
Abstract
Purpose of Review
Patients with relapsed T cell acute lymphoblastic leukemia (T-ALL) have limited therapeutic options and a poor prognosis. Although a variety of salvage chemotherapy regimens may be used, response rates are unsatisfactory. This article summarizes current approaches and promising emerging strategies for the treatment of relapsed T-ALL.
Recent Findings
Although nelarabine is the only agent approved specifically for T-ALL, recent studies have identified a variety of genetic alterations and signaling pathways that are critical in its pathogenesis. Based on these findings, a number of small-molecule inhibitors and other targeted therapies are being studied for relapsed T-ALL, including gamma-secretase inhibitors, BCL-2 inhibitors, cyclin-dependent kinase inhibitors, and mTOR inhibitors. In addition, pre-clinical studies of chimeric antigen receptor T cells targeting CD5 and CD7 as well as the monoclonal antibody daratumumab have shown promising results for T-ALL.
Summary
Relapsed T-ALL currently remains challenging to treat, but recent pre-clinical studies of targeted and immunotherapeutic agents have shown encouraging results. A number of clinical trials investigating these approaches for T-ALL are currently underway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
T cell acute lymphoblastic leukemia (T-ALL) accounts for approximately 15% of ALL cases and is twice as common in males than females [1]. Historically, newly diagnosed T-ALL was associated with substantially worse outcomes compared to B cell ALL (B-ALL), but this is no longer true with modern chemotherapy regimens. Although children with T-ALL continue to experience slightly worse survival than those with B-ALL, the gap has narrowed significantly [2, 3]. In adults, several recent studies have shown equivalent or even superior survival in patients with T-ALL as compared to B-ALL. Among adults with newly diagnosed ALL treated on the Eastern Cooperative Oncology Group (ECOG) E2993/Medical Research Council (MRC) UKALL12 trial, the rate of complete remission (CR) for T-ALL and B-ALL was equivalent (94% vs. 93%; p = 0.5), and there was a trend toward improved 5-year overall survival (OS) in the patients with T-ALL (48% vs. 42%; p = 0.07) [4].
However, for those patients with T-ALL who do experience disease relapse or are refractory to induction therapy, outcomes remain poor. Unlike the treatment of relapsed B-ALL, which has been revolutionized by the recent approvals of blinatumomab, inotuzumab ozogamicin, and chimeric antigen receptor (CAR) T cell therapy, there have been no new agents specifically approved for relapsed T cell ALL since nelarabine was approved in 2005. Important progress has been made, however, in understanding the unique genetics and biology of T-ALL which will hopefully lead to improvements in therapeutic options for these patients. In this article, we review the outcomes and current treatment options for patients with relapsed T-ALL and discuss promising new approaches to therapy, including immunotherapeutic and targeted approaches.
Risk of Relapse
In general, adults with T-ALL continue to have significant rates of relapse, although better outcomes have been reported recently in younger adults treated on pediatric-inspired protocols [5]. For adults with T-ALL treated on the E2993/UKALL12 study who achieved a CR, the incidence of relapse at 5 years was 42% (95% confidence interval (CI), 36–47%) and the 5-year OS was 48% (95% CI, 42–53%) [4]. In addition to age, other factors that have been reported to increase the risk of relapse in patients with T-ALL include central nervous system (CNS) involvement, an initial white blood cell count > 100 K, a complex karyotype (≥ 5 cytogenetic abnormalities), and CD1a-negativity, among others, although the relative impact of these factors has been inconsistent across studies [4, 6]. Some studies have reported improved outcomes in patients with NOTCH1 or FBXW7 mutations, while the presence of a mutation in NRAS, KRAS, or PTEN has been associated with a higher cumulative incidence of relapse, but again these results have not been consistent [7••].
With current chemotherapy regimens, measurable residual disease (MRD) has emerged as the most important factor in predicting relapse [7••, 8, 9]. In a study of 464 children with T-ALL treated in the AIEOP-BFM ALL 2000 study, MRD was measured by polymerase chain reaction (PCR) 33 and 78 days after the initiation of induction therapy [8]. Patients were stratified into three groups based on MRD response: standard risk (MRD-negative at both time points), intermediate risk (MRD-positive at one time point), or high risk (MRD-positive at day 78 at a level greater than 10−3) [8]. Seven-year EFS was 91.1% vs. 80.6% vs. 49.8% in the three groups, respectively (p < 0.001) [8]. Another study in adults with ALL who were treated in the Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL)-2003 and GRAALL-2005 trials evaluated MRD by PCR for T cell receptor gene rearrangement 6 weeks after starting induction therapy. Of the 126 T-ALL patients studied, a higher hazard ratio (HR) for cumulative incidence of relapse was associated with MRD levels ≥ 10−4 (HR 2.93, 95% CI 1.5–5.71; p = 0.002) [7••]. As more data become available, the role of MRD in the treatment of T-ALL is likely to continue to evolve.
Because of the poor outcomes in patients with relapsed T-ALL, a lot of research efforts have focused on improving initial induction and/or consolidation regimens in order to reduce the risk of relapse. The incorporation of polyethylene glycol-conjugated (PEG)-asparaginase or dexamethasone (instead of prednisone) in frontline regimens has been reported to decrease the risk of relapse in T-ALL [10•, 11,12,13,14,15]. In addition, the AALL0434 study for children and young adults with T-ALL recently showed that Capizzi-style methotrexate dosing was associated with superior 5-year OS and DFS and a decreased risk of relapse (in the CNS as well as marrow) in comparison to high-dose methotrexate [3]. Given the activity of nelarabine for relapsed T-ALL, it has also been studied in combination with chemotherapy for newly diagnosed T-ALL [16,17,18]. In addition to the methotrexate randomization, the AALL0434 study also randomized patients with T-ALL to receive or not receive nelarabine in addition to chemotherapy. Four-year disease-free survival (DFS) was improved in the group that received the nelarabine (88.9% vs. 83.3%, p = 0.03) [19•]. The ongoing UKALL14 trial (NCT01085617) is evaluating the benefit of nelarabine in addition to standard chemotherapy in adults with newly diagnosed T-ALL.
Early T Cell Precursor ALL
Early T cell precursor (ETP) ALL was recognized as a new provisional entity in the 2016 update to the World Health Organization classification of acute leukemia and is characterized by a unique immunophenotypic and genetic profile [20, 21]. ETP-ALL expresses CD7, does not express CD1a or CD8, and expresses at least one myeloid or stem cell marker (CD34, CD117, HLA-DR, CD13, CD33, CD11b, or CD65) [20]. CD5 may be either weakly expressed or negative [20]. ETP-ALL was initially identified as a distinct subtype of T-ALL in an analysis of children who were enrolled in clinical trials at St. Jude Children’s Research Hospital between 1992 and 2006. Children with an ETP immunophenotype had a significantly worse 10-year OS (19%; 95% CI 0–92%) than children with non-ETP T-ALL (84%; 95% CI 72–96%) (p < 0.001), as well as a higher risk of induction failure and relapse [22].
However, more recent studies have not found such a significant difference in outcome or relapse risk in patients with ETP-ALL [23, 24]. Patrick et al. analyzed the outcomes of children and young adults with T-ALL who were treated in the UKALL 2003 study [23]. Although there was a trend toward a higher relapse rate (18.6% vs. 9.6%, p = 0.10) and a worse 5-year OS (82.4% vs. 90.9%) in patients with ETP-ALL compared to those with non-ETP-ALL, these differences were not statistically significant [23]. In the Children’s Oncology Group (COG) AALL0434 study, which included 1144 children with T-ALL, the patients with ETP-ALL had higher rates of induction failure than those with non-ETP-ALL (7.8% vs. 1.1%, p < 0.0001), but there was no difference in 5-year EFS (87.0% vs. 86.9%) or OS (93.0% vs. 92.0%) [24].
Treatment of Relapsed T-ALL
Despite the improvements in outcomes that have been observed in patients with newly diagnosed T-ALL, survival remains quite poor for those patients who relapse. Among 123 adult patients with T-ALL treated on the E2993/UKALL12 study who relapsed, only 8 of 123 patients (6.5%) were alive after a median of 5.2 years of follow-up [4]. A small retrospective analysis of children with relapsed ALL who were enrolled in trials of the Austrian Berlin-Frankfurt-Munster Study Group also showed poor outcomes for patients with relapsed T-ALL with a 10-year OS of 21% ± 8% [25]. Ten-year OS was significantly worse for patients with relapsed T-ALL than B-ALL [25].
Although nelarabine and liposomal vincristine are both approved for the treatment of relapsed T-ALL, there is no single standard of care salvage chemotherapy regimen used in this setting. Desjonqueres et al. retrospectively analyzed the salvage chemotherapy regimens selected and treatment outcomes for adults with ALL who relapsed after treatment on the GRAALL-2003 and GRAALL-2005 protocols [26•]. The patients were treated with a wide variety of ALL regimens, including hyper-CVAD (cycles of cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with cycles of high-dose methotrexate), pediatric-inspired regimens, other anthracycline-based or methotrexate-based regimens, and nelarabine, in addition to lower intensity regimens (primarily vincristine and steroids) [26•]. Notably, 56% of the patients with relapsed T-ALL did achieve a second remission with salvage chemotherapy [26•]. A longer duration of first remission (≥ 18 months) and a younger age (≤ 45 years) were associated with an increased likelihood of achieving a second remission (p = 0.009). Potential benefit of a salvage chemotherapy regimen therefore depends on both patient-specific and disease-related factors, including duration of first remission. If a second remission is achieved and the patient is a candidate for allogeneic hematopoietic stem cell transplantation (HSCT), transplant seems to improve outcomes [27].
Nelarabine
Nelarabine is a pro-drug that is rapidly converted to the derivative of deoxyguanosine 9-b-d-arabinofuranosylguanine (ara-G) after intravenous administration [28, 29]. Deoxyguanosine analogues were originally studied as a treatment for T-ALL based on the clinical observation that patients with a congenital deficiency of purine nucleoside phosphorylase have a severe T cell immune deficiency as a result of the buildup of deoxyguanine triphosphate, which is selectively cytotoxic to T cells [28, 30,31,32]. Nelarabine was granted accelerated approval by the U.S. Food and Drug Administration (FDA) in 2005 for the treatment of relapsed and/or refractory T-ALL in adults and children who have received at least two prior lines of chemotherapy, and today it remains the only agent that is approved specifically for relapsed T-ALL [33].
The approval of nelarabine was based on several studies done in children and adults demonstrating efficacy as monotherapy. The phase II COG study P9673 enrolled children with relapsed and/or refractory T cell ALL or non-Hodgkin lymphoma [34]. Of the 94 patients with T-ALL treated with nelarabine, 28 (29.8%) achieved a CR [34]. Following two dose de-escalations due to neurotoxicity, this study established a dose of 650 mg/m2/day for 5 days as the optimal dose for children [34]. The Cancer and Leukemia Group B study 19801 included 39 adults with relapsed and/or refractory T-ALL who were treated with nelarabine at a dose of 1.5 g/m2/day on days 1, 3, and 5 [35]. The CR rate was 31% (95% CI, 17–48%) and the 1-year OS rate was 28% (95% CI, 15–43%) [35]. These results were confirmed in a larger phase II study by the German Multicenter Study Group for Adult ALL (GMALL) which included 126 evaluable adults with relapsed and/or refractory T-ALL [36]. In this study, 45 of 126 patients (36%) achieved a CR and the 1-year OS was 24% [36].
Of note, the FDA label for nelarabine includes a boxed warning regarding the risk of severe neurotoxicity, which was the dose-limiting toxicity in the early-phase studies [32]. Although the neurologic events are typically reversible with cessation of the drug, this is not always the case and treatment-related deaths due to neurotoxicity have been reported. In the GMALL study, 16% of patients overall experienced neurotoxicity (any grade), although only 7% of patients had grade 3 or 4 neurotoxicity [36]. The most common neurologic side effects included dizziness (6%), confusion (4%), and mood alterations (6%) [36]. Seizures, ataxia, coma, peripheral neuropathies, and a Guillan-Barre-like syndrome have also been reported with nelarabine [34, 35]. The primary non-neurologic toxicities of nelarabine are hematologic (neutropenia, anemia, and thrombocytopenia) [35, 36].
Two small retrospective series have also reported the outcomes of nelarabine in combination with cyclophosphamide and etoposide for relapsed and/or refractory T-ALL [37, 38]. In a study of 7 children with relapsed and/or refractory T-ALL who were treated sequentially with nelarabine (650 mg/m2/day × 5 days) and etoposide (100 mg/m2 ×5 days)/cyclophosphamide (440 mg/m2/day × 5 days), 5 of 7 patients achieved CR after 1 to 2 cycles [37]. Nearly all subjects experienced neurotoxicity, primarily grade 2–3 sensory and motor neuropathies, although in most cases this was reversible [37]. A similar analysis of 5 adults treated with the same regimen found that 3 of 5 patients (60%) achieved CR after 1–2 cycles, and two of these patients were successfully bridged to allogeneic HSCT [38]. Notably, however, there were 2 treatment-related deaths, including 1 patient who died from sepsis on day 28 and another patient who developed progressive neuromuscular weakness that led to respiratory failure on day 44, which was attributed to nelarabine-induced neurotoxicity [38]. This patient had received intrathecal chemotherapy in combination with the nelarabine, so the authors suggested caution and consideration for separating nelarabine and intrathecal chemotherapy in future studies [38]. Overall, these studies demonstrate the efficacy of nelarabine monotherapy and provide a rationale to further study nelarabine in combination with chemotherapy for relapsed T-ALL.
Liposomal Vincristine
Liposomal vincristine sulfate (Marqibo) was approved by the U.S. FDA in 2012 for the treatment of adults with T cell or Philadelphia chromosome (Ph)-negative B cell ALL in second or greater relapse or with disease progression following at least 2 prior lines of treatment and is given weekly by intravenous infusion at a dose of 2.25 mg/m2 [39]. The encapsulation of vincristine in liposomal nanoparticles comprised of sphingomyelin and cholesterol has been shown to increase the half-life and area under the curve of vincristine, allowing for a higher concentration of drug in the blood compartment and target organs (bone marrow, lymph nodes, and spleen) without increasing toxicity compared to standard vincristine sulfate [40,41,42]. The FDA approval was based on a phase II trial of liposomal vincristine monotherapy in adults with relapsed Ph-negative ALL [43]. Overall, 11% (7/65) of patients treated achieved CR and an additional 9% (6/65) achieved CR with incomplete count recovery (CRi) [43]. The overall response rate was 35% (23/65). Of note, only 10 patients with T-ALL were included in this trial, of which 2 (20%) achieved either CR or CRi. The toxicity profile and rates of adverse events were similar to standard vincristine sulfate, despite the longer half-life and higher drug levels that are achieved with liposomal vincristine. Although 29% of subjects experienced peripheral neuropathy, only 6% met the criteria for this to be considered a serious adverse event. Other serious adverse events observed with liposomal vincristine included constipation (3%), tumor lysis syndrome (5%), and neutropenic fever (5%) [43]. A subsequent small phase I trial in children with relapsed and/or refractory acute leukemia and solid tumors demonstrated that children are able to safely tolerate the same dose of liposomal vincristine as adults, although it is not currently approved for use in children [44].
T-ALL Pathogenesis
Given the limited options for therapy in patients with relapsed T-ALL, pre-clinical research has focused on understanding biological mechanisms of T-ALL with the goal of identifying targetable signaling pathways and/or genetic lesions [45]. Since the discovery of activating mutations in NOTCH1 in a majority of T-ALL cases 15 years ago, considerable progress has been made in understanding the tremendous diversity of genetic alterations that contribute to T-ALL pathogenesis [45, 46].
Notch Signaling
Notch signaling is an evolutionarily conserved pathway with important roles in normal T cell development, hematopoiesis, and cell growth and proliferation [47, 48]. In normal biology, four Notch genes (NOTCH1–4) encode receptors that, following interaction with their ligand, undergo a series of cleavages by an ADAM metalloprotease and then the gamma-secretase complex [49]. This leads to the release of the intracellular part of the Notch protein, which subsequently translocates to the nucleus and activates transcription of a variety of genes [49]. Over 60% of T-ALL cases harbor mutations in NOTCH1 which cause the constitutive activation of Notch signaling [46, 47, 50••]. In rare cases, t(7;9)(q34;q34.3) chromosomal translocations involving NOTCH1 also occur in T-ALL [46]. Mutations in the ubiquitin ligase FBWX7 also occur in approximately 15% of T-ALL [51]. When FBWX7 is mutated, the proteasomal degradation of active (intracellular) NOTCH1 is inhibited, leading to increased Notch signaling [51]. Of note, the loss of CDKN2A, which encodes the tumor suppressors p16INK4A and p14ARF, often co-occurs with NOTCH1 mutations in T-ALL [45]. Overall, Notch signaling plays an important role in the pathogenesis of a majority of T-ALL and leads to the activation of a number of downstream effectors, including Myc [47].
Genetic Heterogeneity of T-ALL
In a study by Liu et al., samples from 264 children and young adults with T-ALL were analyzed by whole-exome DNA sequencing, RNA sequencing, and single-nucleotide polymorphism microarray-based genotyping [50••]. Driver mutations were identified in 106 genes [50••]. In addition to mutations in genes that were already known to occur in T-ALL, such as NOTCH1, FBXW7, and PTEN, the authors identified 39 recurrently mutated genes that had not previously been described [50••]. Additionally, 255 gene rearrangements leading to the formation of 83 fusion proteins were identified [50••]. The genes most commonly involved in these chromosomal translocations included MLLT10, KMT2A, ABL1, and NUP98 [50••]. By integrating these analyses, the authors identified the following 10 categories of pathways which are frequently aberrant in T-ALL, in descending order of frequency: transcriptional regulation, cell cycle regulation and/or tumor suppression, Notch1 signaling, epigenetic regulation, PI3K-AKT-mTOR, JAK-STAT, Ras signaling, ribosomal function, ubiquitination, and RNA processing [50••]. Another group compared the genetics of ETP-ALL to non-ETP T-ALL and found that ETP-ALL is genetically distinct and is characterized by genes regulating cytokine receptor signaling or Ras signaling, hematopoietic development, or histone modifications [21]. Together, these data highlight the substantial genetic heterogeneity in T-ALL and suggest that an approach of targeting different pathways in different subgroups of patients with T-ALL may lead to improved outcomes.
Targeting Notch
Several strategies to inhibit Notch signaling have been investigated for T-ALL, most notably gamma-secretase inhibitors (GSIs). GSIs prevent the cleavage of Notch from the cellular membrane, thereby preventing translocation to the nucleus and Notch-mediated transcriptional activation [47]. In vitro studies demonstrated that GSIs inhibit cell growth and induce apoptosis in certain T-ALL cell lines, particularly in combination with glucocorticoids [52,53,54, 55]. Despite promising pre-clinical data, early-phase clinical trials of GSIs for relapsed T-ALL were disappointing due to both limited anti-leukemic effects and systemic toxicity [56,57,58]. Dose escalation of GSIs in phase I studies was limited by systemic side effects, namely gastrointestinal toxicity [56,57,58]. It is possible that these side effects prevented dose escalation to levels that sufficiently inhibit Notch, which may be why only occasional clinical responses were observed [56,57,58]. Several mechanisms of resistance to GSIs have also been reported, including loss of PTEN, Notch-independent Myc activation, and epigenetic alterations [47, 59, 60].
Current research aims to identify combination therapies that prevent or overcome resistance to GSIs, inhibit downstream effectors of Notch signaling, and improve the specificity of agents targeting mutant Notch1. Using a systems biology “virtual screening” approach, Sanchez-Martin et al. identified withaferin A, rapamycin, and vorinostat as having synergistic activity in combination with GSIs in vitro [61•]. Pre-clinical studies have also suggested that inhibiting downstream effectors of Notch such as the sarco/endoplasmic reticulum calcium ATPase (SERCA) channels, the coregulator Zmiz1, or c-Myc might inhibit Notch signaling without the systemic side effects that have been associated with GSIs [62,63,64]. Other approaches that have been studied in the pre-clinical setting include selective antibodies targeting individual Notch receptors and direct inhibitors of the Notch transcription factor complex [65,66,67].
Immunotherapeutic Approaches
The development of cellular and antibody-based therapies for T-ALL has lagged significantly behind B-ALL, for which an antibody-drug conjugate (inozutumab ozogamicin), a bispecific T cell engager (blinatumomab), and a CAR T cell therapy (tisagenlecleucel) have all been approved within the past few years. In the case of T-ALL, it has been challenging to identify unique antigens on the surface of leukemic blasts that are not present on normal T cells, which is problematic because of the life-threatening opportunistic infections associated with prolonged T cell aplasia [68]. However, several groups have made significant progress in addressing some of these challenges, as briefly outlined here.
CAR T cells
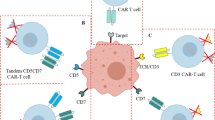
The identification of a unique target on T-ALL cells that is not present on healthy T cells is particularly critical for the development of CAR T cell therapies given the risk of “fratricide,” or that the engineered T cells will target each other [68]. There is also the risk that a CAR T cell product could be contaminated with a patient’s malignant T cells [68]. Mamonkin et al. recently engineered a CAR targeting CD5, which is expressed by a majority of T-ALLs but only a subset of healthy T cells [69••]. They showed that these CAR T cells could be expanded ex vivo, eliminate T-ALL cells in vitro, and slow the progression of disease in a mouse xenograft model of T-ALL with only minimal fratricide, perhaps because CD5 is downregulated after binding with its ligand [68, 69••]. Based on these data, a phase I trial (NCT03081910) of CD5-targeted CAR T cells for patients with relapsed T-ALL and T cell lymphomas has been initiated. A list of selected ongoing clinical trials for relapsed T-ALL is provided in Table 1.
Another potential target that is expressed by most T-ALLs is CD7, which is also expressed on normal T cells. In a creative approach to overcoming the issue of fratricide, several groups have investigated ways to remove or prevent the expression of CD7 on the surface of the engineered T cells. Gomes-Silva et al. used CRISPR/Cas9 gene editing technology to remove CD7 from the surface of T cells engineered to target CD7, and they demonstrated that these CAR T cells could effectively eliminate T-ALL cells both in vitro and in vivo [70••]. A phase I clinical trial (NCT03690011) evaluating this approach for T cell malignancies is currently in progress [70••]. “Off-the-shelf” universal CAR T cells targeting CD7, in which both CD7 and the T cell receptor alpha chain have been edited out, have also been developed and have shown promising pre-clinical results for T-ALL [71•]. A new technology called protein expression blocker (PEBL) has also been studied as a method of preventing the expression of CD7 on the surface of CAR T cells by sequestering it in the endoplasmic reticulum/Golgi apparatus [72]. Other adoptive cellular therapy approaches that are being studied for T-ALL include engineered NK cells (as an alternative to T cells) and shorter-lived mRNA-based CAR T cells, among others [68].
Monoclonal Antibodies
Several antibody-based approaches for T-ALL have been evaluated, most notably daratumumab. Daratumumab is a monoclonal antibody that binds an epitope of CD38 and is approved for the treatment of relapsed and/or refractory multiple myeloma. Bride et al. found that CD38 is typically expressed on T-ALL cells and showed efficacy of daratumumab in 14 of 15 T-ALL patient-derived xenografts studied [73••]. An international multicenter phase II study (NCT03384654) is currently evaluating daratumumab in combination with chemotherapy for children and young adults (≤ 30 years) with relapsed and/or refractory T or B cell ALL. Another potential target to study in T-ALL is CD30. Although CD30 is only expressed in about one-third of T-ALL cases, drugs targeting it have already been developed for other indications (e.g., brentuximab vedotin for Hodgkin lymphoma) and therefore may be worth exploring in the setting of T-ALL [74].
Chemokine Receptors
Inhibition of the chemokine receptor CXCR4 decreases the survival and proliferation of T-ALL cells in vitro and has been shown to inhibit the homing of T-ALL cells to the bone marrow niche (leukemia-initiating activity) in mouse models [75]. Pre-clinical data have also suggested that the inhibition of CXCR4 may impair the interaction of T-ALL cells with the bone marrow microenvironment [75]. Based on this, a short peptide antagonist of CXCR4, BL-8040, is being studied in combination with nelarabine in a phase IIa study for adults with relapsed and/or refractory T-ALL (NCT02763384).
Small-Molecule Inhibitors and Other Targeted Agents
Venetoclax
Venetoclax is an oral inhibitor of the anti-apoptotic protein Bcl-2 that is approved for the treatment of chronic lymphocytic leukemia, as well as for older patients with acute myeloid leukemia in combination with a hypomethylating agent or low-dose cytarabine. Several pre-clinical studies have demonstrated that subsets of T-ALL cell lines and patient samples are sensitive to venetoclax, particularly those with an immature or ETP phenotype [76, 77•]. A phase IB/II ECOG-ACRIN study (NCT03504644) is evaluating venetoclax in combination with liposomal vincristine for adults with relapsed or refractory ALL (T and B cell). An early-phase dose-escalation study (NCT03181126) of venetoclax in combination with navitoclax (a Bcl-2 inhibitor that also inhibits Bcl-XL and Bcl-w) and chemotherapy is also ongoing for children (≥ 4 years old) and adults with relapsed or refractory ALL.
Cyclin-Dependent Kinase Inhibitors
Palbociclib is an inhibitor of the cyclin-dependent kinases CDK4 and CDK6 that is used to treat hormone receptor–positive metastatic breast cancer. Palbociclib has been shown to inhibit cell cycle progression in T-ALL cell lines and primary patient samples and to inhibit disease progression in Notch1-mediated mouse models of T-ALL [78,79,80]. A phase I trial (NCT03515200) is evaluating palbociclib in combination with chemotherapy in children with relapsed and/or refractory T or B cell ALL. Another phase I study (NCT03132454) is studying palbociclib in combination with dexamethasone, sorafenib, or decitabine in adults and adolescents (≥ 15 years old) with ALL. Of note, because palbociclib causes cell cycle arrest in T-ALL cells, there has been a theoretical concern that palbociclib might impair the efficacy of conventional chemotherapeutic agents, many of which are dependent on cell proliferation. To investigate this, Pikman et al. evaluated the CDK4/6 inhibitor LEE011 in combination with various agents that are commonly used to treat T-ALL [80]. They found that LEE011 was synergistic with glucocorticoids and mTOR inhibitors but was antagonistic with several other agents, including methotrexate, mercaptopurine, and l-asparaginase [80]. Whether these in vitro findings will translate to humans is currently unknown but may make sense to consider in the design of future trials or correlative studies of CDK4/6 inhibitors for T-ALL.
PI3K/mTOR Inhibitors
Mutational loss of the tumor suppressor PTEN occurs in approximately 8% of T-ALL cases, which leads to the activation of the PI3K/Akt/mTOR signaling pathway and may also mediate resistance to glucocorticoids, GSIs, and/or chemotherapy [59, 81,82,83]. Dysregulation of PI3K/Akt signaling also occurs via other mechanisms in T-ALL. An analysis of pediatric T-ALL samples by array comparative genomic hybridization, fluorescence in situ hybridization (FISH), and sequencing found that, overall, aberrations in PI3K, AKT, and/or PTEN were present in 21/44 (47.7%) of samples [82]. Pharmacologic inhibition of PI3K, Akt, or mTOR signaling has been shown to inhibit cell proliferation and induce apoptosis in T-ALL cell lines and to inhibit tumor growth in mouse models [83,84,85,86]. Based on these data, ongoing clinical trials are evaluating the mTOR inhibitors everolimus (NCT01523977) and temsirolimus (NCT01614197) in combination with chemotherapy in children with relapsed ALL.
Tyrosine Kinase Inhibitors
NUP215-ABL1 fusions, in which the member of the nuclear pore complex NUP214 and the kinase ABL1 form a constitutively active fusion protein, occur in 3.9–5.8% of T-ALL cases [87, 88]. Of note, NUP214-ABL1 fusions are typically cryptic on metaphase chromosome analysis and therefore need to be identified by either FISH or molecular testing [87]. The tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib, which target the BCR-ABL1 fusion protein, have also been studied for NUP215-ABL1 fusion-positive T-ALL. All three agents induced apoptosis in human NUP214-ABL1-positive T-ALL cell lines, and dasatinib was also shown to have activity in a xenograft model [89]. In a case report, dasatinib induced a complete remission in a young adult with relapsed T-ALL with a NUP215-ABL1 fusion [90]. In a different but similar case, imatinib was used in combination with vincristine and prednisone, although the duration of response in that instance was somewhat brief (6 months) [91]. To better evaluate the potential role for TKIs in this setting, a larger cohort of patients with T-ALL with NUP215-ABL1 fusions will need to be studied.
Our Current Approach
Despite the recent progress that has been made in defining targetable pathways in T-ALL, our approach to patients with relapsed T-ALL currently still relies on chemotherapy with the goal of achieving a second remission followed by allogeneic HSCT for all patients who are eligible and who have not previously undergone transplantation. In general, we consider a clinical trial when possible for patients with relapsed T-ALL. If relapse occurs > 1 year after a first remission was achieved, consideration can also be given to repeating the initial induction regimen (i.e., hyper-CVAD). At our institution, we often select a nelarabine-based regimen (i.e., sequential nelarabine followed by etoposide and cyclophosphamide [38]), and we consider nelarabine or liposomal vincristine monotherapy in patients who are older and/or are unable to tolerate more intensive regimens. Unfortunately, as of January 2019, there is an ongoing national drug shortage of nelarabine, and it is unclear when this supply issue will be resolved.
Conclusions
In summary, outcomes of newly diagnosed T-ALL have improved significantly in recent years with the addition of agents such as PEG-asparaginase and nelarabine, the modification of methotrexate dosing and timing, and changes in the choice of steroids in frontline chemotherapy regimens. Unfortunately, however, outcomes remain unsatisfactory for those patients who do experience disease relapse. Nelarabine and liposomal vincristine are the only drugs that are approved for the treatment of relapsed T-ALL, and a variety of salvage chemotherapy regimens may be used in this setting. Recent pre-clinical and genetic studies have identified recurrent genetic lesions and aberrant pathways that are important in T-ALL pathogenesis. Improved targeting of these pathways and/or the development of novel immunotherapeutic approaches will hopefully lead to improved therapeutic options for patients with relapsed and/or refractory T-ALL, and a number of clinical trials are currently ongoing.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM, Dores GM, et al. Acute leukemia incidence and patient survival among children and adults in the United States, 2001–2007. Blood. 2011;119:34–43.
Hunger SP, Lu X, Devidas M, Camitta BM, Gaynon PS, Winick NJ, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the Children’s Oncology Group. J Clin Oncol. 2012;(30):1663–9.
Winter SS, Dunsmore KP, Devidas M, Wood BL, Esiashvili N, Chen Z, et al. Improved survival for children and young adults with T-lineage acute lymphoblastic leukemia: results from the Children’s Oncology Group AALL0434 methotrexate randomization. J Clin Oncol. 2018;36:2926–34.
Marks DI, Paietta EM, Moorman AV, Richards SM, Buck G, Dewald G, et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood. 2009;114:5136–45.
Stock W, Luger SM, Advani AS, Geyer S, Harvey RC, Mullighan CG, et al. Favorable outcomes for older adolescents and young adults (AYA) with acute lymphoblastic leukemia (ALL): early results of U.S. Intergroup Trial C10403 [Abstract]. Blood. 2014;124:796.
Marks DI, Rowntree C. Management of adults with T-cell lymphoblastic leukemia. Blood. 2017;129:1134–42.
•• Beldjord K, Chevret S, Asnafi V, Boulland M-L, Leguay T, Thomas X, et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. 2016;123:3739–50 Beldjord et al. analyzed MRD levels in patients with T-ALL and B cell precursor ALL following induction chemotherapy and demonstrated that post-induction MRD levels ≥ 10 −4 predicted an increased risk of relapse.
Schrappe M, Valsecchi MG, Bartram CR, Schrauder A, Panzer-Grümayer R, Möricke A, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. 2011;118:2077–84.
Brüggemann M, Raff T, Flohr T, Go N, Nakao M, Droese J, et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood. 2006;107:1116–23.
• Möricke A, Zimmermann M, Valsecchi MG, Stanulla M, Biondi A, Mann G, et al. Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood. 2016;127:2101–12 This study which randomized pediatric patients with ALL to receive dexamethasone vs. prednisone during induction therapy demonstrated a decreased risk of relapse but a higher rate of induction-related deaths in the group that received dexamethasone.
Asselin BL, Devidas M, Wang C, Pullen J, Borowitz MJ, Hutchison R, et al. Effectiveness of high dose methotrexate in T-cell lymphoblastic leukemia and advanced stage lymphoblastic lymphoma: a randomized study by the Children’s Oncology Group (POG 9404). Blood. 2011;118:874–84.
Wetzler M, Sanford BL, Kurtzberg J, DeOliveira D, Frankel SR, Powell BL, et al. Effective asparagine depletion with pegylated asparaginase results in improved outcomes in adult acute lymphoblastic leukemia: Cancer and Leukemia Group B study 9511. Blood. 2007;109:4164–7.
Gökbuget N, Baumann A, Beck J, Brueggemann M, Diedrich H, Huettmann A, et al. PEG-asparaginase intensification in adult acute lymphoblastic leukemia (ALL): significant improvement of outcome with moderate increase of liver toxicity In the German Multicenter Study Group for Adult ALL (GMALL) Study 07/2003 [Abstract]. Blood. 2010;116:494.
DeAngelo DJ, Stevenson KE, Dahlberg SE, Silverman LB, Couban S, Supko JG, et al. Long-term outcome of a pediatric-inspired regimen used for adults aged 18–50 years with newly diagnosed acute lymphoblastic leukemia. Leukemia. 2015;29:526–34.
Hoelzer D, Thiel E, Arnold R, Beck J, Beelen DW, Bornhäuser M, et al. Successful subtype oriented treatment strategies in adult T-ALL: results of 744 patients treated in three consecutive GMALL studies [Abstract]. Blood. 2009;114:324.
Dunsmore KP, Devidas M, Linda SB, Borowitz MJ, Winick N, Hunger SP, et al. Pilot study of nelarabine in combination with intensive chemotherapy in high-risk T-cell acute lymphoblastic leukemia: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30:2753–9.
Jain P, Kantarjian H, Ravandi F, Thomas D, O’Brien S, Kadia T, et al. The combination of hyper-CVAD plus nelarabine as frontline therapy in adult T-cell acute lymphoblastic leukemia and T-lymphoblastic lymphoma: MD Anderson Cancer Center experience. Leukemia. 2014;28:973–5.
Abaza Y, M. Kantarjian H, Faderl S, Jabbour E, Jain N, Thomas D, et al. Hyper-CVAD plus nelarabine in newly diagnosed adult T-cell acute lymphoblastic leukemia and T-lymphoblastic lymphoma. Am J Hematol. 2018;93:91–9.
• Dunsmore KP, Winter S, Devidas M, Wood BL, Esiashvili N, Eisenberg N, et al. COG AALL0434: a randomized trial testing nelarabine in newly diagnosed T-cell malignancy [Abstract]. J Clin Oncol. 2018;36:10500 The initial results of the large COG AALL0434 study which randomized pediatric and young adult patients with newly diagnosed T-ALL to receive or not receive nelarabine demonstrated improved 4-year DFS in the patients that received nelarabine.
Arber DA, Orazi A, Hasserjian R, Borowitz MJ, Le Beau MM, Bloomfield CD, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–406.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63.
Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10:147–56.
Patrick K, Wade R, Goulden N, Mitchell C, Moorman AV, Rowntree C, et al. Outcome for children and young people with early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br J Haematol. 2014;166:421–4.
Wood BL, Winter SS, Dunsmore KP, Devidas M, Chen S, Asselin B, et al. T-lymphoblastic leukemia (T-ALL) shows excellent outcome, lack of significance of the ETP immunophenotype, and validation of the prognostic value of end-induction MRD in COG study AALL0434 [abstract]. Blood 2014;124:1–1.
Reismüller B, Attarbaschi A, Peters C, Dworzak MN, Pötschger U, Urban C, et al. Long-term outcome of initially homogenously treated and relapsed childhood acute lymphoblastic leukaemia in Austria - a population-based report of the Austrian Berlin-Frankfurt-Münster (BFM) Study Group. Br J Haematol. 2009;144:559–70.
• Desjonquères A, Chevallier P, Thomas X, Huguet F, Leguay T, Bernard M, et al. Acute lymphoblastic leukemia relapsing after first-line pediatric-inspired therapy: a retrospective GRAALL study. Blood Cancer J. 2016;6:e504 This large retrospective analysis of adults with relapsed Ph-ALL demonstrated that there is no specific standard of care regimen for relapsed T-ALL.
Fielding AK, Richards SM, Chopra R, Lazarus HM, Litzow MR, Buck G, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109:944–50.
Cohen A, Lee JWW, Gelfand EW. Selective toxicity of deoxyguanosine and arabinosyl guanine for T-leukemic cells. Blood. 1983;61:660–7.
Kisor DF, Plunkett W, Kurtzberg J, Mitchell B, Hodge JP, Ernst T, et al. Pharmacokinetics of nelarabine and 9-beta-D-arabinofuranosyl guanine in pediatric and adult patients during a phase I study of nelarabine for the treatment of refractory hematologic malignancies. J Clin Oncol. 2000;18:995–1003.
Shewach DS, Daddona PE, Ashcroft E, Mitchell BS. Metabolism and selective cytotoxicity of 9-B-D-arabinofuranosyiguanine in human lymphoblasts. Cancer Res. 1985;45:1008–14.
Lambe CU, Averett DR, Paff MT, Reardon JE, Wilson JG, Krenitsky TA. 2-Amino-6-methoxypurine arabinoside: an agent for T-cell malignancies. Cancer Res. 1995;100:3352–6.
Kurtzberg J, Ernst TJ, Keating MJ, Gandhi V, Hodge JP, Kisor DF, et al. Phase I study of 506U78 administered on a consecutive 5-day schedule in children and adults with refractory hematologic malignancies. J Clin Oncol. 2005;23:3396–403.
Cohen MH, Johnson JR, Justice R, Pazdur R. FDA drug approval summary: nelarabine (Arranon) for the treatment of T-cell lymphoblastic leukemia/lymphoma. Oncologist. 2008;13:709–14.
Berg SL, Blaney SM, Devidas M, Lampkin TA, Murgo A, Bernstein M, et al. Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children’s Oncology Group. J Clin Oncol. 2005;23:3376–82.
DeAngelo DJ, Yu D, Johnson JL, Coutre SE, Stone RM, Stopeck AT, et al. Nelarabine induces complete remissions in adults with relapsed or refractory T-lineage acute lymphoblastic leukemia or lymphoblastic lymphoma: Cancer and Leukemia Group B study 19801. Blood. 2007;109:5136–42.
Gökbuget N, Basara N, Baurmann H, Beck J, Brüggemann M, Diedrich H, et al. High single-drug activity of nelarabine in relapsed T-lymphoblastic leukemia/lymphoma offers curative option with subsequent stem cell transplantation. Blood. 2011;118:3504–11.
Commander LA, Seif AE, Insogna IG, Rheingold SR. Salvage therapy with nelarabine, etoposide, and cyclophosphamide in relapsed/refractory paediatric T-cell lymphoblastic leukaemia and lymphoma. Br J Haematol. 2010;150:345–51.
Luskin MR, Ganetsky A, Landsburg DJ, Loren AW, Porter DL, Nasta SD, et al. Nelarabine, cyclosphosphamide and etoposide for adults with relapsed T-cell acute lymphoblastic leukaemia and lymphoma. Br J Haematol. 2016;174:332–4.
Silverman JA, Deitcher SR. Marqibo®(vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharmacol. 2013;71:555–64.
Webb MS, Harasym TO, Masin D, Bally MB, Mayer LD. Sphingomyelin-cholesterol liposomes significantly enhance the pharmacokinetic and therapeutic properties of vincristine in murine and human tumour models. Br J Cancer. 1995;72:896–904.
Webb MS, Logan P, Kanter PM, St.-Onge G, Gelmon K, Harasym T, et al. Preclinical pharmacology, toxicology and efficacy of sphingomyelin/cholesterol liposomal vincristine for therapeutic treatment of cancer. Cancer Chemother Pharmacol. 1998;42:461–70.
Krishna R, Webb MS, St MLD. Liposomal and nonliposomal drug pharmacokinetics after administration of liposome-encapsulated vincristine and their contribution to drug tissue distribution properties. J Pharmacol Exp Ther. 2001;298:1206–12.
O’Brien S, Schiller G, Lister J, Damon L, Goldberg S, Aulitzky W, et al. High-dose vincristine sulfate liposome injection for advanced, relapsed, and refractory adult Philadelphia chromosome-negative acute lymphoblastic leukemia. J Clin Oncol. 2013;31:676–83.
Shah NN, Merchant MS, Cole DE, Jayaprakash N, Bernstein D, Delbrook C, et al. Vincristine sulfate liposomes injection (VSLI, Marqibo): results from a phase I study in children, adolescents, and young adults with refractory solid tumors or leukemias. Pediatr Blood Cancer. 2016;63:997–1005.
Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2016;16:494–507.
Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(80):269–71.
Sanchez-Martin M, Ferrando A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood. 2017;129:1124–33.
Maillard I. Notch and human hematopoietic stem cells. Blood. 2015;123:1116–9.
Paganin M, Ferrando A. Molecular pathogenesis and targeted therapies for NOTCH1-induced T-cell acute lymphoblastic leukemia. Blood Rev. 2011;25:83–90.
•• Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49:1211–8 This genetic and RNA expression study on samples collected from 264 children and young adults with T-ALL identified 106 driver mutations, 39 novel gene rearrangements, and 10 pathways commonly dysregulated in T-ALL, illustrating the tremendous genetic heterogeneity of T-ALL and revealing potentially targetable pathways.
O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to γ-secretase inhibitors. J Exp Med. 2007;204:1813–24.
Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, De Stanchina E, et al. γ-Secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. 2009;15:50–8.
De Keersmaecker K, Lahortiga I, Mentens N, Folens C, Van Neste L, Bekaert S, et al. In vitro validation of γ-secretase inhibitors alone or in combination with other anti-cancer drugs for the treatment of T-cell acute lymphoblastic leukemia. Haematologica. 2008;93:533–42.
Samon JB, Castillo-Martin M, Hadler M, Ambesi-Impiobato A, Paietta E, Racevskis J, et al. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer Ther. 2012;11:1565–75.
Gavai AV, Quesnelle C, Norris D, Han WC, Gill P, Shan W, et al. Discovery of clinical candidate BMS-906024: a potent pan-notch inhibitor for the treatment of leukemia and solid tumors. ACS Med Chem Lett. 2015;6:523–7.
Deangelo DJ, Stone RM, Silverman LB, Stock W, Attar EC, Fearen I, et al. A phase I clinical trial of the Notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias [Abstract]. J Clin Oncol. 2006;24:6585.
Zweidler-McKay PA, Deangelo DJ, Douer D, Dombret H, Ottmann OG, Vey N, et al. The safety and activity of BMS-906024, a gamma secretase inhibitor with anti-Notch activity, in patients with relapsed T-cell acute lymphoblastic leukemia: initial results of a phase 1 trial [Abstract]. Blood. 2014;124:968.
Papayannidis C, DeAngelo DJ, Stock W, Huang B, Shaik MN, Cesari R, et al. A phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015;5:e350–3.
Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203–10.
Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364–70.
• Sanchez-Martin M, Ambesi-Impiombato A, Qin Y, Herranz D, Bansal M, Girardi T, et al. Synergistic antileukemic therapies in NOTCH1 -induced T-ALL. Proc Natl Acad Sci. 2017;114:2006–11 This study identified several drugs that are synergistic with GSIs in the pre-clinical setting for the treatment of T-ALL.
Pinnell N, Yan R, Cho HJ, Keeley T, Murai MJ, Liu Y, et al. The PIAS-like coactivator Zmiz1 is a direct and selective cofactor of Notch1 in T cell development and leukemia. Immunity. 2015;43:870–83.
Roti G, Carlton A, Ross KN, Markstein M, Pajcini K, Su AH, et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell. 2013;23:390–405.
Roderick JE, Tesell J, Shultz LD, Brehm MA, Greiner DL, Harris MH, et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood. 2014;123:1040–51.
Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, et al. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462:182–8.
Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, De Leon GP, Chen Y, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010;464:1052–7.
Agnusdei V, Minuzzo S, Frasson C, Grassi A, Axelrod F, Satyal S, et al. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia. 2014;28:278–88.
Alcantara M, Tesio M, June CH, Houot R, T-cells CAR. CAR T-cells for T-cell malignancies: challenges in distinguishing between therapeutic, normal, and neoplastic T-cells. Leukemia. 2018;32:2307–15.
•• Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell – directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126:983–93 Mamonkin et al. showed that CAR T cells targeting CD5 are effective against T-ALL cells in vitro and in vivo.
•• Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130:285–96 This important proof-of-concept study showed that gene editing to prevent the expression of CD7 in CAR Tcells that were engineered to target CD7 allowed for the efficacious targeting of T-ALL cells in vitro and in a mouse xenograft model with minimal fratricide.
• Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32:1970–83 This study demonstrated efficacy of “off the shelf” CAR T cells targeting CD7 that lack both CD7 and T cell receptor alpha chain expression against T-ALL cells in vitro and in xenograft models.
Png YT, Vinanica N, Kamiya T, Shimasaki N, Coustan-Smith E, Campana D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. 2017;1:2348–60.
•• Bride KL, Vincent TL, Im S-Y, Aplenc R, Barrett DM, Carroll WL, et al. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia (T-ALL). Blood. 2018;131:995–9 This study demonstrated that CD38 is expressed on the majority of T-ALL blasts and that daratumumab is effective in pre-clinical models of T-ALL, which provided the rationale for an ongoing trial of daratumumab for relapsed T-ALL.
Zheng W, Medeiros LJ, Young KH, Goswami M, Powers L, Kantarjian HH, et al. CD30 expression in acute lymphoblastic leukemia as assessed by flow cytometry analysis. Leuk Lymphoma. 2014;55:624–7.
Passaro D, Irigoyen M, Catherinet C, Gachet S, Da Costa De Jesus C, Lasgi C, et al. CXCR4 is required for leukemia-initiating cell activity in T cell acute lymphoblastic leukemia. Cancer Cell. 2015;27:769–79.
Chonghaile TN, Roderick JE, Glenfield C, Ryan J, Sallan SE, Silverman LB, et al. Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov. 2014;4:1074–87.
• Peirs S, Matthijssens F, Goossens S, Van De Walle I, Ruggero K, De Bock CE, et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood. 2015;124:3738–48 This study showed that a subset of T-ALLs, particularly those with an immature immunophenotype, are sensitive to BCL-2 inhibition in vitro and in vivo and provided the pre-clinical rationale for several ongoing studies of venetoclax for relapsed and/or refractory ALL.
Sawai CM, Freund J, Oh P, Ndiaye-Lobry D, Bretz JC, Strikoudis A, et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell Elsevier Inc.; 2012:22:452–65.
Jena N, Sheng J, Hu JK, Li W, Zhou W, Lee G, et al. CDK6-mediated repression of CD25 is required for induction and maintenance of Notch1-induced T-cell acute lymphoblastic leukemia. Leukemia. 2016;30:1033–43.
Pikman Y, Alexe G, Roti G, Conway AS, Furman A, Lee ES, et al. Synergistic drug combinations with a CDK4/6 inhibitor in T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2017;23:1012–24.
Palomero T, Dominguez M, Ferrando AA. The role of the PTEN/AKT pathway in NOTCH1-induced leukemia. Cell Cycle. 2008;7:965–70.
Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114:647–50.
Piovan E, Yu J, Tosello V, Herranz D, Ambesi-Impiombato A, DaSilva AC, et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell. 2013;24:766–76.
Wei G, Twomey D, Lamb J, Schlis K, Agarwal J, Stam RW, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006;10:331–42.
Subramaniam PS, Whye DW, Efimenko E, Chen J, Tosello V, De Keersmaecker K, et al. Targeting nonclassical oncogenes for therapy in T-ALL. Cancer Cell. 2012;21:459–72.
Efimenko E, Davé UP, Lebedeva IV, Shen Y, Sanchez-Quintero MJ, Diolaiti D, et al. PI3Kγ/δ and NOTCH1 cross-regulate pathways that define the T-cell acute lymphoblastic leukemia disease signature. Mol Cancer Ther. 2017;16:2069–82.
Graux C, Cools J, Melotte C, Quentmeier H, Ferrando A, Levine R, et al. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat Genet. 2004;36:1084–9.
Burmeister T, Gökbuget N, Reinhardt R, Rieder H, Hoelzer D, Schwartz S. NUP214-ABL1 in adult T-ALL: the GMALL study group experience. Blood. 2006;108:3556–9.
Quintás-Cardama A, Tong W, Manshouri T, Vega F, Lennon PA, Cools J, et al. Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T cell malignancies. Leukemia. 2008;22:1117–24.
Deenik W, Beverloo HB, van der Poel-van de Luytgaarde SCPAM, Wattel MM, van Esser JWJ, Valk PJM, et al. Rapid complete cytogenetic remission after upfront dasatinib monotherapy in a patient with a NUP214-ABL1-positive T-cell acute lymphoblastic leukemia. Leukemia. 2009;23:627–9.
Clarke S, O’Reilly J, Romeo G, Cooney J. NUP214-ABL1 positive T-cell acute lymphoblastic leukemia patient shows an initial favorable response to imatinib therapy post relapse. Leuk Res. 2011;35:131–3.
Acknowledgements
We would like to thank Ivan P. Maillard, MD, PhD, for discussion regarding recent advances in T cell ALL biology.
Funding
C.M. is supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number TL1TR001880.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Acute Lymphocytic Leukemias
Rights and permissions
About this article

Cite this article
McMahon, C.M., Luger, S.M. Relapsed T Cell ALL: Current Approaches and New Directions. Curr Hematol Malig Rep 14, 83–93 (2019). https://doi.org/10.1007/s11899-019-00501-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-019-00501-3