
Abstract
Chronic myelomonocytic leukemia (CMML) is a clonal stem cell disorder, characterized by peripheral blood monocytosis and overlapping features between myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPNs). Clonal cytogenetic changes are seen in up to 30 % patients, while approximately 90 % have detectable molecular abnormalities. Most patients are diagnosed in the seventh decade of life. Gene mutations in ten-eleven translocation (TET) oncogene family member 2 (TET2) (60 %), SRSF2 (50 %), ASXL1 (40 %), and RAS (20-30 %) are frequent, with only frame shift and nonsense ASXL1 mutations negatively impacting overall survival. With the lack of formal guidelines, management and response criteria are often extrapolated from MDS and MPN. Contemporary molecularly integrated CMML-specific prognostic models include the Groupe Francais des Myelodysplasies (GFM) model and the Molecular Mayo Model, both incorporating ASXL1 mutational status. Hypomethylating agents and allogeneic stem cell transplant remain the two most commonly used treatment strategies, with suboptimal results. Clinical trials exploiting epigenetic and signal pathway abnormalities, frequent in CMML, offer hope and promise.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic myelomonocytic leukemia (CMML) is a clonal hematopoietic stem cell disorder characterized by overlapping features of both myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPNs) [1]. In the 2008 World Health Organization (WHO) classification of hematological malignancies, CMML is categorized as an MDS/MPN overlap syndrome, with other disorders in this group being the following: juvenile myelomonocytic leukemia (JMML), atypical chronic myeloid leukemia (CML), MDS/MPN-unclassifiable, and refractory anemia with ring sideroblasts and thrombocytosis (RARS-T; currently a provisional entity).
The 2008 WHO criteria define CMML as a disorder characterized by the following: (a) persistent peripheral blood (PB) monocytosis >1 × 109/l, (b) absence of the Philadelphia chromosome and the BCR-ABL1 fusion oncogene, (c) absence of the PDGFRA or PDGFRB gene rearrangements, (d) less than 20 % blasts and promonocytes in the PB and bone marrow (BM), and (e) dysplasia involving one or more myeloid lineages [1]. If myelodysplasia is absent or minimal, the diagnosis of CMML can still be made if the other requirements are met and: an acquired, clonal, or molecular genetic abnormality is present in the hematopoietic cells or if the monocytosis has persisted for at least 3 months and other causes of monocytosis have been excluded [1, 2••]. Additionally, CMML is further subclassified into CMML-1 (<5 % circulating blasts and <10 % BM blasts) and CMML-2 (5–19 % circulating blasts, 10–19 % BM blasts, or when Auer rods are present irrespective of the blast count), with the median overall survival (OS) being approximately 20 and 15 months, respectively [3, 4••, 5]. The median age at diagnosis is approximately 71–74 years, with a male preponderance [6–8]. Therapy-related CMML cases have been described and, like their MDS counterparts, are associated with poor outcomes [9].
The platelet-derived growth factor receptors alpha and beta (PDGFRA-chromosome 4q12 and PDGFRB- chromosome 5q31-q32) are type III receptor tyrosine kinases. Abnormal chromosomal translocations involving these growth factor receptors have been associated with myeloid neoplasms characterized by prominent blood eosinophilia and marked responsiveness to imatinib mesylate [10, 11]. At times, PDGFR-rearranged myeloid neoplasms can be associated with monocytosis and BM dysplasia, but given their unique responsiveness to imatinib, these are no longer classified as CMML. Patients presenting with a clinical phenotype of CMML with eosinophilia should be assessed for the t(5;12)(q31-q32;p13), giving rise to the ETV6(TEL)-PDGFRB fusion oncogene [12]. The association between monocytosis and PDGFRA rearrangements is an uncommon occurrence [13].
CMML Biology
Cytogenetic Abnormalities in CMML
Clonal cytogenetic abnormalities are seen in 20–30 % of patients with CMML [3, 7, 14•, 15]. Common alterations include the following: trisomy 8, -Y, abnormalities of chromosome 7, trisomy 21, and complex karyotypes [14•]. Based on these findings, the Spanish cytogenetic risk stratification system was developed, categorizing patients into three groups: high risk (trisomy 8, chromosome 7 abnormalities, or complex karyotype), intermediate risk (all chromosomal abnormalities, except for those in the high- and low-risk categories), and low risk (normal karyotype or -Y), with 5-year OS of 4, 26, and 35 %, respectively [14•]. Similar to MDS, the adverse prognostic impact of monosomal karyotype (MK) in CMML was described, predicting an inferior OS in comparison to complex karyotypes without monosomies [16, 17]. Unlike MDS, sole del(5q) is very infrequent in CMML [18].
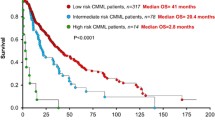
Recently, a single institutional CMML database, with 417 patients, was analyzed for cytogenetic abnormalities [15]. While the Spanish cytogenetic risk stratification system was found to be effective, patients with +8 were found to have a median OS of 22 months, similar to the intermediate-risk group, but significantly better than the high-risk group (14 months). By moving +8 to the intermediate-risk group, they demonstrated a more effective cytogenetic risk stratification system, predicting for both, OS and leukemia-free survival (LFS) [15]. Additionally, in a large international collaborative study, 409 patients with CMML were analyzed for cytogenetic and molecular abnormalities [268 (66 %) and 141 (34 %) from the Mayo Clinic and French Consortium, respectively] [18]. Thirty percent displayed an abnormal karyotype; common abnormalities being +8 (23 %), -Y (20 %), -7/7q- (14 %), 20q- (8 %), +21 (8 %), and der(3q) (8 %) [18]. A stepwise survival analysis resulted in three distinct cytogenetic risk categories: high (complex and MKs), intermediate (all abnormalities not in the high- or low-risk groups), and low (normal, sole -Y and sole der (3q)) with median OS of 3 (hazard ratio (HR) 8.1, 95 % confidence interval (CI) 4.6–14.2), 21 (HR 1.7, 95 % CI 1.2–2.3), and 41 months, respectively [18].
Molecular Abnormalities in CMML
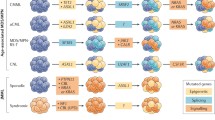
The advent of next-generation sequencing technology has identified molecular abnormalities in approximately 90 % of patients with CMML [19, 20••]. These abnormalities can be classified into the following categories:
-
1.
Mutations involving epigenetic regulator genes: ten-eleven translocation (TET) oncogene family member 2 (TET2) (~60 %), DNMT3A, IDH1, and IDH2
-
2.
Mutations involving chromatin regulation: ASXL1 (~40 %) and enhancer of zeste homolog 2 (EZH2)
-
3.
Mutations involving the splicing machinery: SF3B1, SRSF2 (~50 %), SF3A1 U2AF1, ZRSR2, PRPF30B, SF1
-
4.
Mutations involving DNA damage response genes: Tp53 (<1 %), PFH6
-
5.
Mutations in signal transduction and cellular/receptor tyrosine kinase pathways: JAK2, KRAS, NRAS, CBL, FLT3, RUNX1
TET2 Mutations
TET2 is a tumor suppressor gene on chromosome 4q24 [21]. The incidence of TET2 mutations in CMML is ~60 % [22]. As reported for TET1, TET2 converts 5-methyl-cytosine to 5-hydroxymethyl-cytosine in embryonic stem cells, and thus, mutations of TET2 are proposed to contribute to leukemogenesis by altering epigenetic regulation of transcription through DNA methylation [21]. The exact mechanism and the extent to which TET2 mutations affect DNA methylation remain in question. Ko et al. reported that loss of 5-methyl-cytosine (hypomethylation) was a remarkable characteristic in CMML patients with TET2 mutations and found 2510 differentially hypomethylated regions and only two hypermethylated regions [23]. In contrast, Figueroa et al. studied TET2 mutant leukemic cells and identified a hypermethylation phenotype, including 129 differentially methylated regions [24]. Yamazaki et al., using bisulfite pyrosequencing, confirmed that TET2 mutations affect global methylation in CMML but hypothesized that most of the changes were likely to be outside gene promoter regions [21]. Although TET2 mutations are widely prevalent in CMML, they have not been shown to independently impact either OS or LFS [22]. Similar to MDS, where clonal TET2 mutations in the absence of clonal ASXL1 mutations predict for response to hypomethylating agents (HMA) [25], at least in young CMML patients (age <65 years), there seems to be a similar relationship [26].
ASXL1 and EZH2 Mutations
The ASXL1 (additional sex comb-like 1) gene maps to chromosome 20q11 and regulates chromatin by interacting with the polycomb group repressive complex proteins (PRC1 and PRC2) [27]. ASXL1 mutations are seen in ~40 % of patients with CMML [22, 5]. In a seminal paper, Abdel-Wahab et al. demonstrated that ASXL1 mutations resulted in loss of PRC2-mediated histone H3 lysine 27 (H3K27) trimethylation [28]. Through integration of microarray data with genome-wide histone modification ChIP-Seq (chromatin immunoprecipitation) data, they identified targets of ASXL1 repression including the posterior HOXA cluster that is known to contribute to myeloid transformation. In addition, they showed that ASXL1 associates with PRC2 and that loss of ASXL1 in vivo collaborates with NRASG12D to promote myeloid leukemogenesis [28]. The EZH2 gene, located on chromosome 7q35–q36, encodes for the PRC2 protein, a highly conserved enzyme which serves as a histone H3K27 methyltransferase. EZH2 mutations are infrequent (~5 %) in CMML [22]. Thus far, both of these mutations have been associated with an independent prognostic impact, in some, but not all studies [29, 22, 5]. The specific prognostic role of ASXL1 mutations in CMML will be further discussed.
Spliceosome Component Mutations
Spliceosome component mutations (SRSF2, SF3B1, and U2AF1) affect pre-messenger RNA (mRNA) splicing and result in diverse clinicopathological effects. They are involved in the 3′ splice site recognition of pre-mRNA, including abnormal/alternative splicing. The U2 auxiliary factor that consists of the U2AF65-U2AF1 heterodimer, establishes physical interaction with SF1 and a serine/arginine-rich protein such as SRSF1 or SRSF2, resulting in recognition of the 3′ splice site and its nearby polypyrimidine tract [30]. This leads to the subsequent recruitment of U2 snRNP, containing SF3A1 and SF3B1 to establish the splicing A complex [30]. SRSF2 mutations are very common (~50 %) in CMML and are associated with increased age, less-pronounced anemia, and a diploid karyotype [7]. Thus far, in CMML, SRSF2 mutations have not demonstrated an independent prognostic impact for either OS or LFS [31, 7, 22]. SF3B1 mutations have a high prevalence (~80 %) in patients with MDS and ring sideroblasts (RS) [32] and can also be seen in patients with CMML and RS (<10 %) [7]. However, these mutations do not influence either the OS or LFS [33, 34]. Similarly, U2AF1 mutations are seen in ~10 % of patients with CMML and have thus far lacked an independent prognostic effect [30].
Signal Pathway Mutations
Signal pathway mutations are common in CMML: JAK2V617F (~10–15 %), RAS (KRAS and NRAS ~20–30 %), RUNX1 (~15 %), and CBL (~10–20 %) [35, 22]. RAS mutations are often associated with a MPN-like phenotype with monocytosis [36]. Although univariate analysis studies with RAS mutations have demonstrated inferior outcomes in CMML, these findings have not been substantiated in multivariable models [3, 22]. The CBL gene codes for an E3 ubiquitin ligase involved in degradation of activated receptor tyrosine kinases. RING finger domain (RFD) mutations of CBL are frequently associated with UPD11q (uniparental disomy) and have been reported in 10–20 % of patients with CMML [22, 35]. RUNX1 is essential for normal hematopoiesis, and mutations can be seen in 10–15 % of patients with CMML [22, 35]. Although these mutations do not impact OS, there is a trend toward a higher risk of AML progression [37]. Recently, in vivo studies have demonstrated granulocyte monocyte-colony-stimulating factor (GM-CSF)-dependent pSTAT5 sensitivity in CMML [38].
SETBP1 Mutations
SETBP1, located on chromosome 18q21.1, encodes the SET-binding protein 1, a binding partner for the multifunction SET protein. This protein is involved in apoptosis, transcription, and nucleosome assembly [39]. The proposed functional outcome of this interaction is based on in vitro studies that demonstrate a protection of SET protein from protease cleavage that results in inhibition of protein phosphatase 2A activity, leading to higher rates of cell proliferation. In CMML, SETBP1 mutations have a frequency of 5–10 %, with some [40, 39], but not all studies demonstrating prognostic relevance [4••].
CMML Prognostic Scoring Systems
Numerous prognostic models have attempted to risk stratify patients with CMML. In this regard, the value of Bournemouth, Lille, and the International Prognostic Scoring Systems (IPSS) is limited, as they were designed primarily for patients with MDS, excluding CMML patients with a proliferative phenotype [41, 11, 42]. The MD Anderson Prognostic Scoring System (MDAPS) is CMML-specific and identified a hemoglobin level <12 g/dl, presence of circulating immature myeloid cells (IMC), absolute lymphocyte count (ALC) >2.5 × 109/l, and ≥10 % BM blasts as independent predictors for inferior survival [3]. This model identified four subgroups of patients with median survivals of 24, 15, 8, and 5 months, respectively [3]. The MDAPS was subsequently applied to 212 CMML patients in the Dusseldorf registry; in a univariate analysis, circulating IMC had no prognostic impact, while on multivariable analysis, elevated LDH, BM blast count >10 %, male gender, hemoglobin <12 g/dl, and ALC >2.5 × 109/l were independently prognostic [43].
In 2008, the Global MDAPS was developed for patients with de novo MDS, secondary MDS, and CMML (n = 1915) [44]. On a multivariable analysis, independent prognostic factors included the following: older age, poor performance status, thrombocytopenia, anemia, increased BM blasts, leukocytosis (>20 × 109/l), chromosome 7 or complex cytogenetic abnormalities, and a prior history of red blood cell transfusions [44]. This model identified four prognostic groups with median survivals of 54 (low), 25 (intermediate-1), 14 (intermediate-2), and 6 months (high), respectively [44]. The CMML-specific prognostic scoring system (CPSS) was developed in a large cohort of CMML patients (n = 558) and identified the following four variables as being prognostic for both OS and LFS: French-American-British (FAB) and WHO CMML subtypes, red blood cell transfusion dependency, and the Spanish cytogenetic risk stratification system [14•, 8]. One point was accorded for each variable, with the exception of high-risk cytogenetics which earned two points, and four risk categories were determined: low (0 points), intermediate-1 (1), intermediate-2 (2–3), and high risk (4–5). Median OS was 72, 31, 13, and 5 months for each of the categories, respectively [8].
The discovery of molecular aberrations in CMML has resulted in the development of models inclusive of these abnormalities. A Mayo Clinic study (n = 226) analyzed several parameters, including ASXL1 mutations, and on multivariable analysis, risk factors for survival included hemoglobin <10 g/dl, platelet count <100 × 109/l, absolute monocyte count (AMC) >10 × 109/l, and circulating IMC [5]. ASXL1 mutations did not impact either the OS or the LFS. The study resulted in the development of the Mayo prognostic model, with three risk categories, low (0 risk factor), intermediate (1 risk factor), and high (≥2 risk factors), with median survivals of 32, 18.5, and 10 months, respectively [5]. The Groupe Francais des Myelodysplasies (GFM), however, demonstrated an adverse prognostic effect for ASXL1 mutations in 312 patients with CMML; additional risk factors on multivariable analysis included age >65 years, white blood count (WBC) >15 × 109/l, platelet count <100 × 109/l, and hemoglobin level <10 g/dl in females and <11 g/dl in males [22]. The GFM prognostic model assigns three adverse points for WBC >15 × 109/l and two adverse points for each one of the remaining risk factors, resulting in a three-tiered risk stratification: low (0–4 points), intermediate (5–7), and high (8–12), with respective median survivals of 56, 27.4, and 9.2 months [22]. It should be noted that all nucleotide variations (missense, nonsense, and frame shift) were regarded as ASXL1 mutations in the Mayo study [5], whereas only nonsense and frame shift ASXL1 mutations were considered in the French study [22]. To further clarify the prognostic relevance of ASXL1 mutations, an international collaborative cohort of 466 patients was analyzed [18]. In univariate analysis, survival was adversely affected by ASXL1 (nonsense and frame shift) but not SETBP1 mutations. In multivariable analysis, ASXL1 mutations, AMC >10 × 109/l, hemoglobin <10 g/dl, platelets <100 × 109/l, and circulating IMC were independently predictive of shortened survival. A regression coefficient-based prognostic model based on these five risk factors delineated high (≥3 risk factors; HR 6.2, 95 % CI 3.7–10.4) intermediate-2-risk (two risk factors; HR 3.4, 95 % CI 2.0–5.6), intermediate-1-risk (one risk factor; HR 1.9, 95 % CI 1.1–3.3), and low-risk (no risk factors) categories with median survivals of 16, 31, 59, and 97 months, respectively. This model is referred to as the Molecular Mayo Model.
Management of CMML
Given the inherent similarities with MDS and MPN, management and response evaluation for patients with CMML is often extrapolated from these diseases. The management of cytopenias is similar to lower-risk MDS patients and includes the use of erythropoiesis-stimulating agents (ESA) and transfusion-based supportive care [45•, 46].
Management of Cytopenias
Commercially available ESA include recombinant human erythropoietin (rh-EPO) and darbepoetin. Response rates in lower-risk MDS/CMML patients range from 30 to 60 %, with the median duration of response being ~24 months [47–49]. Parameters predictive of ESA response include the following: low transfusion burden (<2 units a month), use of a fixed-dose versus weight-based EPO regimen, shorter time from diagnosis to starting treatment, and a lower baseline serum EPO level (<500 IU/ml) [47, 48]. Most responses to ESA occur within 8 weeks of treatment. These agents have to be used with caution in CMML patients with a proliferative phenotype, given the inherent risk for spontaneous splenic rupture.
Two first in-class agents targeting late stages of erythropoiesis are currently in development for MDS and CMML. Sotatercept (ACE-011), a recombinant fusion protein containing the extracellular domain of the human activin receptor IIA, binds a variety of TGF-B superfamily ligands [50]. A phase II, dose finding study demonstrated an erythroid response in 40 % of lower-risk transfusion-dependent MDS patients resistant to ESA [51]. Notably, 19 of 44 patients with high transfusion burden responded with a greater than 4 units/8 week RBC transfusion burden reduction. No data has been presented on the CMML subset of this study as of yet. ACE-536 is another recombinant fusion protein containing the extracellular domain of the human activin receptor IIB. A phase II study assessing low- versus high-transfusion-burden MDS patients (<4 transfusion in the preceding 8 weeks versus ≥4) demonstrated that six of seven patients achieved RBC transfusion independence for more than 8 weeks in the low-transfusion-burden group. In the high-transfusion-burden group, 6 of 19 patients had a greater than 50 % reduction in transfusion requirements [52]. These are exciting prospects for transfusion-dependent CMML patients.
Eltrombopag, a small molecule agonist of c-mpl (megakaryocyte receptor), has been investigated in lower-risk MDS, and preliminary results of two phase II placebo-controlled trials have demonstrated a durable platelet response of 24 and 29 % with no evidence of increasing blast percentage [53, 54]. Success in the setting of autoimmune dysfunction and thrombocytopenia has resulted in off-label use of this agent for patients with CMML [55]. The current status and preliminary response data for investigative agents in CMML has been outlined in Table 1.
Management of Proliferative Disease
Hydroxyurea is the mainstay for management of proliferative CMML. In a prospective randomized study, Wattel et al. compared hydroxyurea to oral etoposide in 105 patients [56]. After a median follow-up of 11 months, 60 % of patients in the hydroxyurea arm responded compared to 36 % in the etoposide arm. Median OS was statistically superior in the hydroxyurea arm (20 months versus 9 months). Several other trials evaluated agents such as low-dose cytarabine with or without the use of all-trans retinoic acid [57–59], topotecan [60, 61], 9-nitro-campothecin (a novel topoisomerase inhibitor) [62], valproic acid (histone deacetylase inhibitor) [63], and lonafarnib (farnesyltransferase inhibitor) [64] in the treatment of CMML. Collectively, response rates in these trials were disappointing and treatment was associated with significant toxicities.
Epigenetic Modifying Agents
Hypomethylating agents (HMA) such as 5-azacytidine (AZA) and decitabine (DAC) have been approved for the management of higher-risk MDS patients. Several phase II studies have now been completed using HMA in CMML [6, 34, 65–71]. A complete list of the studies is shown in Table 2. The overall response rates range from 25 to 70 %, and median OS ranges from 12 to 37 months. Unfortunately, the retrospective nature of the vast majority of the studies along with the lack of a comparator arm makes it difficult to draw cross-study conclusions.
Histone deacetylase inhibitors (HDACi) assist in the remodeling of chromatin and play a key role in epigenetic regulation of gene expression [2••]. This has prompted combination therapy with HMA and immunomodulatory agents due to postulated synergistic effects [72]. A randomized phase II intergroup study (S1117) evaluated AZA + lenalidomide, AZA + vorinostat (HDACi), versus AZA monotherapy in MDS and CMML [73]. The median follow-up was 9 months, and no statistically significant difference was seen in response rates across all three arms. Other HDACi currently undergoing clinical trial evaluation include mocetinostat and pracinostat. An additional strategy has been the reformulation of HMA. High levels of cytidine deaminases in the liver and gastrointestinal tract result in rapid elimination of HMA when administered orally. A second-generation HMA, SGI-110, is a reformulation of DAC coupled with deoxyguanosine giving it a significantly prolonged half-life by protection from deamination [74]. A phase II study in treatment-naive and pre-treated patients (20 % CMML) resulted in a complete remission (CR and marrow CR) rate of ~20 % [75]. Red cell transfusion independence of at least 8 weeks was achieved in 32 % of both treatment groups. Alternative mechanisms of cytidine deaminase inhibition are also being investigated with oral DAC and E7727, a novel oral cytidine deaminase inhibitor (NCT02103478-www.clinicaltrials.gov).
Hematopoietic Stem Cell Transplantation
Allogeneic stem cell transplantation (HSCT) remains the only curative option for patients with CMML. This modality is, however, fraught with complications including acute and chronic graft versus host disease (GVHD), nonrelapse mortality, and post-transplant disease relapse. There, unfortunately, exists no prospective data analyzing the risks and befits for HSCT in CMML. The numbers of CMML patients in retrospective series have ranged from 8 to 283, with the median ages ranging from 50 to 56 years. The response rates in these studies have ranged from 17 to 50 % and treatment-related mortality from 12 to 52 % [76–83]. The 10-year OS of 85 patients who underwent HSCT at Fred Hutchinson Cancer Center was 40 %. A multivariable model identified increasing age, higher SCT comorbidity index, and poor-risk cytogenetics to be associated with increased mortality and reduced relapse-free survival (RFS) [76]. The European Group for Blood and Marrow Transplantation (EBMT) reported an OS of 42 % for 283 patients with CMML that underwent HSCT. None of the baseline factors including the conditioning regimen, age, disease status at transplant, cytogenetics, donor-recipient gender match, HLA type of donor, stem cell source, T cell depletion, or the development of GVHD affected the RFS or OS [82]. A recent application of the CPSS in the HSCT setting assessed 209 adult patients from 2001 to 2012 with a median age of 57 years and followed for a median of 51 months [84]. On multivariate analysis, CPSS score, Karnofsky performance status, and graft source were significant predictors of OS.
Conclusion
CMML is an MDS/MPN overlap syndrome, enriched with molecular abnormalities impairing epigenetic and chromatin regulation. Cytogenetic changes are seen in 20–30 % of patients, while molecular abnormalities are seen in ~90 %. Gene mutations involving TET2 (60 %), SRSF2 (50 %), ASXL1 (40 %), and RAS (30 %) are common. Given the lack of formal treatment and response criteria, management is often extrapolated from MDS and MPN, with allogeneic HSCT being the only cure. Given the relatively poor responses to HMA, newer drugs exploiting the aforementioned molecular abnormalities are currently being explored, either in combination with HMA or as single-agent therapies. The developments of uniform response criteria and CMML-specific trials are much needed steps for this otherwise orphan disease.
References
Papers of particular interest, published recently, have been highlighted as : •Of importance •• Of major importance
Swederlow S, Camp E, Harris NL, Jaffe ES, Stefano PA, Stein H, et al., editors. WHO classification of tumors of haematopoietic and lymphoid tissues. Lyon: International Agency for Research on Cancer; 2008.
Patnaik MM, Parikh SA, Hanson CA, Tefferi A. Chronic myelomonocytic leukaemia: a concise clinical and pathophysiological review. Br J Haematol. 2014;165(3):273–86. doi:10.1111/bjh.12756. A concise, up to date review on pathophysiology, genetics, and therapeutic options in chronic myelomonocytic leukemia.
Onida F, Kantarjian HM, Smith TL, Ball G, Keating MJ, Estey EH, et al. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood. 2002;99(3):840–9.
Patnaik MM, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA, et al. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia. 2014;28(11):2206–12. doi:10.1038/leu.2014.125. A comprehensive and collaborative mutation analysis, resulting in the development of the Mayo Clinic Molecular Model for prognostication.
Patnaik MM, Padron E, LaBorde RR, Lasho TL, Finke CM, Hanson CA, et al. Mayo prognostic model for WHO-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes. Leukemia. 2013;27(7):1504–10. doi:10.1038/leu.2013.88.
Ades L, Sekeres MA, Wolfromm A, Teichman ML, Tiu RV, Itzykson R, et al. Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk Res. 2013;37(6):609–13. doi:10.1016/j.leukres.2013.01.004.
Patnaik MM, Lasho TL, Finke CM, Hanson CA, Hodnefield JM, Knudson RA, et al. Spliceosome mutations involving SRSF2, SF3B1, and U2AF35 in chronic myelomonocytic leukemia: prevalence, clinical correlates, and prognostic relevance. Am J Hematol. 2013;88(3):201–6. doi:10.1002/ajh.23373.
Such E, Germing U, Malcovati L, Cervera J, Kuendgen A, Della Porta MG, et al. Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood. 2013;121(15):3005–15. doi:10.1182/blood-2012-08-452938.
Takahashi K, Pemmaraju N, Strati P, Nogueras-Gonzalez G, Ning J, Bueso-Ramos C, et al. Clinical characteristics and outcomes of therapy-related chronic myelomonocytic leukemia. Blood. 2013;122(16):2807–11. doi:10.1182/blood-2013-03-491399. quiz 920.
Apperley JF, Gardembas M, Melo JV, Russell-Jones R, Bain BJ, Baxter EJ, et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med. 2002;347(7):481–7. doi:10.1056/NEJMoa020150.
Pardanani A, Ketterling RP, Li CY, Patnaik MM, Wolanskyj AP, Elliott MA, et al. FIP1L1-PDGFRA in eosinophilic disorders: prevalence in routine clinical practice, long-term experience with imatinib therapy, and a critical review of the literature. Leuk Res. 2006;30(8):965–70. doi:10.1016/j.leukres.2005.11.011.
Gunby RH, Cazzaniga G, Tassi E, Le Coutre P, Pogliani E, Specchia G, et al. Sensitivity to imatinib but low frequency of the TEL/PDGFRbeta fusion protein in chronic myelomonocytic leukemia. Haematologica. 2003;88(4):408–15.
Tefferi A, Gilliland DG. Oncogenes in myeloproliferative disorders. Cell Cycle. 2007;6(5):550–66.
Such E, Cervera J, Costa D, Sole F, Vallespi T, Luno E, et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica. 2011;96(3):375–83. doi:10.3324/haematol.2010.030957. A seminal paper in CMML outlining risk based on a cytogenetic stratification system.
Tang G, Zhang L, Fu B, Hu J, Lu X, Hu S, et al. Cytogenetic risk stratification of 417 patients with chronic myelomonocytic leukemia from a single institution. Am J Hematol. 2014;89(8):813–8. doi:10.1002/ajh.23751.
Alsahlawi A, Alkhateeb H, Patnaik M, Begna K, Elliott M, Hogan WJ, et al. Monosomal karyotype predicts adverse prognosis in patients diagnosed with chronic myelomonocytic leukemia: a single-institution experience. Clin Lymphoma Myeloma Leuk. 2014. doi:10.1016/j.clml.2014.06.007.
Patnaik MM, Hanson CA, Hodnefield J, Knudson R, Van Dyke D, Tefferi A. Monosomal karyotype in myelodysplastic syndromes, with or without monosomy 7 or 5, is prognostically worse than an otherwise complex karyotype. Leukemia. 2010;25(2):266–70.
Wassie EA, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA, et al. Molecular and prognostic correlates of cytogenetic abnormalities in chronic myelomonocytic leukemia: a Mayo Clinic-French Consortium Study. Am J Hematol. 2014;89(12):1111–5. doi:10.1002/ajh.23846.
Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol. 2012;5:12. doi:10.1186/1756-8722-5-12.
Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C, et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121(12):2186–98. doi:10.1182/blood-2012-06-440347. A contemporary prognostic model incorporating the ASXL1 mutation within a large data set.
Yamazaki J, Taby R, Vasanthakumar A, Macrae T, Ostler KR, Shen L, et al. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics. 2012;7(2):201–7. doi:10.4161/epi.7.2.19015.
Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31(19):2428–36. doi:10.1200/JCO.2012.47.3314.
Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468(7325):839–43. doi:10.1038/nature09586.
Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17(1):13–27. doi:10.1016/j.ccr.2009.11.020.
Bejar R, Lord A, Stevenson K, Bar-Natan M, Perez-Ladaga A, Zaneveld J, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;127(17):2705–12. doi:10.1182/blood-2014-06-582809.
Patnaik MM, Wassie EA, Padron E, Onida F, Itzykson R, Lasho TL, et al. Chronic myelomonocytic leukemia in younger patients: molecular and cytogenetic predictors of survival and treatment outcome. Blood Cancer J. 2015;4:e270. doi:10.1038/bcj.2014.90.
Abdel-Wahab O, Pardanani A, Patel J, Wadleigh M, Lasho T, Heguy A, et al. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia. 2011;25(7):1200–2. doi:10.1038/leu.2011.58.
Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22(2):180–93. doi:10.1016/j.ccr.2012.06.032.
Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145(6):788–800. doi:10.1111/j.1365-2141.2009.07697.x.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–9. doi:10.1038/nature10496.
Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood. 2012;120(15):3080–8. doi:10.1182/blood-2012-01-404863.
Patnaik MM, Lasho TL, Hodnefield JM, Knudson RA, Ketterling RP, Garcia-Manero G, et al. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood. 2012;119(2):569–72.
Patnaik MM, Hanson CA, Sulai NH, Hodnefield JM, Knudson RA, Ketterling RP, et al. Prognostic irrelevance of ring sideroblast percentage in World Health Organization-defined myelodysplastic syndromes without excess blasts. Blood. 2012;119(24):5674–7.
Aribi A, Borthakur G, Ravandi F, Shan J, Davisson J, Cortes J, et al. Activity of decitabine, a hypomethylating agent, in chronic myelomonocytic leukemia. Cancer. 2007;109(4):713–7. doi:10.1002/cncr.22457.
Kohlmann A, Grossmann V, Klein HU, Schindela S, Weiss T, Kazak B, et al. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8 % of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol. 2010;28(24):3858–65. doi:10.1200/JCO.2009.27.1361.
Ricci C, Fermo E, Corti S, Molteni M, Faricciotti A, Cortelezzi A, et al. RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res. 2010;16(8):2246–56. doi:10.1158/1078-0432.CCR-09-2112.
Kuo MC, Liang DC, Huang CF, Shih YS, Wu JH, Lin TL, et al. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia. 2009;23(8):1426–31. doi:10.1038/leu.2009.48.
Padron E, Painter JS, Kunigal S, Mailloux AW, McGraw K, McDaniel JM, et al. GM-CSF-dependent pSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia. Blood. 2013;121(25):5068–77. doi:10.1182/blood-2012-10-460170.
Makishima H, Yoshida K, Nguyen N, Przychodzen B, Sanada M, Okuno Y, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45(8):942–6. doi:10.1038/ng.2696.
Laborde RR, Patnaik MM, Lasho TL, Finke CM, Hanson CA, Knudson RA, et al. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia. 2013;27(10):2100–2. doi:10.1038/leu.2013.97.
Mufti GJ, Stevens JR, Oscier DG, Hamblin TJ, Machin D. Myelodysplastic syndromes: a scoring system with prognostic significance. Br J Haematol. 1985;59(3):425–33.
Morel P, Hebbar M, Lai JL, Duhamel A, Preudhomme C, Wattel E, et al. Cytogenetic analysis has strong independent prognostic value in de novo myelodysplastic syndromes and can be incorporated in a new scoring system: a report on 408 cases. Leukemia. 1993;7(9):1315–23.
Germing U, Strupp C, Aivado M, Gattermann N. New prognostic parameters for chronic myelomonocytic leukemia. Blood. 2002;100(2):731–2. author reply 2-3.
Kantarjian H, O’Brien S, Ravandi F, Cortes J, Shan J, Bennett JM, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008;113(6):1351–61. doi:10.1002/cncr.23697.
Malcovati L, Hellstrom-Lindberg E, Bowen D, Ades L, Cermak J, Del Canizo C, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122(17):2943–64. doi:10.1182/blood-2013-03-492884. Consensus recommendations for supportive care management in myelodysplastic system can be gathered for use in CMML, including transfusion support, infection prophylaxis, and growth factor utilization.
Garcia-Manero G. Myelodysplastic syndromes: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol. 2014;89(1):97–108. doi:10.1002/ajh.23642.
Hellstrom-Lindberg E. Efficacy of erythropoietin in the myelodysplastic syndromes: a meta-analysis of 205 patients from 17 studies. Br J Haematol. 1995;89(1):67–71.
Moyo V, Lefebvre P, Duh MS, Yektashenas B, Mundle S. Erythropoiesis-stimulating agents in the treatment of anemia in myelodysplastic syndromes: a meta-analysis. Ann Hematol. 2008;87(7):527–36. doi:10.1007/s00277-008-0450-7.
Jadersten M, Montgomery SM, Dybedal I, Porwit-MacDonald A, Hellstrom-Lindberg E. Long-term outcome of treatment of anemia in MDS with erythropoietin and G-CSF. Blood. 2005;106(3):803–11. doi:10.1182/blood-2004-10-3872.
Carrancio S, Markovics J, Wong P, Leisten J, Castiglioni P, Groza MC, et al. An activin receptor IIA ligand trap promotes erythropoiesis resulting in a rapid induction of red blood cells and haemoglobin. Br J Haematol. 2014;165(6):870–82. doi:10.1111/bjh.12838.
Komrokji R, Garcia-Manero G, Ades L, Laadem A, Vo B, Prebet T, et al. An open-label, phase 2, dose-finding study of sotatercept (ACE-011) in patients with low or intermediate-1 (Int-1)-risk myelodysplastic syndromes (MDS) or non-proliferative chronic myelomonocytic leukemia (CMML) and anemia requiring transfusion. Blood. 2014;124(21):Abstract 3251.
Platzbecker U, Germing U, Giagounidis A, Goetze K, Kiewe P, Mayer K, et al. ACE-536 increases hemoglobin and reduces transfusion burden in patients with low or intermediate-1 risk myelodysplastic syndromes (MDS): preliminary results from a phase 2 study. Blood. 2014;124(21):Abstract 411.
Oliva E, Santini V, Zini G, Palumbo G, Poloni A, Cortelezzi A, et al. Efficacy and safety of eltrombopag for the treatment of thrombocytopenia of low and intermediate-1 IPSS risk myelodysplastic syndromes: interim analysis of a prospective, randomized, single-blind, placebo-controlled trial (EQoL-MDS). Blood. 2012;120:Abstract 923.
Mittelman M, Assouline S, Briasoulis E, Alonso A, Delgado R, O’Gorman P, et al. Eltrombopag treatment of thrombocytopenia in advanced myelodysplastic syndromes and acute myeloid leukemia: results of the 8-week open-label part of an ongoing study. Blood. 2012;120:Abstract 3822.
Modi Y, Shaaban H, Gauchan D, Maroules M. Successful treatment of severe thrombocytopenia with the use of thrombopoeitin receptor agonist eltrombopag in a patient with chronic myelomonocytic leukemia. J Oncol Pharm Pract. 2015;21(1):74–5. doi:10.1177/1078155214544076.
Wattel E, Guerci A, Hecquet B, Economopoulos T, Copplestone A, Mahe B, et al. A randomized trial of hydroxyurea versus VP16 in adult chronic myelomonocytic leukemia. Groupe Francais des Myelodysplasies and European CMML Group. Blood. 1996;88(7):2480–7.
Cambier N, Wattel E, Menot ML, Guerci A, Chomienne C, Fenaux P. All-trans retinoic acid in adult chronic myelomonocytic leukemia: results of a pilot study. Leukemia. 1996;10(7):1164–7.
Gerhartz HH, Marcus R, Delmer A, Zwierzina H, Suciu S, Dardenne M, et al. A randomized phase II study of low-dose cytosine arabinoside (LD-AraC) plus granulocyte-macrophage colony-stimulating factor (rhGM-CSF) in myelodysplastic syndromes (MDS) with a high risk of developing leukemia. EORTC Leukemia Cooperative Group. Leukemia. 1994;8(1):16–23.
Venditti A, Tamburini A, Buccisano F, Scimo MT, Del Poeta G, Maurillo L, et al. A phase-II trial of all trans retinoic acid and low-dose cytosine arabinoside for the treatment of high-risk myelodysplastic syndromes. Ann Hematol. 2000;79(3):138–42.
Beran M, Estey E, O’Brien SM, Giles FJ, Koller CA, Kornblau S, et al. Results of topotecan single-agent therapy in patients with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk Lymphoma. 1998;31(5-6):521–31. doi:10.3109/10428199809057611.
Beran M, Estey E, O’Brien S, Cortes J, Koller CA, Giles FJ, et al. Topotecan and cytarabine is an active combination regimen in myelodysplastic syndromes and chronic myelomonocytic leukemia. J Clin Oncol. 1999;17(9):2819–30.
Quintas-Cardama A, Kantarjian H, O’Brien S, Jabbour E, Giles F, Ravandi F, et al. Activity of 9-nitro-camptothecin, an oral topoisomerase I inhibitor, in myelodysplastic syndrome and chronic myelomonocytic leukemia. Cancer. 2006;107(7):1525–9. doi:10.1002/cncr.22186.
Siitonen T, Timonen T, Juvonen E, Terava V, Kutila A, Honkanen T, et al. Valproic acid combined with 13-cis retinoic acid and 1,25-dihydroxyvitamin D3 in the treatment of patients with myelodysplastic syndromes. Haematologica. 2007;92(8):1119–22.
Feldman EJ, Cortes J, DeAngelo DJ, Holyoake T, Simonsson B, O’Brien SG, et al. On the use of lonafarnib in myelodysplastic syndrome and chronic myelomonocytic leukemia. Leukemia. 2008;22(9):1707–11. doi:10.1038/leu.2008.156.
Braun T, Itzykson R, Renneville A, de Renzis B, Dreyfus F, Laribi K, et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: a phase 2 trial. Blood. 2011;118(14):3824–31. doi:10.1182/blood-2011-05-352039.
Costa R, Abdulhaq H, Haq B, Shadduck RK, Latsko J, Zenati M, et al. Activity of azacitidine in chronic myelomonocytic leukemia. Cancer. 2011;117(12):2690–6. doi:10.1002/cncr.25759.
Fianchi L, Criscuolo M, Breccia M, Maurillo L, Salvi F, Musto P, et al. High rate of remissions in chronic myelomonocytic leukemia treated with 5-azacytidine: results of an Italian retrospective study. Leuk Lymphoma. 2013;54(3):658–61. doi:10.3109/10428194.2012.719617.
Garcia-Manero G, Gore SD, Cogle C, Ward R, Shi T, Macbeth KJ, et al. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol. 2011;29(18):2521–7. doi:10.1200/JCO.2010.34.4226.
Thorpe M, Montalvao A, Pierdomenico F, Moita F, Almeida A. Treatment of chronic myelomonocytic leukemia with 5-azacitidine: a case series and literature review. Leuk Res. 2012;36(8):1071–3. doi:10.1016/j.leukres.2012.04.024.
Wijermans PW, Ruter B, Baer MR, Slack JL, Saba HI, Lubbert M. Efficacy of decitabine in the treatment of patients with chronic myelomonocytic leukemia (CMML). Leuk Res. 2008;32(4):587–91. doi:10.1016/j.leukres.2007.08.004.
Wong E, Seymour JF, Kenealy M, Westerman D, Herbert K, Dickinson M. Treatment of chronic myelomonocytic leukemia with azacitidine. Leuk Lymphoma. 2013;54(4):878–80. doi:10.3109/10428194.2012.730615.
Thurn KT, Thomas S, Moore A, Munster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol. 2011;7(2):263–83. doi:10.2217/fon.11.2.
Sekeres MA, Othus M, List AF, Odenike O, Stone RM, Gore SD, et al. A randomized phase II study of azacitidine combined with lenalidomide or with vorinostat vs. azacitidine monotherapy in higher-risk myelodysplastic syndromes (MDS) and chronic myelomonocytic leukemia (CMML): North American Intergroup Study SWOG S1117. Blood. 2014;124(21):Abstract LBA-5.
Kantarjian H, Jabbour E, Yee K, Kropf P, O’Connell C, Stock W, et al. First clinical results of a randomized phase 2 study of SGI-110, a novel subcutaneous (SQ) hypomethylating agent (HMA), in adult patients with acute myeloid leukemia (AML). Blood. 2013;122:497.
Garcia-Manero G, Ritchie EK, Walsh K, Savona M, Kropf P, O’Connell C, et al. First clinical results of a randomized phase 2 dose-response study of SGI-110, a novel subcutaneous (SC) hypomethylating agent (HMA), in 102 patients with intermediate (Int) or high risk (HR) myelodysplastic syndromes (MDS) or chronic myelomonocytic leukemia (CMML). Blood. 2014;124(21):Abstract 529.
Eissa H, Gooley TA, Sorror ML, Nguyen F, Scott BL, Doney K, et al. Allogeneic hematopoietic cell transplantation for chronic myelomonocytic leukemia: relapse-free survival is determined by karyotype and comorbidities. Biol Blood Marrow Transplant. 2011;17(6):908–15. doi:10.1016/j.bbmt.2010.09.018.
Elliott MA, Tefferi A, Hogan WJ, Letendre L, Gastineau DA, Ansell SM, et al. Allogeneic stem cell transplantation and donor lymphocyte infusions for chronic myelomonocytic leukemia. Bone Marrow Transplant. 2006;37(11):1003–8. doi:10.1038/sj.bmt.1705369.
Krishnamurthy P, Lim ZY, Nagi W, Kenyon M, Mijovic A, Ireland R, et al. Allogeneic haematopoietic SCT for chronic myelomonocytic leukaemia: a single-centre experience. Bone Marrow Transplant. 2010;45(10):1502–7. doi:10.1038/bmt.2009.375.
Kroger N, Zabelina T, Guardiola P, Runde V, Sierra J, Van Biezen A, et al. Allogeneic stem cell transplantation of adult chronic myelomonocytic leukaemia. A report on behalf of the Chronic Leukaemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Br J Haematol. 2002;118(1):67–73.
Mittal P, Saliba RM, Giralt SA, Shahjahan M, Cohen AI, Karandish S, et al. Allogeneic transplantation: a therapeutic option for myelofibrosis, chronic myelomonocytic leukemia and Philadelphia-negative/BCR-ABL-negative chronic myelogenous leukemia. Bone Marrow Transplant. 2004;33(10):1005–9. doi:10.1038/sj.bmt.1704472.
Ocheni S, Kroger N, Zabelina T, Zander AR, Bacher U. Outcome of allo-SCT for chronic myelomonocytic leukemia. Bone Marrow Transplant. 2009;43(8):659–61. doi:10.1038/bmt.2008.366.
Symeonidis A, van Biezen A, Mufti G, Finke J, Beelen D, Bornhauser M, et al. Allogeneic stem cell transplantation in patients with chronic myelomonocytic leukaemia: the impact of WHO classification and of the conditioning regimen on the transplantation outcome. Bone Marrow Transplant. 2010;45(S241):Abstract P803.
Park S, Labopin M, Yakoub-Agha I, Delaunay J, Dhedin N, Deconinck E, et al. Allogeneic stem cell transplantation for chronic myelomonocytic leukemia: a report from the Societe Francaise de Greffe de Moelle et de Therapie Cellulaire. Eur J Haematol. 2013;90(5):355–64. doi:10.1111/ejh.12073.
Duong HK, Akhtari M, Ahn KW, Hu Z-H, Popat UR, Alyea Iii EP, et al. Allogeneic hematopoietic cell transplantation for adult chronic myelomonocytic leukemia. Biol Blood Marrow Transplant. 2015;21(2, Supplement):S30–1. doi:10.1016/j.bbmt.2014.11.022.
Platzbecker U, Wong R, Verma A, Abboud C, Araujo S, Chiou T et al. Placebo-controlled, randomized, phase I/II trial of the thrombopoietin receptor agonist eltrombopag in thrombocytopenic patients with advanced myelodysplastic syndromes or acute myeloid leukemia. Haematologica. 2013;98(s1):Abstract S1108.
Navada S, Garcia-Manero G, Wilhelm F, Hearn K, Odchimar-Reissig R, Demakos E, et al. A phase I/II study of the combination of oral rigosertib and azacitidine in patients with myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML). Blood. 2014;124(21):Abstract 3252.
Garcia-Manero G, Atallah E, Odenike O, Medeiros B, Cortes J, Esquibel V, et al. Pracinostat in combination with azacitidine produces a high rate and rapid onset of disease remission in patients with previously untreated acute myeloid leukemia (AML). Blood. 2014;124(21):Abstract 947.
Stein E, Altman JK, Collins R, DeAngelo DJ, Fathi AT, Flinn I, et al. AG-221, an oral, selective, first-in-class, potent inhibitor of the IDH2 mutant metabolic enzyme, induces durable remissions in a phase I study in patients with IDH2 mutation positive advanced hematologic malignancies. Blood. 2014;124(21):Abstract 115.
Pollyea DA, De Botton S, Fathi AT, Tallman MS, Agresta S, Bowden C, et al. Clinical safety and activity in a phase I trial of AG-120, a first in class, selective, potent inhibitor of the IDH1-mutant protein, in patients with IDH1 mutant positive advanced hematologic malignancies. Eur J Haematol. 2014;50:195.
Pleyer L, Germing U, Sperr WR, Linkesch W, Burgstaller S, Stauder R, et al. Azacitidine in CMML: matched-pair analyses of daily-life patients reveal modest effects on clinical course and survival. Leuk Res. 2014;38(4):475–83.
Drummond MW, Pocock C, Boissinot M, Mills J, Brown J, Cauchy P, et al. A multi-centre phase 2 study of azacitidine in chronic myelomonocytic leukaemia. Leukemia. 2014;28(7):1570–2. doi:10.1038/leu.2014.85.
Acknowledgments
The authors would like to acknowledge the Henry J. Predolin Foundation for Research in Leukemia, Mayo Clinic, Rochester, MN.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Dr. Kristen McCullough and Dr. Mrinal Patnaik each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Myelodysplastic Syndromes
Rights and permissions
About this article

Cite this article
McCullough, K.B., Patnaik, M.M. Chronic Myelomonocytic Leukemia: a Genetic and Clinical Update. Curr Hematol Malig Rep 10, 292–302 (2015). https://doi.org/10.1007/s11899-015-0271-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-015-0271-4