
Abstract
Establishing whether large vessel occlusive disease threatens tissue oxygenation and viability in the post-stenotic kidney is difficult for clinicians. Development of blood oxygen level–dependent (BOLD) MRI methods can allow functional evaluation of regional differences in deoxyhemoglobin levels within the kidney without requiring contrast. The complex renal circulation normally provides a gradient of oxygenation from a highly vascular cortex to much reduced levels in the deep sections of medulla, dependent upon adjustments in renal afferent arterioles, oxygen consumption related to solute transport, and arteriovenous shunting related to the juxtaposition of descending and ascending vasa recta. Studies with BOLD imaging have identified adaptation to substantial reductions in renal blood flow, volume, and glomerular filtration rate in post-stenotic kidneys that preserves medullary and cortical oxygenation during medical therapy. However, extreme vascular compromise overwhelms these adaptive changes and leads to cortical hypoxia and microvascular injury.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Evaluation of renovascular disease, particularly atherosclerotic renal artery stenosis (ARAS), presents daunting challenges for clinicians dealing with secondary hypertension. The prevalence of ARAS is increasing in developed countries, along with aging of the population. When imaging of the renal arteries is included in vascular studies for conditions associated with atherosclerosis, including coronary and peripheral vascular disease, some degree of renal artery stenosis can be identified in 18% to 30% or more of patients [1•]. Remarkable advances in antihypertensive drug therapy, particularly agents that block the renin-angiotensin system, have made it possible to achieve satisfactory blood pressure control for many of these patients and have improved cardiovascular outcomes as compared with treatment not using these agents [2]. As a result, clinical treatment algorithms are controversial, especially in the light of recent publications from clinical trials such the Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) trial [3••] and the STAR study of stent placement in patients with ARAS [4], which question the added benefit of revascularization in ARAS [5].
Determining when occlusive vascular lesions actually threaten the oxygenation and viability of the affected kidney remains an elusive goal, in part because glomerular filtration function normally is associated with a vast oversupply of the blood required for metabolic activity [6]. Paradoxically, the complex anatomy of renal vessels and heterogeneous oxygen consumption within the cortical and medullary regions are recognized to leave some areas, particularly within the deep medulla, at the brink of hypoxia much of the time. Powerful tomographic magnetic resonance tools such as blood oxygen level–dependent (BOLD) MRI, which provide an estimate of in vivo tissue oxygenation, represent an important advance in understanding the pathophysiology of renovascular disease. This review summarizes our current understanding of the use of BOLD MRI as it applies to renovascular hypertension.
Blood Flow and Oxygenation Within the Kidney
The renal circulations and distribution of blood flow are complex. Because renal blood flow is unusually high compared with that of other organs, relative “hyperoxia” and its potential for oxidative free-radical formation is a genuine threat [7••]. As a result, tissue oxygen levels are regulated by a triad of pathways specific to the kidney, namely alterations in afferent arteriolar blood flow, oxygen consumption (primarily governed by tubular solute transport), and arteriovenous shunting related to the apposition of descending and ascending vasa recta [8•]. Studies of microcirculatory regulation indicate that admixture of arterial and venous deoxygenated venous blood in mammalian kidneys produces relatively constant cortical oxygen tension, in the range of 50 mm Hg [8•]. Renal blood flow and pressure show little change until the renal artery lumen cross-sectional area falls by 70% to 80% [9]. When a “critical” degree of stenosis is attained, renal hypoperfusion leads to a cascade of events from initial activation of the renin-angiotensin system to the rarefaction of small renal vessels, kidney fibrosis, loss of function, and atrophy [9]. Renal changes may be reversible up to a certain point, beyond which even technically perfect revascularization often does not restore renal function. No studies have yet completely characterized the nature and duration of the structural alterations that render kidney function unsalvageable.
Tissue damage within the kidney resulting from decreased renal blood flow may differ between locations, possibly because renal structures differ in their vulnerability, on account of heterogeneous basal oxygenation and metabolic activity. Local differences of blood supply and energy-dependent transport produce major differences in oxygen tension between the cortex and inner medullary regions. Furthermore, local differences in oxygen supply and tubular transport activity result in substantial spatial heterogeneity even within the medulla (Fig. 1, bottom). The cortical microcirculation receives a vast surplus of oxygenated blood. Even with substantial reduction of blood flow, cortical tissue has enough oxygen for the metabolic demands. Studies of oxygen delivery and consumption in anesthetized rabbits indicate that acutely reduced filtration and oxygen consumption can leave tissue oxygen levels stable even if cortical blood flow is reduced by up to 40% [10, 11]. A smaller portion of blood flow is delivered to deeper medullary regions of the kidney via post-glomerular vasa recta; in these regions, active metabolic processes dependent upon aerobic energy pathways lead to oxygen consumption and local areas of hypoxia, even within normal kidneys. Local gradients of cortical and medullary oxygenation are closely regulated, sometimes independently from each other. Remarkably, these regions can tolerate reduced blood flow with compensatory changes by numerous vasoactive systems within the circulation. In a rat model, for example, angiotensin II infusion produced a 40% decrease of cortical perfusion, but medullary perfusion was unchanged, apparently protected by prostaglandin E2 synthesis [12]. Quantitative changes in the degree of arteriovenous shunting as a result of changes in renal blood flow appear to maintain the oxygen tension and adjust local areas of blood supply, but also may render some focal regions more susceptible to hypoxia [10].

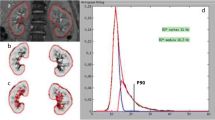
CT angiogram (upper left) showing preserved blood flow to the right kidney and advanced occlusive disease to the left kidney. T2* imaging (upper right) illustrates the gradient of oxygenated blood between cortex and medullary region of both kidneys. R2* parametric maps using blood oxygen level–dependent (BOLD) MRI identifies rising R2* levels in a deep medullary region in the normal kidney (lower left), with a more extensive medullary region of hypoxia and elevated level of cortical R2* as well evident in the kidney with vascular occlusion (lower right). These measurements illustrate the limitations of the post-stenotic kidney in adapting to reduced blood flow, eventually developing overt cortical hypoxia and widening regions of medullary deoxygenation. (From Textor and Lerman [9])
Much of medullary oxygen consumption is determined by solute transport. Cortical O2 levels in anesthetized swine models, for example, average 40 to 50 mm Hg, as measured by tissue oxygen probes (Table 1). Medullary O2 tension in normal animals under the same conditions average 20 to 23 mm Hg but rise to 41 mm Hg after the administration of furosemide, consistent with its action inhibiting solute transport in the Loop of Henle [13••, 14]. Blood returning from hypoxic regions travels adjacent to descending vasa recta with higher oxygen levels. When combined with some degree of arteriovenous mixing from preglomerular shunts, renal venous oxygen returns to inflow levels, sometimes above efferent arteriolar oxygen levels within the cortex [7••]. These characteristics have made it difficult to establish valid estimates for tissue oxygenation within disparate regions of the kidney in the intact organism.
BOLD MRI Applied to the Kidney
MRI allows functional imaging of individual regions of interest within the kidney parenchyma. BOLD imaging takes advantage of the paramagnetic properties of deoxyhemoglobin to generate images whose signal intensity reflects tissue O2. Deoxyhemoglobin generates local magnetic-field inhomogeneities leading to more rapid spin dephasing and a corresponding increase in “relaxivity,” defined as R2*. Oxygenated hemoglobin is diamagnetic and is devoid of these properties. The amount of deoxyhemoglobin functions as a biologic “contrast agent” and can be related to the strength of R2*-weighted pulse sequences. Studies comparing 1.5 and 3.0 Tesla magnetic fields in humans indicate that higher magnetic field strength increases the measurable R2* levels within deoxygenated regions of interest (ROIs), particularly in the medulla [15].
A set of parametric images of R2* is generated from the BOLD sequence data by fitting signal intensity data from each echo on a voxel-by-voxel basis to an exponential function describing the expected signal decay as a function of echo time (TE) and solving for the unknown value of R2*. Parametric maps of R2* over the image illustrate the R2* translation of renal structures. Typically, cortex can be identified by lower R2* values, with a gradient developing to higher R2* levels in the deeper medullary sections (Fig. 1). Parametric imaging makes the selection of ROIs more focused, especially when the deeper parts of the medulla are targeted, and allows the exclusion of adjacent artifacts from areas outside the kidneys. However, variable oxygenation within the kidney makes interpretation of BOLD MRI for specific regions difficult. The selection of ROIs using T2*-weighted gradient echo images sometimes depicts more gradual demarcation of deoxyhemoglobin than can be identified using iodinated contrast. The importance of reliably remaining within cortex and/or medulla for serial measurements highlights the importance of limiting the ROI to well-delineated, small segments in cortex and medulla, as we have proposed [15]. On the other hand, the use of parametric maps may direct ROI selection to areas with the most extreme hypoxia, under-sampling regions within the heterogeneous medulla that are better oxygenated (Fig. 1).
Placement of oxygen-sensing electrodes in experimental animals at various locations within the kidney confirms that tissue oxygen levels indeed follow the pattern of oxygenation gradients observed with BOLD MRI. Average levels of tissue oxygen tension in the mammalian kidney range from about 50 mm Hg in cortex to as low as 15 to 20 mm Hg in the deep sections of the medulla. These levels coincide with a range of hemoglobin oxygen saturation, which falls steeply from 85% to 15%, thereby making this a range favorable for detection using BOLD imaging. Results from experimental studies in rats [16••, 17] and other models [18, 19] using oxygen probes confirm that tissue oxygen levels fall by 45% to 50% in moving from cortex to deep medullary regions.
Examples of rapid changes of tissue oxygenation detectable using BOLD imaging include inhibition of solute transport with diuretics [13••] and prostaglandin inhibition, reduction in blood flow during acute vascular occlusion [19, 20], and models of ureteral obstruction [18]. Many of these studies suggest that alterations in oxygen consumption, rather than changes in blood flow, primarily determine medullary R2* levels. However, studies of local blood flow reduction using inhibitors of nitric oxide synthase in animals and humans illustrate that even small adjustments in blood flow have the potential to raise medullary R2* [16••]. Signal strength for T2* (the reciprocal of R2*) can be affected by other local factors influencing the homogeneity of magnetic dipole alignment, including the volume of tissue and blood (i.e., hemoglobin concentration) contained in a measured volume. Some authors question whether changes in deoxyhemoglobin precisely reflect tissue oxygenation status under all conditions [21].
Study of different oxygenation zones is of clinical relevance [6]. BOLD MRI studies in humans using 1.5 Tesla systems identified the potential to detect alterations within medulla and cortex after administration of nephrotoxic contrast [22], allograft injury [23], water loading, and occlusive renal arterial disease [24, 25••]. Several of these have been assumed to induce local “ischemia” related to reduced blood flow. Remarkably, conditions that also severely limit tubular metabolic activity, such as acute interstitial inflammation associated with transplant rejection, acute tubular necrosis, or even nonfunctioning renal atrophy beyond an occluded vessel, are associated with reduced oxygen consumption. Measured R2* levels (and thus deoxyhemoglobin) are low in these settings.
Acute Changes in Oxygenation
Serial studies in acute experimental models demonstrate a progressive rise in R2* in both cortex and medulla after stepwise reductions in renal blood flow when the kidney is otherwise functional [20]. Animal studies in a swine model show regional decreases in renal oxygenation during graded ARAS that also appear as changes in R2* signal under similar conditions [19]. Temporary renal artery occlusion in rats induces changes in medullary blood flow and reduction in R2* that persist 3 days after the event [26].
Important maneuvers that extend the utility of BOLD MRI include the use of measures that change renal solute transport and oxygenation, such as a water load or the use of diuretics. For example, increasing solute transport by increasing sodium intake in individuals with normal kidney function leads to a rise in oxygen consumption, thereby raising R2* [27••]. Therefore, beyond assessment of basal intrarenal oxygenation levels as reflected in R2*, the use of a functional challenge can provide insight regarding oxygen consumption related to tubular action. Not surprisingly, changes in R2* observed after administering furosemide are blunted in conditions associated with chronic and marked changes in renal function and structure [25••]. This simple maneuver may be useful to assess the viability of the kidney tissue without depending upon the absolute baseline level of R2*.
Can BOLD MRI Be Applied to Renovascular Disease and “Ischemic Nephropathy” in Humans?
Many experimental models of renovascular occlusion have identified a broad array of mechanisms associated with renovascular disease. These include activation of the renin-angiotensin-aldosterone system and numerous other pathways, including oxidative stress, sympatho-adrenergic activation, microvascular rarefaction, and inflammation [28]. As a rule, vascular occlusive disease develops slowly, unlike many experimental models that have used “clip hypertension.” Many of the pressor pathways can become activated at different times in the development of sustained hypertension and progressive vascular compromise [29•]. Rarely is the exact onset of critical renovascular disease known. Whether reductions in renal perfusion pressure truly induce hypoxia within the kidney parenchyma under these conditions has not been elucidated.
To examine tissue oxygenation in human atherosclerotic disease, our group studied individuals with unilateral ARAS using 3.0 Tesla BOLD MRI under carefully controlled conditions with a known, constant sodium intake of 150 mEq per day [25••]. Individual kidney blood flows within both the cortex and medulla were measured using contrast transit time and regional volumes measured by multidetector CT (MDCT). Initial studies were performed in patients with a reasonably preserved glomerular filtration rate (GFR) and serum creatinine less than 1.7 mg/dL and who otherwise met the entry criteria for the Cardiovascular Outcomes for Renal Atherosclerotic Lesions (CORAL) trial [5]. These individuals were compared with age-matched patients with essential hypertension undergoing the same protocol. All were taking agents that blocked the renin-angiotensin system. These studies confirmed that high-grade ARAS sufficient to activate the renin-angiotensin system does lead to decreases in kidney size and blood flow, as well as GFR in the affected kidney. Surprisingly, average levels of cortical and medullary R2* in these ARAS patients did not differ from those of the patients with essential hypertension or from those of the contralateral, normally perfused kidney (Table 2) [25••]. Despite major reductions in blood flow in the stenotic kidneys sufficient to elevate levels of plasma renin activity, levels of R2* were similar to those in the other patients, indicating preservation of the cortical and medullary oxygenation in these patients with preserved kidney function treated with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers. The response to furosemide administration of medullary R2* in the stenotic kidney was blunted, however, compared with the contralateral kidneys, suggesting that oxygen consumption in the stenotic kidney was less a function of solute transport than it was in the contralateral kidney under these conditions. In fact, it is likely that the contralateral kidney increases its metabolic activity as it assumes some of the filtration and reabsorption functions of the stenotic kidney. We interpret these data to indicate a remarkable intrarenal adaptation in the stenotic kidney that effectively preserves oxygenation balance between cortex and medulla despite reduced blood flow and GFR. One explanation is that the fall in GFR reduces solute filtration, thereby reducing the “workload” and metabolic energy requirements in deeper medullary tissues. Our results are consistent with similar findings in a chronic rat model of mild renovascular hypertension, which demonstrated no evident drop-off in “downstream” renal oxygen content as measured by BOLD MRI using 7.0 Tesla magnetic field strength [30]. These observations indicate that differences between acute and chronic vascular compromise reflect compensatory alterations that circumvent true tissue hypoxia for many patients with chronic conditions. Our results provide one explanation for the clinical trial data indicating that patients with ARAS often can be treated using medical therapy alone for many years without worsening renal function [5].
The capacity for circulatory adaptation within the kidney obviously has limits, however. We recently extended our human studies using MDCT and BOLD MRI to patients with more advanced reductions in blood flow, associated with duplex ultrasound above 384 cm/sec and severe cortical atrophy. In these cases, both kidney volume and tissue perfusion in the cortical regions were reduced. Levels of cortical R2* were elevated in this group, suggesting that overt tissue hypoxia was developing. Deep medullary regions continued to have high R2* levels that did not differ from those in otherwise normal kidneys. The extent of medullary tissue with elevated R2* levels was considerably wider, however. These observations are supported by studies using other estimates of cortical perfusion and dimensions [19]. In patients like these, the capacity for changes in volume and filtration to compensate for reduced blood flow appear to have been overwhelmed, leading to cortical hypoxia. These data agree closely with other studies indicating that loss of cortical thickness is a marker of more severe occlusive disease [31•]. Our results demonstrate for the first time that significant kidney damage in humans with ARAS is characterized by localized hypoxia in vivo. Preliminary data using transvenous biopsies from stenotic kidneys indicate that the degree of tissue injury is closely related to the severity of blood flow decrements in such patients [32].
The mechanisms underlying the development of genuine hypoxia in severely damaged kidneys appear to be the loss of intrarenal vessels and the development of scarring. Experimental studies in a swine model of ARAS indicate that extensive reductions in renal blood flow eventually produce microvascular rarefaction in the post-stenotic kidney that is associated with the loss of renal function [33]. These changes produce major disturbances in local cytokines, activation of inflammatory pathways within the affected kidney, and eventual parenchymal fibrosis—changes that are accelerated by an atherogenic milieu. Importantly, studies in the swine model demonstrate that restoration of vascular patency alone does not reverse the process of fibrogenesis [34••, 35], an observation that is substantiated by similar observations in series of human patients treated with renal revascularization. Recent studies from our laboratory and others suggest that implementation of other (or adjunctive) measures such as infusion of endothelial progenitor cells may facilitate repair or prevention of renal parenchymal injury [36••].
Conclusions
How do these data mesh with clinical evaluation of patients with ARAS? We propose that BOLD MRI may provide major improvements in two domains: (1) BOLD studies can identify individuals for whom an observed vascular occlusion has not produced major disruption of tissue oxygenation or specific disruption of cortical to medullary oxygenation gradients. This finding may support ongoing management without apparent risk of “hypoxic” injury, or at least may indicate that the post-stenotic kidney is well preserved from an oxygenation perspective. (2) A kidney in which BOLD imaging demonstrates cortical hypoxia and/or widening medullary regions of hypoxia likely is at risk for additional tissue injury. We propose using the results of BOLD MRI as real-time markers of local blood flow and oxygenation reserve, to more rationally identify individuals who may truly benefit from renal revascularization and/or adjunctive maneuvers to repair kidneys that are at imminent risk of demise.
It is premature to indicate that these goals will be met. Our interest in the development of improved tools to identify kidneys at risk from vascular injury coincides with recent trials that challenge the widespread benefit of renal revascularization. We believe that one outcome of these ambiguous trials will be a tendency to investigate fewer patients early in the course of renovascular disease, leading to the likely appearance in the future of more patients with vascular occlusion that is much further established. Experienced clinicians already encounter patients in whom total renal occlusion has rendered one kidney—and sometimes both kidneys—beyond salvage. We hope that further studies using tools such as BOLD MRI will allow more precise identification of patients before irreversible kidney injury occurs.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• de Mast Q, Beutler JJ. The prevalence of atherosclerotic renal artery stenosis in risk groups: a systematic literature review. J Hypertens 2009; 27:1333–1340. This useful assembly of previous publications identifies ARAS as part of imaging for other indications. Of patients with suspected “renovascular hypertension,” 14.1% were positive for ARAS. Use of a low threshold (50% occlusion) and single publications regarding end-stage renal disease and congestive heart failure have overstated the risk.
Hackam DG, Duong-Hua ML, Mamdani M, Li P, Tobe SW, Spence JD, et al. Angiotensin inhibition in renovascular disease: a population-based cohort study. Am Heart J. 2008;156:549–55.
•• The ASTRAL Investigators. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962. This important but limited study selected 806 subjects by “uncertainty” as to whether they would benefit from revascularization as compared with medical therapy alone. Remarkably stable levels of kidney function overall did not differ between groups. These data emphasize the success and stability of medical therapy alone for many patients with ARAS for several years. More than 40% of the patients were in the category of 50% to 70% stenosis, which likely diluted the power of this trial.
Bax L, Woittiez AJ, Kouwenberg HJ, Mali PTM, Buskens E, Beek FJA, et al. Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function. Ann Intern Med. 2009;150:840–8.
Textor SC, McKusick MA, Misra S, Glockner J. Timing and selection for renal revascularization in an era of negative trials: what to do? Prog Cardiovasc Dis. 2009;52:220–8.
Heyman SN, Khamaisi M, Rosen S, Rosenberger C. Renal parenchymal hypoxia, hypoxia response and the progression of chronic kidney disease. Am J Nephrol. 2008;28:998–1006.
•• O’Connor PM, Evans RG. Structural antioxidant defense mechanisms in the mammalian and nonmammalian kidney: different solutions to the same problem? Am J Physiol Regul Integr Comp Physiol 2010; 299:R723-R727. This is a thoughtful reference regarding complex intrarenal mechanisms to protect oxygenation at both levels—avoiding hyperoxia and hypoxia by using unique characteristics within the kidney.
• Evans RG, Gardiner BS, Smith DW, O’Connor PM. Intrarenal oxygenation: unique challenges and the biophysical basis of homeostasis. Am J Physiol Renal Physiol 2008; 295:F1259-F1270. This important review of pathways of oxygenation regulation within the kidney supports the multiple roles of blood flow, oxygen consumption, and arteriovenous shunting.
Textor SC, Lerman L. Renovascular hypertension and ischemic nephropathy: state of the art. Am J Hypertens. 2010;23:1159–69.
Evans RG, Eppel GA, Michaels S, Burke SL, Nematbakhsh M, Head GA, et al. Multiple mechanisms act to maintain kidney oxygenation during renal ischemia in anesthetized rabbits. Am J Physiol Renal Physiol. 2010;298:F1235–43.
O’Connor PM, Kett MM, Warwick RP, Evans RG. Renal medullary tissue oxygenation is dependent on both cortical and medullary blood flow. Am J Physiol Renal Physiol. 2006;290:F688–94.
Sadowski J, Badzynska B. Specific features and roles of renal circulation: angiotensin II revisited. J Physiol and Pharmacol. 2006;57 Suppl 11:169–78.
•• Warner L, Glockner JF, Woollard J, Textor SC, Romero JC, Lerman LO. Determinations of renal cortical and medullary oxygenation using blood oxygen level-dependent magnetic resonance imaging and selective diuretics. Invest Radiol 2011; 46:41–47. This study of changes in tissue oxygenation after blockade of solute transport with furosemide, acetazolamide, or both emphasizes the role of oxygen consumption as a key determinant of medullary oxygenation in the normal kidney.
Gomez SI, Warner L, Haas JA, Bolterman RJ, Textor SC, Lerman LO, et al. Increased hypoxia and reduced renal tubular response to furosemide detected by BOLD magnetic resonance imaging in swine renovascular hypertension. Am J Physiol Renal Physiol. 2009;297:F981–6.
Gloviczki ML, Glockner J, Gomez SI, Romero JC, Lerman LO, McKusick M, et al. Comparison of 1.5 and 3 T BOLD MR to study oxygenation of kidney cortex and medulla in human renovascular disease. Invest Radiol. 2009;44:566–71.
•• Li L, Lin J, Santos EA, Dunkle E, Pierchala L, Prasad P. Effect of nitric oxide synthase inhibition on intrarenal oxygenation as evaluated by blood oxygenation level-dependent magnetic resonance imaging. Invest Radiol 2009; 44:67–73. This important study demonstrates that a rise in R2* in medullary regions may be induced by alterations in medullary blood flow, in addition to changes in oxygen consumption.
Palm F, Connors SG, Mendonca M, Welch WJ, Wilcox CS. Angiotensin II type 2 receptors and nitric oxide sustain oxygenation in the clipped kidney of early Goldblatt hypertensive rats. Hypertension. 2008;51:1–7.
Pedersen M, Dissing TH, Morkenborg J, Stodkilde-Jorgensen H, Hansen LH, Pedersen LB, et al. Validation of quantitative BOLD MRI measurements in the kidney: application to unilateral ureteral obstruction. Kidney Int. 2005;67:2305–12.
Warner L, Gomez SI, Bolterman R, Haas JA, Bentley MD, Lerman LO, et al. Regional decrease in renal oxygenation during graded acute renal arterial stenosis: a case for renal ischemia. Am J Physiol Regul Integr Comp Physiol. 2009;296:R67–71.
Juillard L, Lerman LO, Kruger DG, Haas JA, Rucker BC, Polzin JA, et al. Blood oxygen level-dependent measurement of acute intra-renal ischemia. Kidney Int. 2004;65:944–50.
Evans RG, Leong CL, Anderson WP, O’Connor PM. Don’t be so BOLD: potential limitations in the use of BOLD MRI for studies of renal oxygenation. Kidney Int. 2007;71:1327–8.
Hofmann L, Simon-Zoula S, Nowak A, Giger A, Vock P, Boesch C, et al. BOLD-MRI for the assessment of renal oxygenation in humans: acute effect of nephrotoxic xenobiotics. Kidney Int. 2006;70:144–50.
Djamali A, Sadowski EA, Muehrer RJ, Reese S, Smavatkul C, Vidyasagar A, et al. BOLD-MRI assessment of intrarenal oxygenation and oxidative stress in patients with chronic kidney allograft dysfunction. Am J Physiol Renal Physiol. 2007;292:F513–22.
Textor SC, Glockner JF, Lerman LO, Misra S, McKusick MA, Riederer SJ, et al. The use of magnetic resonance to evaluate tissue oxygenation in renal artery stenosis. J Am Soc Nephrol. 2008;19:780–8.
•• Gloviczki ML, Glockner JF, Lerman LO, McKusick MA, Misra S, Grande JP, et al. Preserved oxygenation despite reduced blood flow in poststenotic kidneys in human atherosclerotic renal artery stenosis. Hypertension 2010; 55:961–966. Data from carefully conducted studies in humans with ARAS apply BOLD MRI to demonstrate well-preserved cortical and medullary oxygen levels despite substantial reduction in blood flow and kidney size for most individuals. These observations partly explain the stability of kidney function in patients treated medically in the ASTRAL trial.
Pedersen M, Lausten C, Perot V, Basseau F, Moonen C, Grenier N. Renal hemodynamics and oxygenation in transient renal artery occluded rats evaluated with iron-oxide particles and oxygenation-sensitive imaging. Z Med Phys. 2010;20:134–42.
•• Pruijm M, Hofmann L, Maillard M, Tremblay S, Glatz N, Wuerzner G, et al. Effect of sodium loading/depletion on renal oxygenation and young normotensive and hypertensive men. Hypertension 2010; 55:1116–1122. This important study used repeated BOLD MRI studies to identify effects of changing sodium intake. The results demonstrated a major fall in R2* in medullary segments during low sodium intake, consistent with reduced medullary oxygen consumption related to reduced sodium transport.
Lerman LO, Textor SC, Grande JP. The mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis. 2009;52:196–203.
• Cheng J, Zhou W, Warner GM, Knudsen BE, Garovic VD, Gray CE, et al. Temporal analysis of signaling pathways activated in a murine model of two-kidney, one-clip hypertension. Am J Physiol Renal Physiol 2009; 297:F1055-F1068. This study used a murine model of renal artery stenosis to demonstrate a sequential increase of p-ERK pathways, proliferating cell markers, and transforming growth factor-beta (TGF-β) in poststenotic kidneys preceding the development of interstitial fibrosis and irreversible tissue injury.
Rognant N, Guebre-Egziabher F, Bachetta J, Janier M, Hiba B, Langlois JB, et al. Evolution of renal oxygen content measured by BOLD MRI downstream in a chronic renal artery stenosis. Nephrol Dial Transplant. 2011;26:1205–10.
• Cheung CM, Chrysochou C, Shurrab AE, Buckley DL, Cowie A, Kalra PA. Effects of renal volume and single-kidney glomerular filtration rate on renal functional outcome in atherosclerotic renal artery stenosis. Nephrol Dial Transplant 2010; 25:1133–1140. This interesting study combines measurement of preserved kidney volume (by MR) and single-kidney GFR (by radioisotope scanning). These authors demonstrate greater likelihood of recovering GFR after revascularizing the kidney when a discrepancy is observed between preserved volume and reduced GFR, suggesting “hibernating” kidney function in renal artery stenosis.
Gloviczki ML, Keddis MT, McKusick MA, Misra S, Grande JP, Lerman LO, et al. Transvenous biopsies of kidneys beyond renal artery stenosis demonstrate fibrosis correlated to reduced single kidney blood flow, but not urinary or blood TGF-beta levels [abstract]. American Society of Nephrology 2010; abstract F-P01703
Zhu XY, Chade AR, Rodriguez-Porcel M, Bentley MD, Ritman EL, Lerman A, et al. Cortical microvascular remodeling in the stenotic kidney: role of increased oxidative stress. Arterioscler Thromb Vasc Biol. 2004;24:1854–9.
•• Favreau F, Zhu XY, Krier JD, Lin J, Warner L, Textor SC, Lerman LO. Revascularization of swine renal artery stenosis improves renal function but not the changes in vascular structure. Kidney Int 2010; 78:1110–1118. This important paper identifies a failure of microvascular structures and angiogenic cytokines to normalize despite restoration of patent arteries in experimental renal artery stenosis.
Eirin A, Zhu XY, Urbieta-Caceres VH, Grande JP, Lerman A, Textor SC, et al. Persistent kidney dysfunction in swine renal artery stenosis correlates with outer cortical microvascular remodeling. Am J Physiol Renal Physiol. 2011;300:F1394–1401.
•• Chade AR, Zhu X, Lavi R, Krier JD, Pislaru S, Simari RD, et al. Endothelial progenitor cells restore renal function in chronic experimental renovascular disease. Circulation 2009; 119:547–557. This important experimental study demonstrates the potential for circulating pluripotent progenitor cells to shape the recovery of viable blood vessels, improve filtration, and restore tubular function in kidneys that are undergoing vascular rarefaction beyond a renal artery stenosis. These data provide a rationale for considering stem cells as an adjunctive maneuver in recovering kidney structure in ischemic renal disease.
Welch WJ, Blau J, Xie H, Chabrashvili T, Wilcox CS. Angiotensin-induced defects in renal oxygenation: role of oxidative stress. Am J Physiol Heart Circ Physiol. 2005;288:H22–8.
Lubbers DW, Baumgartl H. Heterogeneities and profiles of oxygen tension in brain and kidney as examples of PO2 distribution in the living tissue. Kidney Int. 1997;51:372–80.
Prasad P, Li LP, Halter SH, Cabray J, Ye M, Battle D. Evaluation of renal hypoxia in diabetic mice by BOLD MRI. Invest Radiol. 2010;45:819–22.
Li L, Vu AT, Li BSY, Dunkle E, Prasad PV. Evaluation of intrarenal oxygenation by BOLD MRI at 3.0 T. J Magn Reson Imaging. 2004;20:901–4.
Tumkur SM, Vu AT, Li LP, Pierchala L, Prasad PV. Evaluation of intra-renal oxygenation during water diuresis: a time-resolved study using BOLD MRI. Kidney Int. 2006;70:139–43.
Acknowledgments
The projects described were supported by Award Number PO1HL85307 from the National Heart, Lung and Blood Institute and NIH/NCRR CTSA Grant Number UL1 RR024150. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gloviczki, M.L., Lerman, L.O. & Textor, S.C. Blood Oxygen Level–Dependent (BOLD) MRI in Renovascular Hypertension. Curr Hypertens Rep 13, 370–377 (2011). https://doi.org/10.1007/s11906-011-0218-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-011-0218-7