
Abstract
Elevated resting heart rate has been linked to poor outcomes in patients with chronic systolic heart failure. Blockade of funny current channel with ivabradine reduces heart rate without inotropic effects. Ivabradine was recently approved by US Food and Drug Administration for patients with stable, symptomatic chronic heart failure (HF) with left ventricular ejection fraction (LVEF) ≤35 %, who are in sinus rhythm with resting heart rate (HR) ≥ 70 bpm and either are on maximally tolerated doses of beta-blockers, or have a contraindication to beta-blockers. This article will review and evaluate the data supporting the use of ivabradine in patients with HF and explore its mechanisms and physiologic effects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite the widespread use of disease-modifying therapies that have led to increased survival of patients with heart failure with reduced ejection fraction (HFrEF), there still remain too many patients who fail to respond [1, 2]. For the last three decades, the bedrock of current pharmacologic treatment of HFrEF has been blockade of the renin–angiotensin–aldosterone and sympathetic nervous systems. In addition to the many proven benefits of this strategy, it has been suggested that part of the reductions in mortality and hospitalizations observed with beta-blockade [3, 4] may be attributed to heart rate (HR) reduction.
Resting HR is a valuable prognosticator of cardiovascular outcomes, which is easily measured in clinical practice [5]. In a study of 2798 subjects enrolled in the Copenhagen male study and followed for 16 years, increasing resting HR was highly associated with overall mortality in a graded manner independent of cardiovascular risk factors and physical fitness. Risk of mortality increased by 16 % per 10 bpm increase in resting HR [6]. Patients with stable coronary artery disease (CAD) and left ventricular dysfunction, and a HR ≥ 70 bpm, have a 34, 53, 46, and 38 % higher risk of cardiovascular death, chronic heart failure, acute myocardial infarction, and coronary revascularization, respectively, compared to those with lower HR [7]. In HF, HR is often elevated to maintain cardiac output and compensate for low stroke volume (SV). Whereas higher HR in normal hearts increases contractility, in HF patients, it is associated with reduced contractility [8]. The CIBIS [9], COMET [10], and CIBIS II [9] trials showed that the magnitude of HR reduction with beta-blockers was an important mediator of beta-blocker effect. Two recent meta-analyses of beta-blocker trials confirmed the relationship between magnitude of HR reduction, survival benefit, and improvement in left ventricular ejection fraction (LVEF) [11, 12].
It is apparent that HR is not well controlled in HF patients and that there exists an unmet need for ivabradine. For example, in a non-randomized cohort of all-comer outpatient HF population, 53 % of patients had inadequate HR control (HR ≥ 75 bpm) despite beta-blocker therapy [13]. Five-year event-free survival was significantly lower among patients with HR ≥70 bpm as compared to those with >70 bpm (p < 0.05) [14]. The clinical need for a HR-lowering drug such as ivabradine was assessed in a group of Scottish patients previously hospitalized with Acute decompensated heart failure (ADHF) [15], of whom 19 % met the indication for ivabradine. In this population, <15 % of patients achieved target doses of beta-blockers [15]. In a UK study, the proportion of patients with chronic heart failure who were suitable for ivabradine was about 14 % at 12 months following titration of standard medical therapy [16].
Ivabradine (Corlanor®, Amgen, Thousand Oaks, California) was recently approved by US Food and Drug Administration for patients with stable, symptomatic chronic HF with LVEF ≤35 %, who are in sinus rhythm with resting HR ≥70 bpm and either are on maximally tolerated doses of beta-blockers, or have a contraindication to beta-blockers [17]. This article will review and evaluate the data supporting the use of ivabradine in patients with HF.
Mechanisms of Action and Adverse Reactions
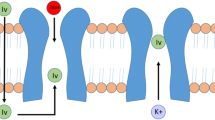
Ivabradine is a hyperpolarization-activated cyclic nucleotide-gated channel blocker that blunts spontaneous pacemaker activity of the sinus node by selectively inhibiting the Ifcurrent, resulting in HR reduction [18] (Fig. 1). At therapeutic doses, the drug does not act on other cardiac ion currents and has no direct effects on myocardial contractility or relaxation, cardiac output, coronary hemodynamics, blood pressure, or peripheral resistance in humans. HR reduction with ivabradine is approximately 10 bpm at rest and during exercise, and the size of its effect is dependent on baseline HR. The starting dose is 2.5–5 mg twice daily up to a maximum of 7.5 mg twice daily. If symptoms of bradycardia appear or HR < 50 bpm, the dose needs to be decreased by 2.5 mg twice daily or stopped [19].

a Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels allow the passage of the funny current. b Current-dependent block of HCN channels by ivabradine
Most common adverse events are bradycardia 10 %, HTN 8.9 %, atrial fibrillation 8.3 %, and phosphenes/visual brightness 2.8 % [19]. Ivabradine is contraindicated in patients with resting HR < 60 bpm, acute decompensated HF, hypotension <90/50 mmHg, sick sinus syndrome, sinoatrial block, third degree AV block, severe hepatic impairment, pacemaker dependence, and a concomitant use of strong CYP3A4 inhibitors (azole antifungals, macrolide, clarithromycin, HIV protease inhibitors).
Risk factors for bradycardia include sinus node dysfunction, conduction abnormalities, and the use of other negative chronotropes such as verapamil or diltiazem. In pooled data from BEAUTIFUL and SHIFT, atrial fibrillation (AF) was reported as an adverse event in 8 % of patients on ivabradine and 7 % of patients on placebo (p < 0.001) [20••]. More recently, in 2014, a meta-analysis of all ivabradine clinical trial data (including SHIFT and BEAUTIFUL) found that ivabradine treatment is associated with a 15 % increase in the relative risk (RR) of AF: in other words, 208 patient-years of treatment would be required to cause one new case of AF [21••]. Interestingly, AF incidence was greater in patients with higher baseline HR, raising the possibility that those with the most to gain from ivabradine HR reduction, also are at highest risk of developing AF [21••]. Figure 2 shows the physiologic impact of ivabradine.

Physiologic effects of ivabradine
Preclinical Studies
Preclinical studies with ivabradine (known as S-16257 during development) focused on establishing the direct cellular and physiologic effects in animal models. In diabetic mice, treatment with ivabradine for 3 months attenuated matrix metalloproteinase 2 (MMP2) expression, induced dephosphorylation of caspase 3, and enhanced phosphorylation of NF-kB, resulting in improvement in cardiac function via inhibition of cardiomyocyte apoptotic pathways [22]. Apoptosis was also inhibited by ivabradine in mice models of HF induced by angiotensin II injections [23]. Ivabradine also reduced LV expression of hypoxia-induced factor (HIF), increased endothelial cell proliferation, endothelial NOS expression, and improved coronary vasodilation in HF murine models [24].
Physiologically, treatment with ivabradine improved filling time, and reduced isovolumic contraction and relaxation times in conscious pig models [25]. This also led to decreased production of immune activation markers (TNF-α and IL-6) [26] and decreased myocardial fibrosis in murine models of viral myocarditis [27]. Ivabradine, but not metoprolol, decreased LV remodeling and hypertrophy in hypertension murine models [23]. Ivabradine also attenuates angiotensin pathway [24], via decrease in protein expression of angiotensin converting enzyme (ACE) and angiotensin I (AT-1) receptors leading to decreased remodeling in HF models [28]. In acute myocardial ischemia models, ivabradine improved reperfusion, decreased LV hypoxic lesion size, but was also associated with a 2.9-fold increase in threshold for ventricular fibrillation [29, 30]. Interestingly, in coronary artery ligation rabbit models treated with ivabradine or placebo and followed for 28 days, mortality was improved in the treatment group [31].
Taken together, the data suggest that ivabradine has effects beyond heart rate reduction, by inhibiting apoptotic pathways, decreasing inflammatory markers, enhancing reperfusion after myocardial ischemia, and possibly improving mortality. It is unclear, however, whether these effects are direct effects of ivabradine or effects resulting from HR reduction.
Early Clinical Studies
The initial promising results of animal studies led to human investigations in multiple early human trials focusing on safety and efficacy of the drug. Phase I studies in humans established that the plasma levels of ivabradine and its active metabolite (N-desmethylivabradine) were dose dependent in a linear fashion [32, 33]. For example, in a study of 36 healthy volunteers treated with ivabradine for 6 days, linear Cmax and area under the curve of plasma levels showed linear increase with dose and time. The mean heart rate reduction was 12.5 and 20.5 bpm for the 5 and 20 mg, without changes in blood pressures [33]. Similarly, in another placebo-controlled trial of 123 patients presenting with ST segment elevation myocardial infarction (STEMI), intravenous ivabradine reduced heart rate by 22 bpm and LV volumes, without affecting blood pressure, cardiac biomarkers or LVEF [34].
Longer-term studies in patients with HF also proved encouraging. In a single-arm study of 35 patients with dilated cardiomyopathy and EF < 40 %, HR > 70 on OMT, escalating doses of ivabradine for 3 months reduced HR by 25.9 % without changing BP, reduced PVCs, and was generally well tolerated [35]. Studies comparing ivabradine with beta-blockers, however, showed conflicting results. In a randomized trial of 121 patients with HF assigned to carvedilol, ivabradine, or combination, ivabradine-treated patients had better 6MWT, exercise time, peak VO2, and quality of life than those on carvedilol alone [36]. Similarly, in 221 patients with systolic HF randomized to 1-month treatment with ivabradine (5 mg twice daily) of beta-blockers (carvedilol or bisoprolol), the ivabradine group showed improved physical and emotional functioning and mental health scales than beta-blockers despite minimal decrease in heart rate (63 in ivabradine vs 67 beta-blockers bpm) [37].
As a consequence of initial preclinical and clinical experience, larger multicenter randomized trials focusing on major adverse cardiovascular outcomes have been performed and will be discussed below. It must be noted, however, that there still remains a lack of human studies confirming the pleiotropic effects of ivabradine seen in animal models.
Pivotal Clinical Trials
SHIFT (Systolic Heart failure treatment with the If inhibitor ivabradine Trial) was a randomized, double-blind, placebo-controlled, international trial that included 6505 patients with symptomatic chronic HF, New York Heart Association (NYHA) classes II–IV, LVEF ≤35 % and in sinus rhythm with HR ≥70 bpm that tested the hypothesis that HR reduction per se could improve cardiovascular outcomes [38, 39]. This study showed that for every 1-beat increase in HR, the risk for the primary composite of cardiovascular mortality and hospital admission for worsening HF increased by 3 % and for every 5-bpm increase in HR, the risk increased by 16 % in the placebo group [38]. Patients with baseline HR ≥87 bpm were at more than twofold increased risk for the primary composite compared with those with HR 70 to 72 bpm (HR = 2 · 34, 95 % CI 1 · 84–2 · 98, p < 0 · 0001) and had the greatest reductions in HR with ivabradine (22.5 bpm) [38]. HR in patients on ivabradine fell by a mean 15 · 4 bpm at 28 days compared with pretreatment. Patients with HR < 60 bpm at 28 days on treatment had fewer primary composite endpoint events during the study (event rate 17 · 4 %, 95 % CI 15 · 3–19 · 6) than did patients with higher HR. The neutralization of the treatment effect study after adjustment for change in HR at 28 days (HR 0 · 95, 0 · 85–1 · 06, p = 0 · 352) showed that the effect of ivabradine could be accounted for by heart rate reduction [38].
Hospital admissions for worsening HF occurred in 21 % of patients on placebo versus 16 % of those taking ivabradine (HR 0 · 74, 95 % CI 0 · 66–0 · 83, p < 0 · 0001). HF deaths but not cardiovascular deaths were significantly reduced in the ivabradine group (HR 0 · 74, 95 % CI 0 · 58–0 · 94, p = 0 · 014). Although small, there was a significant improvement in NYHA class, 28 % of patients on ivabradine improved versus 24 % of patients on placebo (p = 0 · 001), Figs. 3 and 4.

Kaplan-Meier cumulative event curves for a death from heart failure and b all-cause death (reproduced with permission from Swedberg et al. 2010)

Kaplan-Meier cumulative event curves for a the primary composite endpoint of cardiovascular death or hospital admission for worsening heart failure and b hospital admission for worsening heart failure (reproduced with permission from Swedberg et al. 2010)
Serious adverse events occurred at a lower rate in the ivabradine group than in the placebo group (p = 0 · 025). Symptomatic and asymptomatic bradycardia was recorded in about 10 % of patients, but only in 1 % it did lead to study withdrawal. There were no difficulties in initiation, uptitration, or continuation of β-blocker treatment, proving that ivabradine is well tolerated in HF patients on β-blockers [38].
In BEAUTIFUL (Morbidity–Mortality Evaluation of the If Inhibitor Ivabradine in Patients with Coronary Artery Disease and Left Ventricular Systolic Dysfunction), trial ivabradine treatment did not affect the primary composite outcome cardiovascular death, or admission to hospital for new-onset or worsening HF in patients with CAD and LVEF < 40 %. These findings were explained by insufficient reductions in HR (6 bpm at 12 months and 5 bpm at 24 months) and/or from low HR at baseline [40]. In a subset of patients with baseline HR of 70 bpm and LVEF < 40 %, ivabradine was associated with a 36 % decrease in hospital admissions secondary to fatal and nonfatal myocardial infarction and a 30 % decrease in coronary revascularization. These findings suggest that ivabradine can be given safely in conjunction with β-blockers and can improve outcomes in CAD patients with HR ≥ 70 bpm.
In the INTENSIFY study (PractIcal daily effectiveNess and TolEraNce of ivabradine in chronic SystolIc heart Failure in GermanY), over 4 months of treatment, ivabradine effectively reduced HR and symptoms in CHF patients [41]. The proportion of patients with a LVEF ≤35 % decreased from 26.6 to 17.4 %, ADHF declined from 22.7 to 5.4 %, and the proportion of patients with BNP level ≥400 ng/mL decreased from 53.9 to 26.5 % over a 4-month period. The mean value of the QOL EQ-5D sum score index improved to 0.79 ± 0.21 from 0.64 ± 0.28 at baseline. Lastly, physician investigators rated effectiveness of ivabradine as “very good” in 54.9 % and “good” in 41.5 % of patients.
In the INITIATIVE trial (INternatIonal TrIal of the AnTi-anginal effects of IVabradinE), ivabradine was compared directly to atenolol in doses selected to achieve similar HR reductions [42]. HR decrement at peak exercise was greater with atenolol (14 bpm) than with ivabradine (8.6–10.3 bpm). However, ivabradine was non-inferior to atenolol for all exercise parameters, showing that it induced similar or greater improvement in exercise capacity than atenolol with comparatively smaller reductions in rate pressure product and HR [42]. Table 1 summarizes these studies.
Echocardiographic and Hemodynamic Effects
In a dog model of HF produced by intracoronary microembolizations (LVEF = 35 %), ivabradine prevented LV dilatation, significantly decreased end-systolic volumes and increased stroke volumes, improving LVEF and fractional shortening, while also reducing NT-pro BNP and pro-ANP [43]. In addition, LV diastolic function and SR calcium handling were improved by increased E’/A’, prolonged deceleration time, and lower LV end-diastolic circumferential wall stress [43].
These animal results were replicated by a large Echo substudy of the SHIFT trial that analyzed 208 patients on ivabradine and 203 on placebo at baseline and after 8 months, with the primary endpoint of change of left ventricular systolic volume index (LVESVI) [44]. Ivabradine treatment resulted in a 7 mL/m2 reduction of LVESVI, as compared with 0.9 mL/m2 in the placebo group, and an increase in LVEF of 2.4 %, with no change in the placebo group [44]. A total of 38 % of patients on ivabradine had a decrease in LVESVI of at least 15 %, and 36 % had an increase of LVEF of at least 5 % after 8 months. Larger decreases in HR were associated with greater increases in LVEF (r = −0.17, P = 0.0006). No significant changes in left-atrial end-systolic volume index, RV myocardial performance index, or mitral regurgitation were observed in either group, although RV s wave peak velocity increased over with ivabradine and decreased with placebo. LV diastolic function improved in 22 % of ivabradine patients by at least one grade, versus only in 10 % of those on placebo (P = 0.02).
Still, about 50 % of patients taking additional ivabradine on a very well optimized population (92 % were on beta-blockers and 94 % on a RAS antagonist) experienced no change in LVESVI. However, in an analysis that used 275 of the 411 patients of the SHIFT Echo substudy, the significant HR reduction in the ivabradine group was accompanied by marked reduction in effective arterial elastance (p < 0.0001)—a parameter representing the pulsatile and mean load of the left ventricle [45•]. Ivabradine also improved total arterial compliance defined as the ratio of SV and pulse pressure (p = 0.004). Although contractility remained unchanged, ventricular-arterial coupling was markedly improved (p = 0.002), resulting in a higher SV (p < 0.0001) in the ivabradine-treated patients [45•].
Ivabradine in HFpEF
In addition to other abnormalities, patients with heart failure with preserved ejection fraction (HFpEF) have excessive tachycardia during exercise likely due to impaired stroke volume reserve and overreliance on heart rate to augment cardiac output [46]. The role of therapeutic bradycardia in HFpEF is controversial due to inconsistent results of beta-blockers on LV diastolic function and exercise tolerance in these patients. Because ivabradine can delay diastolic filling time by HR reduction, augmenting stroke volume and cardiac output without affecting inotropy, it could be a putative option in HFpEF patients. Supporting this hypothesis, a recent study of 61 patients with HFpEF randomly assigned to ivabradine 5 mg twice daily or placebo for 7 days demonstrated significant improvement of exercise capacity (4.2 ± 1.8 METs vs 5.7 ± 1.9 METs, p = 0.001) and peak oxygen uptake (14.0 ± 6.1 ml/min/kg vs 17.0 ± 3.3 ml/min/kg, p = 0.001), with simultaneous reduction in exercise-induced increase in the ratio of peak early diastolic mitral flow velocity to peak early diastolic mitral annular velocity (3.1 ± 2.7 vs 1.3 ± 2.0, p = 0.004) [46]. While these results are promising, these observations need to be replicated in larger cohorts of patients.
Ivabradine in Cardiogenic Shock
Several case reports and small observational case series suggest beneficial effects of ivabradine in the management of cardiogenic shock. For example, in 58 ADHF patients requiring inotropic support with LVEF < 35 %, ivabradine prevented dobutamine-induced tachycardia and its untoward effects [47]. Also, ivabradine successfully reduced HR and improved cardiogenic shock in a patient with anterior STEMI who had persistent sinus tachycardia and cardiogenic shock despite revascularization and IABP [48]. Similarly, Zwicker et al. reported a case of cardiogenic shock due to tachycardia-induced cardiomyopathy after heart transplantation that was treated effectively with ivabradine [49]. Roubille et al. reported a case of acute idiopathic heart failure evolving to cardiogenic shock that also resolved with ivabradine [50]. To verify these anecdotal reports, the MODI(f)Y trial is a prospective single-center open-label randomized controlled phase II-trial that has been designed to evaluate the effect of ivabradine on patients with newly diagnosed multi-organ dysfunction syndrome, who will be treated for 4 days and followed for 6 months [51].
Inappropriate Sinus Tachycardia and Tachyarrhythmia-Induced Cardiomyopathy
Inappropriate sinus tachycardia (IST) is a clinical syndrome with a relative or absolute increase in sinus rate out of proportion to physiologic need [52] that can occasionally result in tachyarrhythmia-induced cardiomyopathy. Whereas, beta-blockers and non-dihydropyridine calcium channel blockers are considered first-line therapy for isolated IST [52], patients with heart failure and/or low blood pressure may not tolerate these drugs. Eighteen symptomatic with inappropriate sinus tachycardia who underwent 24-h Holter ECG, and exercise ECG at baseline and at 3 and 6 months, showed a significant reduction of medium HR (P = 0.001) and maximal HR (p = 0.001, basal vs 3–6 months; p = 0.02, 3 vs 6 months) [53]. Exercise ECG showed a significant decrease of basal HR and of HR reached at maximal load, suggesting an increased tolerance to physical stress following ivabradine and confirmed by a progressive increase of maximal load reached during stress test at 3 and 6 months. Another group of 24 IST patients treated with ivabradine showed both HR normalization and quality-of-life improvement maintained in the long-term follow-up. Stopping ivabradine after 1 year unexpectedly showed that HR remained in the normal limits in 80 % of the patients [54].
This drug could represent a second-line therapy in patients with IST who are refractory or intolerant to beta-blockers and non-dihydropyridine calcium channel blockers.
Ivabradine in Heart Transplant
In 30 patients with heart transplant and marked symptomatic sinus tachycardia, ivabradine reduced the mean HR from 96.2+/−8.6 bpm at baseline to 80.9+/−8.1 bpm follow-up (p < 0.0001) [55]. A statistically significant effect of HR reduction on left ventricular mass index was observed (104.3+/−22.7 g at baseline vs. 95.9+/−18.5 g at follow-up at 12 months, p = 0.04). Ivabradine remained effective and safe in chronic stable patients after heart transplantation during 36-month long-term follow-up [56] and after 1.13 years, associated with improvement in symptoms [57]. Sixteen heart transplant (HTX) recipients with sinus rhythm >90/min were prospectively studied while treated with ivabradine and performed cardiopulmonary exercise testing (CPET), using a cycle ergometer and a modified Naughton protocol after a median of 6 weeks. A significant reduction in HR was observed in HTX recipients with ivabradine at rest and at maximum exercise but the functional parameters in CPET were unaffected.
Conclusions
In patients with systolic HF and elevated HR > 70 bpm despite being on beta-blockers or intolerant of beta-blockers, ivabradine can be of value in decreasing heart rate, HF hospitalizations and HF death. The magnitude of HR reduction beyond what is achieved by a beta-blocker, rather than background beta-blocker dose, primarily determines subsequent outcome [58]. Also, greater reduction in HR is associated with greater improvements in LVEF [59, 60]. Whether HR reduction may serve as a useful surrogate marker for magnitude of LVEF improvement needs to be explored. The HR reduction with ivabradine also significantly decreases NT proBNP, cystatin-C and CA-125 [61].
There is accumulating evidence that HF treatment targeted to beta-blocker doses may not be sufficient to achieve clinical outcomes but rather should be aimed at cumulative HR reduction [11, 12, 58, 62, 63]. A Japanese study showed that the delta HR decrease from admission to discharge was the strongest predictor of cardiac events in patients with ADHF in patients receiving beta-blockers [63].
The clear benefit observed with lowering the HR in patients with HF but not in those with stable CAD may reflect the fact that an elevated HR is due to different pathophysiological mechanisms in these two conditions. In HF, the possible explanations for the benefits of lowering HR are decreased energy expenditure, increased blood supply by prolonging diastole, and improvement in vivo and in vitro force-frequency associations, unloading the ventricle, and decreased mechanical dyssynchrony.
In conclusion, HR plays an important role in the pathophysiology and progression of HF and should therefore be considered as a therapeutic target. The addition of ivabradine to patients whose HR remain above 70 bpm despite being on maximally tolerated doses of beta-blockers, or in those intolerant to them, is associated with favorable echocardiographic, hemodynamic and clinical outcomes.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16):e240–327.
Heidenreich PA, Sahay A, Kapoor JR, Pham MX, Massie B. Divergent trends in survival and readmission following a hospitalization for heart failure in the veterans affairs health care system 2002 to 2006. J Am Coll Cardiol. 2010;56(5):362–8.
Rush CJ, Campbell RT, Jhund PS, et al. Falling cardiovascular mortality in heart failure with reduced ejection fraction and implications for clinical trials. JACC Heart Fail. 2015;3(8):603–14.
Foody JM, Farrell MH, Krumholz HM. beta-Blocker therapy in heart failure: scientific review. JAMA. 2002;287(7):883–9.
Tardif JC. Heart rate as a treatable cardiovascular risk factor. Br Med Bull. 2009;90:71–84.
Jensen MT, Suadicani P, Hein HO, Gyntelberg F. Elevated resting heart rate, physical fitness and all-cause mortality: a 16-year follow-up in the Copenhagen male study. Heart. 2013;99(12):882–7.
Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R. Heart rate as a prognostic risk factor in patients with coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a subgroup analysis of a randomised controlled trial. Lancet. 2008;372(9641):817–21.
Mulieri LA, Hasenfuss G, Leavitt B, Allen PD, Alpert NR. Altered myocardial force-frequency relation in human heart failure. Circulation. 1992;85(5):1743–50.
Lechat P, Hulot JS, Escolano S, et al. Heart rate and cardiac rhythm relationships with bisoprolol benefit in chronic heart failure in CIBIS II trial. Circulation. 2001;103(10):1428–33.
Metra M, Torp-Pedersen C, Swedberg K, et al. Influence of heart rate, blood pressure, and beta-blocker dose on outcome and the differences in outcome between carvedilol and metoprolol tartrate in patients with chronic heart failure: results from the COMET trial. Eur Heart J. 2005;26(21):2259–68.
McAlister FA, Wiebe N, Ezekowitz JA, Leung AA, Armstrong PW. Meta-analysis: beta-blocker dose, heart rate reduction, and death in patients with heart failure. Ann Intern Med. 2009;150(11):784–94.
Flannery G, Gehrig-Mills R, Billah B, Krum H. Analysis of randomized controlled trials on the effect of magnitude of heart rate reduction on clinical outcomes in patients with systolic chronic heart failure receiving beta-blockers. Am J Cardiol. 2008;101(6):865–9.
Franke J, Wolter JS, Meme L, et al. Optimization of pharmacotherapy in chronic heart failure: is heart rate adequately addressed? Clin Res Cardiol Off J German Card Soc. 2013;102(1):23–31.
Franke J, Schmahl D, Lehrke S, et al. Adjuvant use of ivabradine in acute heart failure due to myocarditis. Case Rep Med. 2011;2011:203690.
Elder DH, Mohan M, Cochrane L, Charles H, Lang CC. Characterizing patients with chronic heart failure in community care after hospitalization: a potential role for ivabradine. Cardiovasc Ther. 2015;33(3):104–8.
Cullington D, Goode KM, Cleland JG, Clark AL. Limited role for ivabradine in the treatment of chronic heart failure. Heart. 2011;97(23):1961–6.
FDA approves Corlanor to treat heart failure. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm442978.htm. Accessed 2015/9/15, 2015.
Sulfi S, Timmis AD. Ivabradine—the first selective sinus node I(f) channel inhibitor in the treatment of stable angina. Int J Clin Pract. 2006;60(2):222–8. 10//received 12//accepted.
Amgen. Corlanor prescribing information. 2015.
Fox K, Komajda M, Ford I, et al. Effect of ivabradine in patients with left-ventricular systolic dysfunction: a pooled analysis of individual patient data from the BEAUTIFUL and SHIFT trials. Eur Heart J. 2013;34(29):2263–70. This is a pooled analysis of two major trials of Ivabradine in left ventricular systolic dysfunction.
Martin RI, Pogoryelova O, Koref MS, Bourke JP, Teare MD, Keavney BD. Atrial fibrillation associated with ivabradine treatment: meta-analysis of randomised controlled trials. Heart. 2014;100(19):1506–10. This is a metaanalysis of atrial fibrillation associated wit ivabradine.
Chen SL, Hu ZY, Zuo GF, Li MH, Li B. I(f) current channel inhibitor (ivabradine) deserves cardioprotective effect via down-regulating the expression of matrix metalloproteinase (MMP)-2 and attenuating apoptosis in diabetic mice. BMC Cardiovasc Disord. 2014;14:150.
Becher PM, Lindner D, Miteva K, et al. Role of heart rate reduction in the prevention of experimental heart failure: comparison between If-channel blockade and beta-receptor blockade. Hypertension. 2012;59(5):949–57.
Fang Y, Debunne M, Vercauteren M, et al. Heart rate reduction induced by the if current inhibitor ivabradine improves diastolic function and attenuates cardiac tissue hypoxia. J Cardiovasc Pharmacol. 2012;59(3):260–7.
Rienzo M, Melka J, Bize A, et al. Ivabradine improves left ventricular function during chronic hypertension in conscious pigs. Hypertension. 2015;65(1):122–9.
Yue-Chun L, Teng Z, Na-Dan Z, et al. Comparison of effects of ivabradine versus carvedilol in murine model with the coxsackievirus B3-induced viral myocarditis. PLoS One. 2012;7(6):e39394.
Li YC, Luo Q, Ge LS, et al. Ivabradine inhibits the production of proinflammatory cytokines and inducible nitric oxide synthase in acute coxsackievirus B3-induced myocarditis. Biochem Biophys Res Commun. 2013;431(3):450–5.
Milliez P, Messaoudi S, Nehme J, Rodriguez C, Samuel JL, Delcayre C. Beneficial effects of delayed ivabradine treatment on cardiac anatomical and electrical remodeling in rat severe chronic heart failure. Am J Physiol Heart Circ Physiol. 2009;296(2):H435–41.
Vaillant F, Timour Q, Descotes J, et al. Ivabradine induces an increase in ventricular fibrillation threshold during acute myocardial ischemia: an experimental study. J Cardiovasc Pharmacol. 2008;52(6):548–54.
Vaillant F, Dehina L, Mazzadi A, et al. Heart rate reduction with ivabradine increases ischaemia-induced ventricular fibrillation threshold: role of myocyte structure and myocardial perfusion. Resuscitation. 2011;82(8):1092–9.
Langenbach MR, Schmitz-Spanke S, Brockert M, et al. Comparison of a beta-blocker and an If current inhibitor in rabbits with myocardial infarction. J Cardiovasc Surg. 2006;47(6):719–25.
Choi HY, Noh YH, Cho SH, et al. Evaluation of pharmacokinetic and pharmacodynamic profiles and tolerability after single (2.5, 5, or 10 mg) and repeated (2.5, 5, or 10 mg bid for 4.5 days) oral administration of ivabradine in healthy male Korean volunteers. Clin Ther. 2013;35(6):819–35.
Jiang J, Tian L, Huang Y, Li Y, Xu L. Pharmacokinetic and safety profile of ivabradine in healthy Chinese men: a phase I, randomized, open-label, increasing single- and multiple-dose study. Clin Ther. 2013;35(12):1933–45.
Steg P, Lopez-de-Sa E, Schiele F, et al. Safety of intravenous ivabradine in acute ST-segment elevation myocardial infarction patients treated with primary percutaneous coronary intervention: a randomized, placebo-controlled, double-blind, pilot study. Eur Heart J Acute Cardiovasc Care. 2013;2(3):270–9.
Rayan M, Tawfik M, Alabd A, Gamal A. Ivabradine, a novel heart rate slower: is it a sword of double blades in patients with idiopathic dilated cardiomyopathy? Anadolu Kardiyoloji Dergisi: AKD Anatol J Cardiol. 2011;11(5):402–6.
Volterrani M, Cice G, Caminiti G, et al. Effect of carvedilol, ivabradine or their combination on exercise capacity in patients with heart failure (the CARVIVA HF trial). Int J Cardiol. 2011;151(2):218–24.
Riccioni G, Masciocco L, Benvenuto A, et al. Ivabradine improves quality of life in subjects with chronic heart failure compared to treatment with beta-blockers: results of a multicentric observational APULIA study. Pharmacology. 2013;92(5–6):276–80.
Swedberg K, Komajda M, Bohm M, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376(9744):875–85.
Swedberg K, Komajda M, Bohm M, Borer JS, Ford I, Tavazzi L. Rationale and design of a randomized, double-blind, placebo-controlled outcome trial of ivabradine in chronic heart failure: the Systolic Heart Failure Treatment with the I(f) Inhibitor Ivabradine Trial (SHIFT). Eur J Heart Fail. 2010;12(1):75–81.
Fox K, Ford I, Steg PG, Tendera M, Ferrari R, Investigators B. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372(9641):807–16.
Zugck C, Martinka P, Stockl G. Ivabradine treatment in a chronic heart failure patient cohort: symptom reduction and improvement in quality of life in clinical practice. Adv Ther. 2014;31(9):961–74.
Tardif JC, Ford I, Tendera M, Bourassa MG, Fox K. Efficacy of ivabradine, a new selective I(f) inhibitor, compared with atenolol in patients with chronic stable angina. Eur Heart J. 2005;26(23):2529–36.
Sabbah HN, Gupta RC, Kohli S, Wang M, Zhang K, Rastogi S. Heart rate reduction with ivabradine improves left ventricular function and reverses multiple pathological maladaptations in dogs with chronic heart failure. ESC Heart Fail. 2014;1(2):94–102.
Tardif JC, O’Meara E, Komajda M, et al. Effects of selective heart rate reduction with ivabradine on left ventricular remodelling and function: results from the SHIFT echocardiography substudy. Eur Heart J. 2011;32(20):2507–15.
Reil JC, Tardif JC, Ford I, et al. Selective heart rate reduction with ivabradine unloads the left ventricle in heart failure patients. J Am Coll Cardiol. 2013;62(21):1977–85. This substudy of SHIFT provides evidence for the effects of ivabradine on LV unloading.
Kosmala W, Holland DJ, Rojek A, Wright L, Przewlocka-Kosmala M, Marwick TH. Effect of If-channel inhibition on hemodynamic status and exercise tolerance in heart failure with preserved ejection fraction: a randomized trial. J Am Coll Cardiol. 2013;62(15):1330–8.
Cavusoglu Y, Mert U, Nadir A, Mutlu F, Morrad B, Ulus T. Ivabradine treatment prevents dobutamine-induced increase in heart rate in patients with acute decompensated heart failure. J Cardiovasc Med. 2015;16(9):603–9.
Bonadei I, Sciatti E, Vizzardi E, D’Aloia A, Metra M. Ivabradine during cardiogenic shock: a clinical case and review of the literature. Heart Lung J Crit Care. 2015;44(1):57–8.
Zwicker C, Becker M, Lepper W, Koch KC, Westenfeld R. Cardiogenic shock due to tachycardiomyopathy after heart transplantation: successful treatment with ivabradine. Cardiology. 2010;116(3):174–7.
Roubille F, Lattuca B, Busseuil D, et al. Is ivabradine suitable to control undesirable tachycardia induced by dobutamine in cardiogenic shock treatment? Med Hypotheses. 2013;81(2):202–6.
Nuding S, Ebelt H, Hoke RS, et al. Reducing elevated heart rate in patients with multiple organ dysfunction syndrome by the I (f) (funny channel current) inhibitor ivabradine: MODI (f)Y trial. Clin Res Cardiol Off J German Card Soc. 2011;100(10):915–23.
Krahn AD, Yee R, Klein GJ, Morillo C. Inappropriate sinus tachycardia: evaluation and therapy. J Cardiovasc Electrophysiol. 1995;6(12):1124–8.
Calo L, Rebecchi M, Sette A, et al. Efficacy of ivabradine administration in patients affected by inappropriate sinus tachycardia. Heart Rhythm Off J Heart Rhythm Soc. 2010;7(9):1318–23.
Benezet-Mazuecos J, Rubio JM, Farre J, Quinones MA, Sanchez-Borque P, Macia E. Long-term outcomes of ivabradine in inappropriate sinus tachycardia patients: appropriate efficacy or inappropriate patients. Pacing Clin Electrophysiol PACE. 2013;36(7):830–6.
Doesch AO, Ammon K, Konstandin M, et al. Heart rate reduction for 12 months with ivabradine reduces left ventricular mass in cardiac allograft recipients. Transplantation. 2009;88(6):835–41.
Doesch AO, Mueller S, Erbel C, et al. Heart rate reduction for 36 months with ivabradine reduces left ventricular mass in cardiac allograft recipients: a long-term follow-up study. Drug Des Dev Ther. 2013;7:1323–8.
Lage-Galle E, Romero-Rodriguez N, Nevado-Portero J, et al. Safety and effectiveness of ivabradine after cardiac transplantation. Transplant Proc. 2010;42(8):3191–2.
Swedberg K, Komajda M, Bohm M, et al. Effects on outcomes of heart rate reduction by ivabradine in patients with congestive heart failure: is there an influence of beta-blocker dose?: findings from the SHIFT (Systolic Heart failure treatment with the I(f) inhibitor ivabradine Trial) study. J Am Coll Cardiol. 2012;59(22):1938–45.
Metra M, Nodari S, Parrinello G, Giubbini R, Manca C, Dei Cas L. Marked improvement in left ventricular ejection fraction during long-term beta-blockade in patients with chronic heart failure: clinical correlates and prognostic significance. Am Heart J. 2003;145(2):292–9.
Packer M, Colucci WS, Sackner-Bernstein JD, et al. Double-blind, placebo-controlled study of the effects of carvedilol in patients with moderate to severe heart failure. The PRECISE trial. Prospective Randomized Evaluation of Carvedilol on Symptoms and Exercise. Circulation. 1996;94(11):2793–9.
Ordu S, Yildiz BS, Alihanoglu YI, et al. Effects of ivabradine therapy on heart failure biomarkers. Cardiol J. 2015;22(5):501–9.
Cullington D, Goode KM, Clark AL, Cleland JG. Heart rate achieved or beta-blocker dose in patients with chronic heart failure: which is the better target? Eur J Heart Fail. 2012;14(7):737–47.
Takahama H, Yokoyama H, Kada A, et al. Extent of heart rate reduction during hospitalization using beta-blockers, not the achieved heart rate itself at discharge, predicts the clinical outcome in patients with acute heart failure syndromes. J Cardiol. 2013;61(1):58–64.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Pharmacologic Therapy
Rights and permissions
About this article

Cite this article
Orasanu, G., Al-Kindi, S.G. & Oliveira, G.H. Ivabradine in Management of Heart Failure: a Critical Appraisal. Curr Heart Fail Rep 13, 60–69 (2016). https://doi.org/10.1007/s11897-016-0276-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-016-0276-x