyperaldosteronism, it was assumed that renal damage was mostly due to chronic hypertension in these patients. Animal experiments have now convincingly demonstrated that renal failure, proteinuria, and histologic renal lesions can be prevented by mineralocorticoid receptor antagonists, even in the presence of high blood pressure [41, 42]. Also in other models of chronic kidney diseases (nephron reduction, diabetic nephropathy, glomerulopathies) mineralocorticoid receptor antagonists exert a positive effect [43, 44]. Recent observations show that mineralocorticoid receptor antagonists can reduce cyclosporine-induced nephrotoxicity [45, 46] and spironolactone improves transplant vasculopathy in a rat renal transplant model [47]. Even acute kidney injury such as ischemia reperfusion injury [48] can be diminished by mineralocorticoid receptor antagonists administered before or directly after ischemia [49, 50]. Like other therapeutic strategies for acute kidney injury, mineralocorticoid receptor antagonists also showed long-term beneficial effects and prevented the delayed occurrence of chronic renal failure and interstitial fibrosis following acute kidney injury [51].
These positive effects of mineralocorticoid receptor antagonists on the kidney seem to be mediated by several cell types. In addition to functional mineralocorticoid receptors in the distal tubular cells, there are also mineralocorticoid receptors expressed in endothelial cells and in vascular smooth muscle cells of renal arteries [52]. Under pathological conditions, the expression of mineralocorticoid receptors may be up-regulated in glomerular cells [53] and expression of mineralocorticoid receptors has been demonstrated ex vivo in cultured podocytes, mesangial cells, and renal fibroblasts [54, 55]. This phenomen of mineralocorticoid receptor upregulation has been documented in patients with chronic renal disease [56]. The importance of inflammatory cells and activation of mineralocorticoid receptor in macrophages has been described above. Huang et al. have shown that renal disease induced by antiglomerular basement membrane antibody is blunted in mice with mineralocorticoid receptor deletion in the myeloid lineage [57].
In clinical studies in patients with chronic kidney disease, two important issues have to be adressed: (1) effects of mineralocorticoid receptor antagonists on the progression of renal disease and (2) reduction of the increased cardiovascular event rate in these patients.
For the medical management of patients with chronic kidney disease, reduction of albuminuria has become a primary aim, in addition to optimizing control of hyperglycaemia and blood pressure. Guidelines currently recommend treatment with angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin II receptor blockers (ARBs) for diabetic patients with hypertension and high (formerly known as macro-) or low grade albuminuria [58]. Both ACEIs and ARBs act via inhibition of the renin–angiotensin system (RAS). Despite initial down-regulation of plasma aldosterone levels, up to 50 % of patients treated with a RAS blocker experience elevations of the hormone within a year after treatment initiation [59]. This “aldosterone breakthrough/escape” is associated with impairment of albuminuria and kidney function. In subjects with CKD, systematic reviews of small studies have suggested that even when added to ACEIs and ARBs, mineralocorticoid receptor antagonists substantially reduce proteinuria [60, 61]. In several clinical studies, a potential role for the steroidal mineralocorticoid antagonist spironolactone and eplerenone as antiproteinuric agents has been identified [62–65]. A meta-analysis supported that mineralocorticoid receptor antagonists, in addition to ACEIs and ARBs, reduced proteinuria [66]. It has to be acknowledged that several studies have questioned the role of aldosterone and the mineralocorticoid receptor in proteinuria and in the progression of chronic kidney disease [43, 44, 67]. A small decrease in estimated glomerular filtration rate (eGFR) during treatment has been reported [68, 69]. This finding occured mostly in diabetic patients, probably reflecting reversal of hyperfiltration [66].
As there is a high risk of cardiovascular-associated mortality and morbidity in patients with diabetic nephropathy, it is possible that mineralocorticoid receptor antagonists may also help preventing cardiovascular events in this population. [70]. It is noteworthy, that in the 4D study, aldosterone levels were associated with sudden death in hemodialyzed patients with type 2 diabetes [71]. A positive impact of mineralocorticoid receptor antagonists on cardiovascular events has been reported in hemodialysis patients [72].
When using mineralocorticoid receptor antagonists in patients prone to hyperkalemia, a major issue is the safety concern regarding an increase in plasma potassium concentration. Hyperkalemia is often observed in patients treated with steroid mineralocorticoid receptor antagonists, reflecting efficacy of mineralocorticoid receptor antagonists in reducing urinary K+ secretion. Life-threatening hyperkalemia has been reported, particularly when using a high dosage of mineralocorticoid receptor antagonists [67]. Therefore, close monitoring of serum potassium levels in patients with impaired renal function is required to avoid clinically relevant hyperkalemia above 5.5 mmol/l. Hyperkalemia is a major problem limiting the use of mineralocorticoid receptor antagonists in patients with chronic kidney disaese, unfortunately at the same time the patient group where they are most needed.
Steroid Mineralocorticoid Receptor Antagonists
Spironolactone was the first steroidal mineralocorticoid receptor antagonist developed in 1959 [73]. Interestingly, this compound was developed 30 years before the molecular characterization of the mineralocorticoid receptor [74]. Spironolactone was approved as a diuretic and natriuretic drug for the management of hypertension and primary hyperaldosteronism and later on to treat congestive heart failure [73]. Spironolactone is a potent competitive mineralocorticoid receptor antagonist but is poorly selective because it also inhibits the androgen and progesterone receptors, leading to side effects such as gynecomastia, impotence, and menstrual irregularities [75•]. At high concentrations, it may also interfere with the glucocorticoid receptor [75•, 76].
The second mineralocorticoid receptor antagonist, eplerenone (9-11a-epoxymexrenone), was developed in 2002 for the treatment of hypertension and heart failure [73, 74]. This compound is much more selective for the mineralocorticoid receptor than spironolactone, but less potent (40× less), requiring higher dosage to achieve similar mineralocorticoid receptor antagonism. The different pharmacokinetic and pharmacodynamic properties of spironolactone and eplerenone, however, result in a difference in efficacy in the treatment of hypertension: 100 mg eplerenone is almost 50 to 75 % as potent as 100 mg spironolactone in patients with essential hypertension [77].
It has been reported that spironolactone (at micromolar concentrations) inhibits aldosterone biosynthesis [78] and blocks enzymes involved in steroidogenesis [79, 80]. In contrast, eplerenone (1–30 μM) did not impair basal and angiotensin II-induced cortisol and aldosterone production in human adrenocortical H295R cells [79]. Thus spironolactone and eplerenone have differential effects on steroidogenesis.
As mentioned above, hyperkalemia is a potential life-threatening side effect of mineralocorticoid receptor antagonists., Indeed these drugs are potassium-sparing diuretics, and their “propensity” to lead to hyperkalemia has been observed in most clinical studies, especially when renal function is impaired. The risk of hyperkalemia clearly accounts for the underuse of this efficient therapeutic class [80, 81].
Third-Generation Non-Steroidal Mineralocorticoid Receptor Antagonists—Finerenone
In the last decade, the search for novel mineralocorticoid receptor antagonists with higher selectivity, higher potency, and, if possible, a reduced risk of hyperkalemia has been intensified. Several nonsteroidal compounds acting as mineralocorticoid receptor antagonists were identified. Dihydropyridines (L-type calcium channel antagonists) can act as mineralocorticoid receptor antagonists in vitro and in vivo [75•]. Based on these molecules successful optimization of antagonists devoid of L-type calcium channel antagonist properties led to several new compounds and derivatives. One example is the Bayer compound BAY 94–8862 (finerenone) which was identified as a potent and selective mineralocorticoid receptor antagonist [82]. Its mode of action differs from classic steroidal mineralocorticoid receptor antagonist. Steroidal mineralocorticoid receptor antagonists bind in the ligand binding domain of the receptor, while finerenone has a bulky side chain which induces conformational changes within the mineralocorticoid receptor. The bulky molecule leads to the protrusion of mineralocorticoid receptor helix 12. This domain plays a major role in the activation of the receptor by binding of important co-activators [83]. The finerenone-induced changes alter recruitment of co-activators and co-repressors, thus changing the stability, nuclear translocation, and activation of the receptor. Thereby, finerenone causes the formation of an unstable receptor complex unable to recruit coregulators which is then rapidly degraded. The third-generation mineralocorticoid receptor antagonist finerenone was used in phase IIa clinical trials [84, 85] in patients with heart failure and mild renal dysfunction. Finerenone was shown to be safe and led to a similar effect on the N-terminal-proBNP surrogate marker compared with spironolactone. However, the rate of hyperkalemia was found to be lower [84, 85]. The relative benefit regarding decreased plasma potassium levels of finerenone over the steroidal antagonists may rely on a differential tissue distribution of the two compounds; while spironolactone and eplerenone accumulated three- to sixfold more in the kidney than in the heart, the distribution of finerenone appears equivalent in rat heart and kidney [75•]. Therefore, low doses may allow sufficient mineralocorticoid receptor antagonism outside of the kidney with less renal MR blockade and reduced potassium sparing effect. Two large clinical trials have been launched recently in heart failure (ARTS-HF) [86••] and in diabetic nephropathy (ARTS-DN) [87, 88••] to further test safety and efficacy in phase IIb trials.
Effect of Finerenone on Albuminuria in Patients with Diabetic Nephropathy (ARTS-DN)
ARTS-DN was a multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase IIB study designed to compare the effects of finerenone, 1.25 to 20 mg once daily, with placebo added to standard of care with RAS blockade. The primary outcome of the study was the ratio of the urinary albumin-creatinine ratio (UACR) at day 90 versus at baseline. Safety end points were (1) changes from baseline in serum potassium and (2) eGFR. Because of the diabetic study population and the inclusion criteria, UACRs and levels of glycated hemoglobin were elevated above the normal range. Average systolic blood pressure was also slightly higher than normal, as expected in patients with type 2 diabetes and diabetic nephropathy. Despite the high prevalence of hypertension in the study population, and because of the exclusion of patients with severe hypertension from this study, patients generally had good blood pressure control at baseline. A substantial proportion (ca 40 %) of patients in ARTS-DN had a history of cardiovascular comorbidity. The percentage of patients using lipid-lowering agents in the study was in line with that seen in other studies of patients with type 2 diabetes who are at increased cardiovascular risk. The study was initiated in June 2013 and clinically completed in August 2014. The results have been published recently [88••].
Finerenone demonstrated a dose-dependent reduction in UACR. Treatment reduced the placebo-corrected UACR at day 90 in a dose-dependent manner, with a significant reduction in UACR ranging from 21 to 38 % in the finerenone dosage groups of 7.5 to 20 mg/d compared with placebo. The placebo corrected mean differences in systolic blood pressure were small and post hoc analysis showed that no meaningful correlation was observed across all treatment groups between the ratio of UACR and the change in systolic blood pressure or change in eGFR from baseline to day 90.
Since control of potassium levels was one of the important safety issues in the study, serum potassium levels were carefully analyzed. No advice on dietary sodium or potassium restrictions was given during the study, and patients maintained their normal diet. With the exception of non–potassium-sparing diuretics, starting treatment with potassium-lowering agents was not permitted during treatment with the study drug. If hyperkalemia occurred during study treatment, the treatment was discontinued prior to starting a potassium lowering agent. Any potassium supplementation was stopped prior to randomization if potassium levels were within the normal range. If potassium levels were low at randomization or at any of the following visits, potassium supplementation was continued or restarted until potassium values were within the normal range again. Important was a prespecified secondary outcome of hyperkalemia leading to discontinuation of the study. Such an endpoint was not observed in the placebo and finerenone 10- mg/d groups. The incidences in the finerenone 7.5-, 15-, and 20-mg/d groups were 2.1, 3.2, and 1.7 %, respectively. There were no differences in the incidence of the prespecified secondary outcome of an eGFR decrease of 30 % or more or in incidences of adverse events and serious adverse events between the placebo and finerenone groups.
Previous studies have shown conflicting results regarding the incidence of hyperkalemia in patients with diabetes receiving steroidal mineralocorticoid receptor antagonists. A systematic review documented an increased incidence of hyperkalemia in patients with diabetic nephropathy receiving steroidal mineralocorticoid receptor antagonists with RAS blockers compared with RAS blockade alone [89] with dropout rates due to hyperkalemia between 8 and 17 %, respectively. Clinically significant hyperkalemia (serum potassium level > 6.0 mmol/L) was noted in 52 % of patients treated with high-dose ACE inhibitors plus low-dose spironolactone [90]. However, in other studies much lower rates of hyperkalemia have been reported. It may be that the higher dosage of ACE inhibitors may have contributed to higher rates of hyperkalemia in some studies. In ARTS-DN, hyperkalemia and subsequent discontinuation of the study drug occurred in 1.8 % of patients receiving finerenone compared with no cases in the placebo group. Three cases of hyperkalemia with serum potassium of more than 6.0 mmol/L were observed overall. It may well be that the lack of a significant decrease in eGFR has been a contributing factor to the low risk of hyperkalemia in ARTS-DN.
It is important to note that ARTS-DN is a dose-finding study that lacks an active control group. Another limitation is that 60 % of patients had an eGFR above 60 mL/min/1.73 m2, thus putting them at lower risk of hyperkalemia in general. Moreover, while reductions in albuminuria are highly correlated with slowed progression of CKD, they are not a validated surrogate marker for renal outcomes. Last but not least, the short duration of the study did not allow assessment of the long-term effects of finerenone on CKD progression or assessment of antifibrotic or anti-inflammatory effects.
Conclusions
Activation of the mineralocorticoid receptor in cardiovascular tissue induces inflammation, oxidative stress and organ fibrosis thereby contributing to the pathogenesis of cardiovascular injury. This role of tissue-specific mineralocorticoid receptors in cardiovascular and renal injury has been shown in knockout animals and in clinical studies. Steroidal mineralocorticoid receptor antagonists have important therapeutic effects but are hampered by side effects, such as hyperkaliemia. The novel non-steroidal mineralocorticoid receptor antagonist finererone demonstrate effectiveness in early clinical trials and a safety profile similar to that in the placebo group. Whether this new mineralocorticoid receptor antagonist class shows relevant clinical end points like reduction in cardiovascular or renal event rate has to be demonostrated in prospective clinical studies.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Fuller PJ, Young MJ. Mechanisms of mineralocorticoid action. Hypertension. 2005;46:1227–35. Comprehensive review on the molecular mechanisms of mineralocorticoids.
Fuller PJ, Yao Y, Yang J, Young MJ. Mechanisms of ligand specificity of the mineralocorticoid receptor. J Endocrinol. 2012;213:15–24.
Lombes M, Alfaidy N, Eugene E, Lessana A, Farman N, Bonvalet J-P. Prerequisite for cardiac aldosterone action: mineralocorticoid receptor and 11β-hydroxysteroid dehydrogenase in the human heart. Circulation. 1995;92(2):175–82.
Viengchareun S, Le Menuet D, Martinerie L, Munier M, Pascual-Le Tallec L, Lombès M. The mineralocorticoid receptor: insights into its molecular and (patho)physiological biology. Nucl Recept Signal. 2007;5, e012.
Bader M. Tissue renin-angiotensin-aldosterone systems: Targets for pharmacological therapy. Annu Rev Pharmacol Toxicol. 2010;50:439–65. This review by an international expert on RASS system gives a comprehensive overview on established and future therapeutic targets. Provides insight into molecular studies of the RAAS system.
Taves MD, Gomez-Sanchez CE, Soma KK. Extra-adrenal glucocorticoids and mineralocorticoids: evidence for local synthesis, regulation, and function. Am J Physiol Endocrinol Metab. 2011;301:E11–24.
Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 2006;20:1546–8.
Park YM, Lim BH, Touyz RM, Park JB. Expression of NAD(P)H oxidase subunits and their contribution to cardiovascular damage in aldosterone/salt-induced hypertensive rat. J Korean Med Sci. 2008;23(6):1039–45.
Michea L, Villagrán A, Urzúa A, Kuntsmann S, Venegas P, Carrasco L, et al. Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and prevents oxidative stress in uremic rats. Hypertension. 2008;52(2):295–300.
Mayyas F, Alzoubi KH, Van Wagoner DR. Impact of aldosterone antagonists on the substrate for atrial fibrillation: aldosterone promotes oxidative stress and atrial structural/electrical remodeling. Int J Cardiol. 2013;168:5135–42.
Karbach S, Wenzel P, Waisman A, Munzel T, Daiber A. eNOS uncoupling in cardiovascular diseases--the role of oxidative stress and inflammation. Curr Pharm Des. 2014;20:3579–94. This review explains the intricate relationship between inflammation and oxidative stress and unravels the complicated mechanisms of oxidative stress in cardiovascular disease.
Schiffrin EL. The immune system: role in hypertension. Can J Cardiol. 2013;29:543–8. Schiffrin gives an overview on the recent devlopments in this novel and intriguing research field in cardiovascular medicine.
Bene NC, Alcaide P, Wortis HH, Jaffe IZ. Mineralocorticoid receptors in immune cells: emerging role in cardiovascular disease. Steroids. 2014;91:38–45.
Kasal DA, Schiffrin EL. Angiotensin II, aldosterone, and anti-inflammatory lymphocytes: interplay and therapeutic opportunities. Int J Hypertens. 2012;2012:829786.
De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol. 2005;25:2106–13.
Rickard AJ, Morgan J, Chrissobolis S, Miller AA, Sobey CG, Young MJ. Endothelial cell mineralocorticoid receptors regulate DOC/salt-mediated cardiac remodeling and vascular reactivity, but not blood pressure. Hypertension. 2014;63:1033–40.
Liu Y, Hirooka K, Nishiyama A, Lei B, Nakamura T, Itano T, et al. Activation of the aldosterone/mineralocorticoid receptor system and protective effects of mineralocorticoid receptor antagonism in retinal ischemia-reperfusion injury. Exp Eye Res. 2012;96:116–23.
Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schütz G, et al. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. 2010;120:3350–64.
Marzolla V, Armani A, Feraco A, De Martino MU, Fabbri A, Rosano G, et al. Mineralocorticoid receptor in adipocytes and macrophages: a promising target to fight metabolic syndrome. Steroids. 2014;91:46–53.
Herrada AA, Campino C, Amador CA, Michea LF, Fardella CE, Kalergis AM. Aldosterone as a modulator of immunity: implications in the organ damage. J Hypertens. 2011;29:1684–92. Review of the literature to explain how aldosterone affects the immune system and which implication ensue for cardiovascular disease.
Herrada AA, Contreras FJ, Marini NP, Amador CA, González PA, Cortés CM, et al. Aldosterone promotes autoimmune damage by enhancing Th17-mediated immunity. J Immunol. 2010;184:191–202.
Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, et al. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension. 2012;59:324–30.
Guo C, Ricchiuti V, Lian BQ, Yao TM, Coutinho P, Romero JR, et al. Mineralocorticoid receptor blockade reverses obesity-related changes in expression of adiponectin, peroxisome proliferator-activated receptor-gamma, and proinflammatory adipokines. Circulation. 2008;117:2253–61.
Anders HJ, Schaefer L. Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J Am Soc Nephrol. 2014;25:1387–400. Important revew on the role of the innate immune system in renal and cardiovascular disease. Provides also insight into mechanisms of inflammasome and cardiovascular disease.
Frantz S, Monaco C, Arslan F. Danger signals in cardiovascular disease. Mediat Inflamm. 2014;2014:395278.
Thomas W, Dooley R, Harvey BJ. Aldosterone as a renal growth factor. Steroids. 2010;75:550–4.
Dooley R, Harvey BJ, Thomas W. The regulation of cell growth and survival by aldosterone. Front Biosci (Landmark Ed). 2011;16:440–57.
Dooley R, Harvey BJ, Thomas W. Non-genomic actions of aldosterone: from receptors and signals to membrane targets. Mol Cell Endocrinol. 2012;350:223–34.
Iraqi W, Rossignol P, Angioi M, Fay R, Nuée J, Ketelslegers JM, et al. Extracellular cardiac matrix biomarkers in patients with acute myocardial infarction complicated by left ventricular dysfunction and heart failure: insights from the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) study. Circulation. 2009;119:2471–9.
Lacolley P, Labat C, Pujol A, Delcayre C, Benetos A, Safar M. Increased carotid wall elastic modulus and fibronectin in aldosterone-salt-treated rats: effects of eplerenone. Circulation. 2002;106:2848–53.
Bauersachs J, Jaisser F, Toto R. Mineralocorticoid receptor activation and mineralocorticoid receptor antagonist treatment in cardiac and renal diseases. Hypertension. 2015;65:257–63.
Vazquez-Rangel A, Soto V, Escalona M, Toledo RG, Castillo EA, Polanco Flores NA, et al. Spironolactone to prevent peritoneal fibrosis in peritoneal dialysis patients: a randomized controlled trial. Am J Kidney Dis. 2014;63:1072–4.
Yelken B, Gorgulu N, Gursu M, Yazici H, Caliskan Y, Telci A, et al. Effects of spironolactone on residual renal function and peritoneal function in peritoneal dialysis patients. Adv Perit Dial. 2014;30:5–10.
Zhang L, Hao JB, Ren LS, Ding JL, Hao LR. The aldosterone receptor antagonist spironolactone prevents peritoneal inflammation and fibrosis. Lab Invest. 2014;94:839–50.
Pizarro M, Solis N, Quintero P, Barrera F, Cabrera D, Rojas-de Santiago P, et al. Beneficial effects of mineralocorticoid receptor blockade in experimental non-alcoholic steatohepatitis. Liver Int. 2015;35:2129–38.
Mitts TF, Bunda S, Wang Y, Hinek A. Aldosterone and mineralocorticoid receptor antagonists modulate elastin and collagen deposition in human skin. J Invest Dermatol. 2010;130:2396–406.
Marney AM, Brown NJ. Aldosterone and end-organ damage. Clin Sci (Lond). 2007;113:267–78.
Rautureau Y, Paradis P, Schiffrin EL. Cross-talk between aldosterone and angiotensin signaling in vascular smooth muscle cells. Steroids. 2011;76:834–9.
Lemarié CA, Simeone SM, Nikonova A, Ebrahimian T, Deschênes ME, Coffman TM, et al. Aldosterone-induced activation of signaling pathways requires activity of angiotensin type 1a receptors. Circ Res. 2009;105:852–9.
Virdis A, Neves MF, Amiri F, Viel E, Touyz RM, Schiffrin EL. Spironolactone improves angiotensin-induced vascular changes and oxidative stress. Hypertension. 2002;40:504–10.
Kobayashi N, Hara K, Tojo A, Onozato ML, Honda T, Yoshida K, et al. Eplerenone shows renoprotective effect by reducing LOX-1-mediated adhesion molecule, PKCepsilon-MAPK-p90RSK, and Rho-kinase pathway. Hypertension. 2005;45:538–44.
Nagase M, Fujita T. Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. Nat Rev Nephrol. 2013;9:86–98.
Bertocchio JP, Warnock DG, Jaisser F. Mineralocorticoid receptor activation and blockade: an emerging paradigm in chronic kidney disease. Kidney Int. 2011;79:1051–60.
Ritz E, Tomaschitz A. Aldosterone and the kidney: a rapidly moving frontier (an update). Nephrol Dial Transplant. 2014;29:2012–9.
Bobadilla NA, Gamba G. New insights into the pathophysiology of cyclosporine nephrotoxicity: a role of aldosterone. Am J Physiol Renal Physiol. 2007;293:F2–9.
Amador CA, Barrientos V, Peña J, Herrada AA, González M, Valdés S, et al. Spironolactone decreases DOCA-salt-induced organ damage by blocking the activation of T helper 17 and the downregulation of regulatory T lymphocytes. Hypertension. 2014;63:797–803.
Waanders F, Rienstra H, Boer MW, Zandvoort A, Rozing J, Navis G, et al. Spironolactone ameliorates transplant vasculopathy in renal chronic transplant dysfunction in rats. Am J Physiol Renal Physiol. 2009;296:F1072–9.
Juncos LA, Juncos LI (2015) Mineralocorticoid receptor antagonism in AKI: a new hope? J Am Soc Nephrol
Barrera-Chimal J, Pérez-Villalva R, Rodríguez-Romo R, Reyna J, Uribe N, Gamba G, et al. Spironolactone prevents chronic kidney disease caused by ischemic acute kidney injury. Kidney Int. 2013;83:93–103.
Sánchez-Pozos K, Barrera-Chimal J, Garzón-Muvdi J, Pérez-Villalva R, Rodríguez-Romo R, Cruz C, et al. Recovery from ischemic acute kidney injury by spironolactone administration. Nephrol Dial Transplant. 2012;27:3160–9.
Barrera-Chimal J, Pérez-Villalva R, Ortega JA, Sánchez A, Rodríguez-Romo R, Durand M, et al. Mild ischemic injury leads to long-term alterations in the kidney: amelioration by spironolactone administration. Int J Biol Sci. 2015;11:892–900.
Nguyen Dinh Cat A, Griol-Charhbili V, Loufrani L, Labat C, Benjamin L, Farman N, et al. The endothelial mineralocorticoid receptor regulates vasoconstrictor tone and blood pressure. FASEB J. 2010;24:2454–63.
Farman N, Vandewalle A, Bonvalet JP. Autoradiographic study of aldosterone and dexamethasone binding in isolated glomeruli of rabbit kidney. Am J Physiol. 1982;243:F235–42.
Lee HA, Song MJ, Seok YM, Kang SH, Kim SY, Kim I. Histone deacetylase 3 and 4 complex stimulates the transcriptional activity of the mineralocorticoid receptor. PLoS ONE. 2015;10, e0136801.
Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008;14:1370–6.
Quinkler M, Zehnder D, Eardley KS, Lepenies J, Howie AJ, Hughes SV, et al. Increased expression of mineralocorticoid effector mechanisms in kidney biopsies of patients with heavy proteinuria. Circulation. 2005;112:1435–43.
Huang LL, Nikolic-Paterson DJ, Han Y, Ozols E, Ma FY, Young MJ, et al. Myeloid mineralocorticoid receptor activation contributes to progressive kidney disease. J Am Soc Nephrol. 2014;25:2231–40.
Standards of medical care in diabetes 2014. Diabetes Care 2014;37 Suppl 1:S14-80
Bomback AS, Klemmer PJ. The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol. 2007;3:486–92.
Navaneethan SD, Nigwekar SU, Sehgal AR, Strippoli GF. Aldosterone antagonists for preventing the progression of chronic kidney disease. Cochrane Database Syst Rev 2009:CD007004.
Bomback AS, Kshirsagar AV, Amamoo MA, Klemmer PJ. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. Am J Kidney Dis. 2008;51:199–211.
Chrysostomou A, Becker G. Spironolactone in addition to ACE inhibition to reduce proteinuria in patients with chronic renal disease. N Engl J Med. 2001;345:925–6.
Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. Diabetes Care. 2005;28:2106–12.
Sato A, Hayashi K, Naruse M, Saruta T. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension. 2003;41:64–8.
Epstein M, Williams GH, Weinberger M, et al. Selective aldosterone blockade with eplerenone reduces albuminuria in patients with type 2 diabetes. Clin J Am Soc Nephrol. 2006;1:940–51.
Bolignano D, Palmer SC, Navaneethan SD, Strippoli GF. Aldosterone antagonists for preventing the progression of chronic kidney disease. Cochrane Database Syst Rev. 2014;4, CD007004.
Schwenk MH, Hirsch JS, Bomback AS. Aldosterone blockade in CKD: emphasis on pharmacology. Adv Chronic Kidney Dis. 2015;22:123–32.
Vardeny O, Wu DH, Desai A, Rossignol P, Zannad F, Pitt B, et al. Influence of baseline and worsening renal function on efficacy of spironolactone in patients with severe heart failure: insights from RALES (Randomized Aldactone Evaluation Study). J Am Coll Cardiol. 2012;60:2082–9.
Rossignol P, Cleland JG, Bhandari S, Tala S, Gustafsson F, Fay R, et al. Determinants and consequences of renal function variations with aldosterone blocker therapy in heart failure patients after myocardial infarction: insights from the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study. Circulation. 2012;125:271–9.
Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–17.
Drechsler C, Ritz E, Tomaschitz A, Pilz S, Schönfeld S, Blouin K, et al. Aldosterone and cortisol affect the risk of sudden cardiac death in haemodialysis patients. Eur Heart J. 2013;34:578–87.
Matsumoto Y, Mori Y, Kageyama S, Arihara K, Sugiyama T, Ohmura H, et al. Spironolactone reduces cardiovascular and cerebrovascular morbidity and mortality in hemodialysis patients. J Am Coll Cardiol. 2014;63:528–36.
Menard J. The 45-year story of the development of an anti-aldosterone more specific than spironolactone. Mol Cell Endocrinol. 2004;217:45–52.
Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268–75.
Kolkhof P, Borden SA. Molecular pharmacology of the mineralocorticoid receptor: Prospects for novel therapeutics. Mol Cell Endocrinol. 2012;350:310–7. This review provides an excellent overview of early non-steroidal development and rationale from an industry perspective.
Rossier BC, Baker ME, Studer RA. Epithelial sodium transport and its control by aldosterone: the story of our internal environment revisited. Physiol Rev. 2015;95:297–340.
Parthasarathy HK, Ménard J, White WB, Young Jr WF, Williams GH, Williams B, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens. 2011;29:980–90.
Netchitailo P, Delarue C, Perroteau I, Leboulenger F, Capron MH, Vaudry H. Relative inhibitory potency of five mineralocorticoid antagonists on aldosterone biosynthesis in vitro. Biochem Pharmacol. 1985;34:189–94.
Ye P, Yamashita T, Pollock DM, Sasano H, Rainey WE. Contrasting effects of eplerenone and spironolactone on adrenal cell steroidogenesis. Horm Metab Res. 2009;41:35–9.
Penhoat A, Darbeida H, Bernier M, Saez JM, Durand P. Inhibition of hormonal-induced cAMP and steroid production by inhibitors of pregnenolone metabolism in adrenal and Leydig cells. Mol Cell Endocrinol. 1988;60:55–60.
Albert NM, Yancy CW, Liang L, Zhao X, Hernandez AF, Peterson ED, et al. Use of aldosterone antagonists in heart failure. JAMA. 2009;302:1658–65.
Amazit L, Le Billan F, Kolkhof P, Lamribet K, Viengchareun S, Fay MR, et al. Finerenone impedes aldosterone-dependent nuclear import of the mineralocorticoid receptor and prevents genomic recruitment of steroid receptor coactivator-1. J Biol Chem. 2015;290(36):21876–89.
Bärfacker L, Kuhl A, Hillisch A, Grosser R, Figueroa-Pérez S, Heckroth H, et al. Kolkhof P Discovery of BAY 94–8862: a nonsteroidal antagonist of the mineralocorticoid receptor for the treatment of cardiorenal diseases. ChemMedChem. 2012;7(8):1385–403.
Pitt B, Kober L, Ponikowski P, et al. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist bay 94–8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: A randomized, double-blind trial. Eur Heart J. 2013;34:2453–63.
Pitt B, Filippatos G, Gheorghiade M, et al. Rationale and design of arts: A randomized, double-blind study of bay 94–8862 in patients with chronic heart failure and mild or moderate chronic kidney disease. Eur J Heart Fail. 2012;14:668–75.
Pitt B, Pfeffer MA, Assmann SF, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370:1383–92. This publication is the first to demonstrate effectiveness of non-steroidal MR antagonists in humans.
Ruilope LM, Agarwal R, Chan JC, Cooper ME, Gansevoort RT, Haller H, et al. Rationale, design, and baseline characteristics of ARTS-DN: a randomized study to assess the safety and efficacy of finerenone in patients with type 2 diabetes mellitus and a clinical diagnosis of diabetic nephropathy. Am J Nephrol. 2014;40:572–81.
Bakris GL, Agarwal R, Chan JC, Cooper ME, Gansevoort RT, Haller H, et al. Effect of finerenone on albuminuria in patients with diabetic nephropathy: a randomized clinical trial. JAMA. 2015;314:884–94. First study to demonstrate anti-proteinuric effects of finererone in diabetic patients with nephropathy.
Mavrakanas TA, Gariani K, Martin PY. Mineralocorticoid receptor blockade in addition to angiotensin converting enzyme inhibitor or angiotensin II receptor blocker treatment: an emerging paradigm in diabetic nephropathy. Eur J Int Med. 2014;25(2):173–6.
Mehdi UF, Adams-Huet B, Raskin P, et al. Addition of angiotensin receptor blockade or mineralocorticoid antagonism to maximal angiotensin-converting enzyme inhibition in diabetic nephropathy. J Am Soc Nephrol. 2009;20(12):2641–50.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Drs. Haller, Bertram, Stahl, and Menne declare no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Key points
• Although the established physiological function of the renal mineralocorticoid receptor is regulation of sodium reabsorption and potassium excretion, activation of the mineralocorticoid receptor in extra-renal tissues also contributes to the pathogenesis of cardiovascular injury.
• The important role of the tissue-specific mineralocorticoid receptors in cardiovascular and renal injury has been shown in knockout animals and in clinical studies.
• The established steroidal mineralocorticoid receptor antagonists show important therapeutic effects but are hampered by a variety of side effects, most importantly clinically significant hyperkaliemia, which limits their use especially in patients with chronic kidney disease and reduced glomerular filtration rates.
• Selective non-steroidal mineralocorticoid receptor antagonists have been recently developed and demonstrate effectiveness in early clinical trials.
• The significant reduction in UACR in patients receiving finerenone, combined with a safety profile similar to that in the placebo group, holds promise for the future application of this new mineralocorticoid receptor antagonist class in patients with chronic kidney disease. However, further long-term studies investigating relevant clinical end points like reduction in cardiovascular or renal event rate are warranted.
This article is part of the Topical Collection on Therapeutic Trials
Rights and permissions
About this article

Cite this article
Haller, H., Bertram, A., Stahl, K. et al. Finerenone: a New Mineralocorticoid Receptor Antagonist Without Hyperkalemia: an Opportunity in Patients with CKD?. Curr Hypertens Rep 18, 41 (2016). https://doi.org/10.1007/s11906-016-0649-2
Published:
DOI: https://doi.org/10.1007/s11906-016-0649-2