
Abstract
Purpose of Review
While commonly associated with pulmonary manifestations, cystic fibrosis (CF) is a systemic disease with wide-ranging effects on the gastrointestinal (GI) tract. This article reviews major recent updates in gastroenterological CF care and research.
Recent Findings
The high burden of GI symptoms in CF has led to recent studies assessing GI-specific symptom questionnaires and scoring systems. Intestinal dysbiosis potentially contributes to gastrointestinal symptoms in patients with CF and an increased risk of gastrointestinal cancers in CF. An increased incidence of colorectal cancer (CRC) has led to CF-specific CRC screening and surveillance recommendations. Pharmacologic therapies targeting specific cystic fibrosis transmembrane conductance regulator (CFTR) mutations have shown promise in treating GI manifestations of CF.
Summary
New research has highlighted the importance of intestinal dysbiosis in CF. Future studies should assess whether CFTR modulators affect the gut microbiome and whether altering the gut microbiome will impact GI symptoms and GI cancer risk.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cystic Fibrosis (CF) is an autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR), an anion channel expressed in epithelial cells. Mutations in CFTR result in impaired Cl− and HCO3− transport in the lungs, pancreas, gastrointestinal tract, and sweat glands [1••]. CFTR mutations can be classified by the resultant functional defect (Table 1). CF affects approximately 80,000 people worldwide [2]. Although known for its pulmonary complications, cystic fibrosis is associated with a high burden of gastrointestinal (GI) symptoms, and was in fact named for its pancreatic manifestations. Patients with CF commonly report abdominal pain [3] and other GI symptoms [4] which may impact quality of life and treatment adherence.
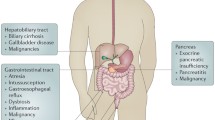
CF is associated with many gastrointestinal manifestations (Fig. 1). Effects of CFTR dysfunction on the digestive system are evident early and feature prominently. CF mouse models display poor growth and potentially lethal bowel obstruction [5]. CF may present in infancy with gastrointestinal obstruction (meconium ileus), poor digestive function due to inability to neutralize gastric acid, and exocrine pancreatic insufficiency (EPI) leading to nutritional deficiencies and failure to thrive. About 85% of the CF population develops EPI during the first year of life [6]. Pancreatic sufficient patients are at risk for pancreatitis. Although endocrine pancreatic function is relatively preserved early in life, the gradual destruction of islet cells in many patients with CF leads to a high prevalence of cystic fibrosis-related diabetes (CFRD) in adults. Gastroesophageal reflux, constipation, gastroparesis, cholelithiasis, and small intestinal bacterial overgrowth are common. Potentially life-threatening complications include liver disease and distal intestinal obstruction syndrome (DIOS), in which inspissated stool in the ileocecum leads to bowel obstruction. CF patients have an increased risk for gastrointestinal cancers [7] as detailed below.

Potential impact of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) modulators on gastrointestinal manifestations of cystic fibrosis (CF). The boxed panel on the left highlights gastrointestinal complications of cystic fibrosis, grouped primarily by organ. The inset graphic in the lower right represents alterations in the fecal microbiome (dysbiosis). Inhibitory arrows (dashed because they are predominantly theoretical at this time) represent potential effects of CFTR modulators on gastrointestinal symptoms, dysbiosis, and other gastrointestinal complications of CF. GI, Gastrointestinal
The purpose of this review is to highlight topics of current importance in the gastroenterological realm of CF. While not exhaustive, the following topics encompass important recent scholarship or emerging therapies. We will discuss the assessment of GI-specific symptom questionnaires and scoring systems in CF, followed by the potential role of intestinal dysbiosis in GI manifestations of CF. Recent screening recommendations address the increased risk of colon cancer in CF, which likely results in part from intestinal dysbiosis. We will conclude with a discussion of CFTR modulators and their potential role in altering GI symptoms or complications in CF. Figure 1 depicts the themes covered in this literature review and their potential interdependence.
Patient-Reported CF Gastrointestinal Symptom Evaluations
Although often considered primarily a pulmonary disease, CF is associated with a higher burden of GI complications compared to the general population [8]. While the GI manifestations of CF may begin in infancy with the classic presentation of meconium ileus, the development of newer therapies has enabled survival of CF patients well into adulthood—among people with CF born in the United States between 2013 and 2017, half are predicted to live to 44 years old or more [9]. Despite this, the overall burden of CF on GI-specific symptoms and overall quality of life (QOL) is not well understood. A major focus of recent scholarship has been the development of patient-centered systems to quantitatively and qualitatively assess CF-related GI manifestations that have increasingly become chronic diseases of CF.
In one of the first efforts to validate a patient-reported abdominal-specific symptom assessment, Tabori et al. recruited 131 pediatric and adult patients to complete the JenAbdomen-CF Score 1.0, a questionnaire addressing GI symptoms from the preceding three months. Symptoms were grouped into four domains: abdominal pain, non-pain symptoms (e.g. nausea), subjective evaluation of feces’ frequency, form, and color, and disorders of eating and appetite [4]. The most commonly reported symptoms were lack of appetite, loss of taste, abdominal pain, flatulence, and distention. The authors also identified 7 conditions which were associated with significantly increased abdominal symptoms: history of rectal prolapse, distal intestinal obstruction syndrome, history of laparotomy, meconium ileus, pancreatic insufficiency, or small bowel resection, and intermittent colonization with P. aeruginosa.
Hayee et al. recruited 107 consecutive (i.e. non-selected) patients attending CF specialist appointments to complete pre-existing symptom surveys—the GI Symptom Rating Scale (GSRS); Irritable Bowel Syndrome Symptom Severity Score (IBS-SSS) and Cystic Fibrosis Questionnaire-Revised (CFQ-R)—in order to assess the burden of chronic or functional bowel symptoms in CF [10••]. This cohort was comprised of adult patients (mean age 27.8 years), 88% of whom were pancreatic insufficient. Excluding symptoms of pancreatic insufficiency, 65% reported significant GI symptoms. Lower GI tract symptoms (bloating, borbyrygmi, flatulence, and abdominal pain) were most common [10••].
This study suggests that many symptoms reported by CF patients are reminiscent of those seen in IBS. In fact, more CF patients had symptoms that met the Rome IV criteria for IBS than would be expected in the general population [10••]. Of the 2 scales used in the study, a group of patients reported significant symptoms in GSRS but not the SSS, suggesting that the GSRS may be more sensitive for this population [10••]. Due to similarities between CF-related GI symptoms and IBS, it has been posited that some therapies that have proven successful in IBS (e.g. linaclotide for the treatment of IBS-C) may be effective in CF [11].
A multicenter European consortium led by Boon and colleagues integrated a pre-existing GI symptom scale, the Pediatric Quality of Life Inventory, Gastrointestinal Symptoms Scales and Module (PedsQL GI), into a mobile application in order to validate the PedsQL GI in CF [12••]. The PedsQL GI had previously been validated in other pediatric populations (inflammatory bowel disease, gastroesophageal reflux disease, and functional GI disorders), but not in CF [13, 14]. Based on administration of the PedsQL GI in 248 pancreatic insufficient patients with CF age 24 months to 18 years, investigators found that the PedsQL GI was a valid, applicable instrument to assess GI related quality of life (QOL) in children with CF.
A UK-based group evaluated 276 responses to an online survey focused on the impact of gastrointestinal symptoms in cystic fibrosis [15•]. Distributed through online platforms including the Cystic Fibrosis Foundation, the United Kingdom CF Trust, and Twitter, the survey received anonymous responses from CF patients (n = 90), close family/friends (n = 79), and CF healthcare providers (n = 107). Healthcare providers reported that the most common symptoms described by CF patients or their caregivers were reduced appetite, bloating and constipation, while lay respondents most commonly reported stomach cramps/pain, bloating and a ‘combination of symptoms.’ Although 94% of healthcare providers felt that pharmacologic therapy helped to relieve GI symptoms, only 58% of lay respondents agreed [15•].
The aforementioned studies illustrate the power of mobile and web-based applications in performing CF symptom assessments, and may facilitate further research using digital platforms to assess CF symptoms. In addition, the Cystic Fibrosis Foundation-sponsored GALAXY study, currently underway, is a multicenter study which utilizes pre-existing patient-reported outcome measures—PAC-SYM, PAGI-SYM, PAC-QOL, and the Bristol Stool scale—with three additional symptom-specific questions [16•]. The GALAXY study aims to assess gastrointestinal symptoms in patients with CF and to develop an objective endpoint for future studies.
The Microbiome in CF
The successful development of Next Generation Sequencing technologies (NGS) has enabled assessment of the relationship between the intestinal microbiome and systemic disease [17]. Knowledge in this arena is rapidly expanding, and it is now widely accepted that the microbiome influences many diseases, including CF, in which mucous accumulation within the GI tract results in abnormal microbial colonization [18].
Multiple recent studies have shown that by infancy, the gut microbiome is altered in CF compared to healthy controls, both in terms of microbial diversity and overall composition [19, 20, 21••, 22, 23, 24]. A functioning pancreas does not seem to affect this dysbiosis [22, 25]. Interestingly, both the lung and gut microbiomes play a crucial role in the expression of CF, and one may influence the other. Madan et al. and Hoen et al. have shown a relationship between the development of the gut and lung microbiomes [26, 27]. Intestinal microbial dysbiosis may be further exacerbated by antibiotic therapy commonly used to treat pulmonary infections in CF [28].
The clinical impact of intestinal dysbiosis in CF is unclear, but a recent body of evidence suggests that it may play a role in disease. Antosca et al. showed a lower predominance of Bacteroides spp in CF, a species which they postulate is associated with healthy immune modulation [19]. Burke et al. showed a high prevalence of virulent strains of C. difficile in the fecal analysis of asymptomatic adult patients with CF, suggesting that they may play a role in nosocomial transmission of the disease [29]. Small intestinal bacterial overgrowth (SIBO) is thought to affect 30–40% of individuals with CF and is associated with abdominal pain, bloating, flatulence weight loss, and nutrient malabsorption including vitamin B12, iron, bile acids, vitamin D, and folate that consequently can cause anemia [30]. Progressive fecal dysbiosis from birth has been associated with growth failure [31]. Multiple studies have demonstrated higher levels of fecal calprotectin in CF, a marker of gut inflammation derived from neutrophils, suggesting an inflammatory component of CF enteropathy [32,33,34] that may be influenced by the microbiome. Using 16S rRNA sequencing, Enaud et al. showed similarities between the CF microbiome and Crohn’s disease [35]. Finally, Flass et al. demonstrated that compared to CF subjects without liver disease, CF subjects with cirrhosis are more likely to have intestinal mucosal lesions, a relative scarcity of Bacteroides, and a relative abundance of Clostridium[36].
Probiotics have been used to target CF dysbiosis with varying degrees of success. Systematic reviews have evaluated the clinical use of probiotics in children and adults with CF [37,38,39,40•], including a 2020 Cochrane review. The Cochrane review found that probiotics reduced fecal calprotectin, but did not improve overall lung function, growth measures, hospitalizations, or quality of life measures. Adverse events were rare. Results were limited by lack of uniformity of probiotic composition and dosage [40•].
Colorectal Cancer Screening Recommendations
Another consequence of increased longevity in CF has been the revelation that CF patients are at higher risk of GI malignancy than age-matched counterparts. Demonstrated by multiple longitudinal studies [7, 41,42,43], CF patients have an increased risk of digestive tract cancer, particularly following solid organ transplantation. CF patients appear to have a 5–tenfold increased risk of colon cancer compared with the general population, and advanced adenomas present more frequently and at a younger age [7, 44••]. Mechanisms proposed for this increased risk include the identification of CFTR as a tumor suppressor gene [45]. In addition, increased intestinal cell turnover as reflected in elevated fecal M2-pyruvate kinase in children with CF has been postulated as a mechanism [46]. Finally, Dayama et al. [47] evaluated colonic mucosal gene expression and the mucosal microbiome in patients with CF and healthy controls. They reported downregulation of 15-hydroxyprostaglandin dehydrogenase (15-PGDH), an enzyme in the cyclooxygenase-2 pathway that acts as a tumor suppressor in colorectal neoplasia [48]. Dayama et al. identified patterns of gene expression and alterations in the microbiome that have been previously linked to colorectal cancer. For example, gene expression of LCN2 was correlated with a paucity of Ruminococcaceae, which is depleted in CRC, while expression of DUOX2 was correlated with an abundance of Veillonella, which was previously identified as a pro-inflammatory bacteria in the CRC microbiome [49].
Based on the increased risk of colon cancer, the Cystic Fibrosis Foundation and the American Gastroenterological Association (AGA) established a CF colorectal cancer (CRC) screening task force to evaluate available evidence and formulate new CRC screening recommendations in CF. Published in 2018, these CRC screening recommendations are unique to the CF population [44••]. The task force recommends that decisions regarding CRC screening and surveillance be based on shared decision making between the provider and patient, with consideration of treatment, comorbid conditions (e.g. severity of lung disease), safety, and quality of life. The task force recommends CRC screening by colonoscopy (and not by non-endoscopic methods) to begin at age 40 in the non-transplant population and at 30 in those patients who have undergone solid organ transplant. Rescreening is recommended every 5 years in all transplant patients. Surveillance colonoscopy is recommended at 3 years for adenomatous polyps. Finally, it is recommended that all adults undergoing colonoscopy receive intensive bowel preparation regimens.
CFTR Modulators
The most important recent pharmacologic advancements in CF have been the approval of CFTR modulating therapies over the last 10 years. Dubbed a “cause for celebration” by National Institutes of Health (NIH) director Francis Collins, these medications function by targeting specific mutations in CFTR [50]. CF is caused by mutations in CFTR that affect the quantity of the protein that reaches the cell surface or the function of CFTR channels at the cell surface (Table 1). Unlike previous therapies which sought to alleviate the symptoms of CF, these molecularly targeted therapies treat the mechanism of disease by targeting specific mutations (Tables 1, 2).
The first of these medications, ivacaftor (Vertex Pharmaceuticals, Boston, MA), was approved by the Food and Drug Administration (FDA) in 2012. A so-called “potentiator,” ivacaftor potentiates the activity dysfunctioning of gating membranes in the CFTR protein. Ivacaftor primarily targets the G551D CFTR missense mutation, present in 4–5% of patients with CF. Ivacaftor has been shown in multiple randomized controlled trials (RCTs) to increase the time that activated CFTR channels at the cell surface remain open, and may lead to significant pulmonary improvement for adult and pediatric patients with the G551D mutation [51,52,53,54]. Secondary outcomes from these trials suggest ivacaftor improves nutritional status in both adult and pediatric patients and exocrine pancreatic function (as measured by increasing fecal elastase and decreasing immunoreactive trypsinogen) in pediatric patients [51, 54]. A large follow-up study extending an RCT population of 2–5 year-olds to 84 weeks showed maintenance, but not improvement of, gains made in body mass index (BMI) z scores and pancreatic function during the original 24-week study period [55]. Analysis of combined data from two RCTs including patients age 6 and older with the G551D mutation revealed improved nutritional status at 48 weeks [56].
The most important target of these therapies is the F508del CFTR mutation, the most common mutation associated with CF [50]. Although hundreds of different disease-causing CFTR mutations have been identified, nearly 90% of individuals with cystic fibrosis have at least one copy of F508del [1••,57]. The F508del mutation leads to a marked reduction in the quantity [1••,58, 59] and quality of CFTR protein at the surface of epithelial cells [1••,58] (Table 1).
Lumacaftor was the first drug to specifically target the F508del mutation, and works by helping to correct F508del CFTR misprocessing and increase the amount of CFTR at the cell surface. FDA approved in 2015 for use in F508del homozygotes aged ≥ 2 years as a combination pill with ivacaftor, lumacaftor-ivacaftor (Vertex Pharmaceuticals, Boston, MA) expanded on the promise shown by ivacaftor alone. As with previous trials that focused primarily on pulmonary outcomes, GI-specific endpoints from large trials are secondary. The TRANSPORT trial, conducted in patients aged 12 or older, demonstrated modest BMI increases at 24 weeks statistically significant compared to placebo [60]. Long-term follow up of these patients to 96 weeks suggests modest but persistent increase in BMI [60, 61]. Despite the success of lumacaftor-ivacaftor, the drug is associated with pulmonary complications including acute pulmonary events such as dyspnea, chest tightness, and a decrease in forced expiratory volume in 1 s (FEV1) in up to 20% of patients [60, 62,63,64]. In addition, lumacaftor causes CYP3A4 induction in some patients, leading to prohibitive drug-drug interactions (e.g. inactivation of hormonal contraception) and potentially limiting efficacy of the drug itself [65].
Developed as an alternative to lumacaftor-ivacaftor, tezacaftor as a fixed dose combination with ivacaftor (Vertex Pharmaceuticals, Boston, MA) was FDA approved in 2018 for CF patients 6 or older who are homozygous for the F508del mutation. Like lumacaftor, tezacaftor is a “corrector,” and improves processing and trafficking of mutant F508del CFTR proteins. Two large randomized placebo-controlled clinical trials were designed to evaluate the efficacy and safety of tezacaftor-ivacaftor. The first, EVOLVE, was a 24-week trial studying tezacaftor-ivacaftor in patients homozygous for the F508del mutation. The second, EXPAND, was a multicenter, crossover trial evaluating tezacaftor-ivacaftor in patients heterozygous for F508del and a residual function mutation. Although both trials demonstrated improvement in FEV1, the primary endpoint, neither achieved statistical significance in BMI improvement [66, 67]. Compared to lumacaftor-ivacaftor, tezacaftor-ivacaftor has demonstrated superiority in side-effect profile and medication interactions [68], though concomitant use of tezacaftor-ivacaftor with strong CYP3A inducers may require dosage adjustments [68, 69].
With the combination of ivacaftor plus either lumacaftor or tezacaftor, approximately 50% of patients with CF were eligible for CFTR modulating therapy [70]. In 2019, a new triple combination therapy containing ivacaftor, tezacaftor, and the novel drug elexacaftor (Vertex Pharmaceuticals, Boston, MA) was FDA approved for CF patients aged ≥ 12 with at least 1 F508del mutation. Elexacaftor is a CFTR corrector that binds CFTR at a different site than tezacaftor to facilitate processing and trafficking of the CFTR protein to the cell membrane [71]. The concomitant use of elexacaftor, tezacaftor, and ivacaftor improve the function of F508del mutated CFTR protein at the cell surface. The addition of triple combination therapy to the arsenal of CFTR modulators has increased eligibility for CFTR modulating therapy to approximately 90% of CF genotypes [70].
Two trials evaluated the safety and efficacy of elaxacaftor-tezacaftor-ivacaftor combination therapy. The first compared the study drug to tezacaftor-ivacaftor in patients aged ≥ 12 who were homozygous for the F508del mutation. After 4 weeks, those patients in the interventional arm had an absolute improvement of 10% predicted FEV1 versus the standard of care. While not included as a primary or secondary outcome, the trial noted a 4-week increase in BMI [72••]. The second trial, VX17-445-102 Study, compared the study drug to placebo in a similarly aged population of F508del heterozygotes. At 4 weeks, the interventional group showed a 13.8% absolute increase in predicted FEV1 versus the standard of care. At 24 weeks, the mean difference in BMI (a secondary outcome) between the interventional group and placebo group was 1.04 kg/m2, which was statistically significant [1••]. Among gastrointestinal side effects in the second trial, diarrhea was reported in 12.9% of patients in the interventional group versus 7.0% in the placebo group. Use of elexacaftor-tezacaftor-ivacaftor in younger children age 6–11 has also been assessed in a 24-week open-label study which demonstrated safety and efficacy as well as increase in BMI for age z-score over the study period [73].
Notably, all of the CFTR modulators have a risk of elevation of liver enzymes. For patients without pre-existing liver disease or liver function test abnormalities, liver function testing should be performed at baseline, every 3 months during the first year of treatment, and annually thereafter. In addition, dosing should be interrupted in patients with significant elevations of transaminases (e.g., ALT or AST > 5 × upper limit of normal [ULN], or ALT or AST > 3 × ULN with bilirubin > 2 × ULN) and laboratory tests should be closely followed until abnormalities resolve [74]. The manufacturer does not recommend use of elexacaftor-tezacaftor-ivacaftor in Child–Pugh Class B or C liver disease; if used despite the risks in Child–Pugh Class B liver disease, dose adjustment is recommended due to an association with bilirubin elevation in a small clinical study [74]. Patients with pre-existing liver disease or history of liver function test abnormalities may need more frequent lab monitoring. In the VX17-445–102 study, one patient with pre-existing cirrhosis in the interventional arm discontinued the drug due to portal hypertension. [1••] Otherwise, data regarding liver injury linked to these medications is sparse, but more data may emerge as their use increases [75]. Further complicating the picture, elexacaftor inhibits uptake by OATP1B1 and OATP1B3, anion transporting polypeptides expressed in hepatocytes [74]. Since bilirubin is a substrate of OATP1B1 and OATP1B3, one might expect hyperbilirubinemia as a result of this mechanism. Indeed, 5% of patients in the VX17-445–102 study receiving the drug developed hyperbilirubinemia, compared to 1% in the placebo group. [1••]
Most of the data regarding GI outcomes of CFTR modulators come in the form of observational studies and case reports. In addition, because of the recent entry into the market of tezacaftor-ivacaftor and elexacaftor-tezacaftor-ivacaftor, much of the current data assessing GI-specific outcomes of CFTR modulators pertains to ivacaftor and lumacaftor-ivacaftor. However, more data should emerge as investigators begin to evaluate the GI effects of these newer medications.
Lumacaftor and ivacaftor have shown promise in treating CF-related hepatobiliary complications. Though causality cannot be established due to its observational nature, a review of U.S. and U.K. CF registry data indicated that ivacaftor treatment was associated with fewer hepatobiliary complications compared with no CFTR modulator therapy [76, 77]. In a cross-sectional study of 20 subjects with CF, lumacaftor-ivacaftor use was associated with reduced hepatic steatosis (as measured by magnetic resonance imaging [MRI] proton density fat fraction [PDFF]) [78]. The drug was also associated with lower total bilirubin. Finally, ivacaftor may help restore disruption of enterohepatic circulation of bile acids in CF patients with S1251N and G551D gating mutations [79].
Lumacaftor and ivacaftor may also improve CF-related pancreatic disease. A small case series suggests that use of ivacaftor may reduce episodes of pancreatitis in pancreatic sufficient CF patients with recurrent pancreatitis [80]. While modest promise has been shown in early markers of CFRD, such as glucose tolerance and insulin response, permanent improvements in CFRD have proven elusive [77, 81,82,83,84,85]. For both ivacaftor alone and lumacaftor-ivacaftor, studies reporting a variety of pancreatic exocrine function outcomes (fecal elastase, lipase supplementation dosage, and serum immunoreactive trypsinogen) suggest improvement of EPI, but limited conclusions can be drawn due to study limitations, including small sample size, lack of control group, and unknown variability of outcome measures over time [51, 54, 77, 86, 87].
Lumacaftor and ivacaftor may improve additional GI-related illness. Ooi and colleagues evaluated the effects of ivacaftor on fecal calprotectin and intestinal microbial communities (using 16SrRNA variable 3 gene region amplicon sequencing) in 16 patients [88••]. Ivacaftor was associated with an increase in Akkermansia spp, and a decrease in Enterobacteriaceae which correlated with a decrease in fecal calprotectin. The clinical impact of these findings is not known. Other studies have shown promise in nutritional status [56], extra-esophageal reflux [89], symptomatic celiac disease (CD) in pediatric patients (case series) [90], and proximal small intestine pH profile [91].
Conclusions
As new therapies emerge and patient longevity increases in CF, research has increasingly focused on GI manifestations. Four recent developments provide a major boost to the emerging field of CF gastroenterology. The development of patient-reported GI symptom scoring systems has helped standardize symptom reporting and identify areas for intervention. Studies attempting to characterize the microbiome in CF demonstrate its potential role in GI symptoms and raise the possibility of targeting the microbiome for therapeutic benefit. Recognition of the heightened risk of colorectal cancer in the CF population has led to recommendations for screening colonoscopy beginning at age 40 in the general CF population and 30 in transplant recipients. Finally, CFTR modulators are the first targeted pharmacologic therapies for CF. Because these new medications were originally studied for their impact on pulmonary complications, data regarding their impact on GI manifestations of CF are just beginning to emerge. Future research should assess the impact of CFTR modulators on the gut microbiome, GI symptoms, and GI cancer risk.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381:1809–19. https://doi.org/10.1056/NEJMoa1908639. This trial demonstrated efficacy of elexacaftor-tezacaftor-ivacaftor in patients with a F508del mutation and minimal function mutation in CFTR.
Brown SD, White R, Tobin P. Keep them breathing. J Am Acad Physician Assist. 2017;30:23–7. https://doi.org/10.1097/01.JAA.0000515540.36581.92.
Festini F, Ballarin S, Codamo T, Doro R, Loganes C. Prevalence of pain in adults with cystic fibrosis. J Cyst Fibros. 2004;3:51–7. https://doi.org/10.1016/j.jcf.2003.12.001.
Tabori H, Arnold C, Jaudszus A, Mentzel HJ, Renz DM, Reinsch S, et al. Abdominal symptoms in cystic fibrosis and their relation to genotype, history, clinical and laboratory findings. PLoS ONE. 2017;12(5): e0174463. https://doi.org/10.1371/journal.pone.0174463.
De Lisle RC, Borowitz D. The cystic fibrosis intestine. Cold Spring Harb Perspect Med. 2013;3(9): a009753. https://doi.org/10.1101/cshperspect.a009753.
Singh VK, Schwarzenberg SJ. Pancreatic insufficiency in Cystic Fibrosis. J Cyst Fibros. 2017;16:S70–8. https://doi.org/10.1016/j.jcf.2017.06.011.
Maisonneuve P, Marshall BC, Knapp EA, Lowenfels AB. Cancer risk in cystic fibrosis: A 20-year nationwide study from the United States. J Natl Cancer Inst. 2013;105:122–9. https://doi.org/10.1093/jnci/djs481.
Gelfond D, Borowitz D. Gastrointestinal complications of cystic fibrosis. Clin Gastroenterol Hepatol. 2013;11:333–42. https://doi.org/10.1016/j.cgh.2012.11.006.
2017 Cystic Fibrosis Foundation Patient Registry Highlights. Cystic Fibrosis Foundation. 2018. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-Cystic-Fibrosis-Foundation-Patient-Registry-Highlights.pdf. Accessed 8 May 2021.
Hayee B, Watson KL, Campbell S, Simpson A, Farrell E, Hutchings P, et al. A high prevalence of chronic gastrointestinal symptoms in adults with cystic fibrosis is detected using tools already validated in other GI disorders. United Eur Gastroenterol J. 2019;7:881–8. https://doi.org/10.1177/2050640619841545. Using validated GI tools, this study demonstrated that significant GI symptoms are common in adult patients with CF.
Suzuki H. To establish a treatment for GI symptoms of cystic fibrosis, it is necessary to develop a symptom evaluation tool United. Eur Gastroenterol J. 2019;7:873–4. https://doi.org/10.1177/2050640619856188.
Boon M, Claes I, Havermans T, Fornés-Ferrer V, Calvo-Lerma J, Asseiceira I, et al. Assessing gastro-intestinal related quality of life in cystic fibrosis: Validation of PedsQL GI in children and their parents. PLoS One. 2019;14(12):e0225004. https://doi.org/10.1371/journal.pone.0225004. This multicenter study validated a symptom measurement instrument gastrointestinal-related quality of life in children with CF.
Varni JW, Bendo CB, Denham J, Shulman RJ, Self MM, Neigut DA, et al. PedsQLTM Gastrointestinal Symptoms Scales and Gastrointestinal Worry Scales in pediatric patients with functional and organic gastrointestinal diseases in comparison to healthy controls. Qual Life Res. 2015;24:363–78. https://doi.org/10.1007/s11136-014-0781-x.
Varni JW, Franciosi JP, Shulman RJ, Saeed S, Nurko S, Neigut DA, et al. PedsQL gastrointestinal symptoms scales and gastrointestinal worry scales in pediatric patients with inflammatory bowel disease in comparison with healthy controls. Inflamm Bowel Dis. 2015;21:1115–24. https://doi.org/10.1097/MIB.0000000000000351.
Smith S, Rowbotham N, Davies G, Gathercole K, Collins SJ, Elliott Z, et al. How can we relieve gastrointestinal symptoms in people with cystic fibrosis? An international qualitative survey. BMJ Open Respir Res. 2020;7:e000614. https://doi.org/10.1136/bmjresp-2020-000614. This survey study captured the burden of gastrointestinal symptoms in cystic fibrosis and their importance as a research priority.
Freeman AJ, Sathe M, Aliaj E, Borowitz D, Fogarty B, Goss CH, et al. Designing the GALAXY study: Partnering with the cystic fibrosis community to optimize assessment of gastrointestinal symptoms. J Cyst Fibros. 2021 Jan 13 [Epub ahead of print]. https://doi.org/10.1016/j.jcf.2020.12.021. This article describes a longitudinal study to assess the prevalence of gastointestinal symptoms in people with cystic fibrosis.
Héry-Arnaud G, Boutin S, Cuthbertson L, Elborn SJ, Tunney MM. The lung and gut microbiome: what has to be taken into consideration for cystic fibrosis? J Cyst Fibros. 2019;18:13–21. https://doi.org/10.1016/j.jcf.2018.11.003.
Schnapp Z, Hartman C, Livnat G, Shteinberg M, Elenberg Y. Decreased fecal calprotectin levels in cystic fibrosis patients after antibiotic treatment for respiratory exacerbation. J Pediatr Gastroenterol Nutr. 2019;68:282–4. https://doi.org/10.1097/MPG.0000000000002197.
Antosca KM, Chernikova DA, Price CE, Ruoff KL, Li K, Guill MF, et al. Altered stool microbiota of infants with cystic fibrosis shows a reduction in genera associated with immune programming from birth. J Bacteriol. 2019;16:e00274-19. https://doi.org/10.1128/JB.00274-19.
Fouhy F, Ronan NJ, O’Sullivan O, McCarthy Y, Walsh AM, Murphy DM, et al. A pilot study demonstrating the altered gut microbiota functionality in stable adults with Cystic Fibrosis. Sci Rep. 2017;7:6685. https://doi.org/10.1038/s41598-017-06880-y.
Coffey MJ, Nielsen S, Wemheuer B, Kaakoush NO, Garg M, Needham B, et al. Gut Microbiota in Children With Cystic Fibrosis: A Taxonomic and Functional Dysbiosis. Sci Rep. 2019;9:18593. https://doi.org/10.1038/s41598-019-55028-7. This study demonstrated intestinal dysbiosis in children with cystic fibrosis compared with healthy controls and associations between the intestinal microbiome and both growth parameters and lung function.
Vernocchi P, Del CF, Russo A, Majo F, Rossitto M, Valerio M, et al. Gut microbiota signatures in cystic fibrosis: Loss of host CFTR function drives the microbiota enterophenotype. PLoS One Public Lib Sci. 2018;13(12): e0208171. https://doi.org/10.1371/journal.pone.0208171.
Burke DG, Fouhy F, Harrison MJ, Rea MC, Cotter PD, O’Sullivan O, et al. The altered gut microbiota in adults with cystic fibrosis. BMC Microbiol. 2017;17:58. https://doi.org/10.1186/s12866-017-0968-8.
Duytschaever G, Huys G, Bekaert M, Boulanger L, De Boeck K, Vandamme P. Cross-sectional and longitudinal comparisons of the predominant fecal microbiota compositions of a group of pediatric patients with cystic fibrosis and their healthy siblings. Appl Environ Microbiol. 2011;77:8015–24. https://doi.org/10.1128/AEM.05933-11.
Nielsen S, Needham B, Leach ST, Day AS, Jaffe A, Thomas T, et al. Disrupted progression of the intestinal microbiota with age in children with cystic fibrosis. Sci Rep. 2016;6:24857. https://doi.org/10.1038/srep24857.
Hoen AG, Li J, Moulton LA, O’Toole GA, Housman ML, Koestler DC, et al. Associations between gut microbial colonization in early life and respiratory outcomes in cystic fibrosis. J Pediatr. 2015;167:138-147.e3. https://doi.org/10.1016/j.jpeds.2015.02.049.
Madan JC, Koestle DC, Stanton BA, Davidson L, Moulton LA, Housman ML, et al. Serial analysis of the gut and respiratory microbiome in cystic fibrosis in infancy: interaction between intestinal and respiratory tracts and impact of nutritional exposures. mBio. 2012;3(4):e00251-1251. https://doi.org/10.1128/mBio.00251-12.
Kristensen M, Prevaes SMPJ, Kalkman G, Tramper-Stranders GA, Hasrat R, de Winter, de Groot KM, et al. Development of the gut microbiota in early life: The impact of cystic fibrosis and antibiotic treatment. J Cyst Fibros. 2020;19:553–61. https://doi.org/10.1016/j.jcf.2020.04.007.
Burke DG, Harrison MJ, Fleming C, McCarthy M, Shortt C, Sulaiman I, et al. Clostridium difficile carriage in adult cystic fibrosis (CF); implications for patients with CF and the potential for transmission of nosocomial infection. J Cyst Fibros. 2017;16:291–8. https://doi.org/10.1016/j.jcf.2016.09.008.
Dorsey J, Gonska T. Bacterial overgrowth, dysbiosis, inflammation, and dysmotility in the Cystic Fibrosis intestine. J Cyst Fibros. 2017;16:S14-23. https://doi.org/10.1016/j.jcf.2017.07.014.
Hayden HS, Eng A, Pope CE, Brittnacher MJ, Vo AT, Weiss EJ, et al. Fecal dysbiosis in infants with cystic fibrosis is associated with early linear growth failure. Nat Med. 2020;26:215–21. https://doi.org/10.1038/s41591-019-0714-x.
Adriaanse MPM, Van Der Sande LJTM, Van Den Neucker AM, Menheere PPCA, Dompeling E, Buurman WA, et al. Evidence for a cystic fibrosis enteropathy. PLoS ONE. 2015;10(10):e0138062. https://doi.org/10.1371/journal.pone.0138062.
Parisi GF, Papale M, Rotolo N, Aloisio D, Tardino L, Scuderi MG, et al. Severe disease in cystic fibrosis and fecal calprotectin levels. Immunobiology. 2017;222:582–6. https://doi.org/10.1016/j.imbio.2016.11.005.
Ellemunter H, Engelhardt A, Schüller K, Steinkamp G. Fecal calprotectin in cystic fibrosis and its relation to disease parameters: a longitudinal analysis for 12 years. J Pediatr Gastroenterol Nutr. 2017;65(4):438–42. https://doi.org/10.1097/MPG.0000000000001544.
Enaud R, Hooks KB, Barre A, Barnetche T, Hubert C, Massot M, et al. Intestinal inflammation in children with cystic fibrosis is associated with Crohn’s-like microbiota disturbances. J Clin Med. 2019;8:645. https://doi.org/10.3390/jcm8050645.
Flass T, Tong S, Frank DN, Wagner BD, Robertson CE, Kotter CV, et al. 2015 Intestinal Lesions are associated with altered intestinal microbiome and are more frequent in children and young adults with cystic fibrosis and cirrhosis. PLoS One. 2015;10:e0116967. https://doi.org/10.1371/journal.pone.0116967.
Ananthan A, Balasubramanian H, Rao S, Patole S. Probiotic supplementation in children with cystic fibrosis—a systematic review. Eur J Pediatr. 2016;175(10):1255–66. https://doi.org/10.1007/s00431-016-2769-8.
Anderson JL, Miles C, Tierney AC. Effect of probiotics on respiratory, gastrointestinal and nutritional outcomes in patients with cystic fibrosis: a systematic review. J Cyst Fibros. 2017;16(2):186–97. https://doi.org/10.1016/j.jcf.2016.09.004.
Neri LDCL, Taminato M, Da Silva FLVRF. Systematic review of probiotics for cystic fibrosis patients: Moving forward. J Pediatr Gastroenterol Nutr. 2019;68(3):394–9. https://doi.org/10.1097/MPG.0000000000002185.
Coffey MJ, Garg M, Homaira N, Jaffe A, Ooi CY. Probiotics for people with cystic fibrosis. Cochrane Database Syst Rev. 2020; Issue 1. Art. No.: CD012949. https://doi.org/10.1002/14651858.CD012949.pub2. This systematic review assessed the evidence for the use of probiotics in people with cystic fibrosis.
Maisonneuve P, FitzSimmons SC, Neglia JP, Campbell PW, Lowenfels AB. Cancer risk in nontransplanted and transplanted cystic fibrosis patients: A 10-year study. J Natl Cancer Inst. 2003;95:381–7. https://doi.org/10.1093/jnci/95.5.381.
Fink AK, Yanik EL, Marshall BC, Wilschanski M, Lynch CF, Austin AA, et al. Cancer risk among lung transplant recipients with cystic fibrosis. J Cyst Fibros. 2017;16:91–7. https://doi.org/10.1016/j.jcf.2016.07.011.
Neglia JP, Fitzsimmons SC, Maisonneuve P, Schöni MH, Schöni-Affolter F, Corey M, et al. The risk of cancer among patients with cystic fibrosis. N Engl J Med. 1995;332:494–9. https://doi.org/10.1056/NEJM199502233320803.
Hadjiliadis D, Khoruts A, Zauber AG, Hempstead SE, Maisonneuve P, Lowenfels AB, et al. Cystic Fibrosis Colorectal Cancer Screening Consensus Recommendations. Gastroenterology. 2018;154:736–745.e14. https://doi.org/10.1053/j.gastro.2017.12.012. This article provided consensus recommendations for colorectal cancer screening in patients with colorectal cancer.
Than BLN, Linnekamp JF, Starr TK, Largaespada DA, Rod A, Zhang Y, et al. CFTR is a tumor suppressor gene in murine and human intestinal cancer. Oncogene. 2016;35:4191–9. https://doi.org/10.1038/onc.2015.483.
Pang T, Leach ST, Katz T, Jaffe A, Day AS, Ooi CY. Elevated fecal M2-pyruvate kinase in children with cystic fibrosis: A clue to the increased risk of intestinal malignancy in adulthood? J Gastroenterol Hepatol. 2015;30:866–71. https://doi.org/10.1111/jgh.12842.
Dayama G, Priya S, Niccum DE, Khoruts A, Blekhman R. Interactions between the gut microbiome and host gene regulation in cystic fibrosis. Genome Med. 2020;12:12. https://doi.org/10.1186/s13073-020-0710-2.
Myung SJ, Rerko RM, Yan M, Platzer P, Guda K, Dotson A, et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci U S A. 2006;103:12098–102. https://doi.org/10.1073/pnas.0603235103.
Geng J, Song Q, Tang X, Liang X, Fan H, Peng H, et al. Co-occurrence of driver and passenger bacteria in human colorectal cancer. Gut Pathog. 2014;6:26. https://doi.org/10.1186/1757-4749-6-26.
Collins FS. Realizing the DREAM OF MOLECULARLY TARGETED THERAPIES FOR CYSTIC FIBRosis. N Engl J Med. 2019;381:1863–5. https://doi.org/10.1056/NEJMe1911602.
Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-years with cystic fibrosis and a CFTR gating mutation (KIWI): An open-label, single-arm study. Lancet Respir Med. 2016;4:107–15. https://doi.org/10.1016/S2213-2600(15)00545-7.
Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N Engl J Med. 2011;365:1663–72. https://doi.org/10.1056/NEJMoa1105185.
Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187:1219–25. https://doi.org/10.1164/rccm.201301-0153OC.
Rosenfeld M, Wainwright CE, Higgins M, Wang LT, McKee C, Campbell D, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir. 2018;6:545–53. https://doi.org/10.1016/S2213-2600(18)30202-9.
Rosenfeld M, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2–5 years (KLIMB). J Cyst Fibros. 2019;18:838–43. https://doi.org/10.1016/j.jcf.2019.03.009.
Borowitz D, Lubarsky B, Wilschanski M, Munck A, Gelfond D, Bodewes F, et al. Nutritional Status Improved in Cystic Fibrosis Patients with the G551D Mutation After Treatment with Ivacaftor. Dig Dis Sci. 2016;61:198–207. https://doi.org/10.1007/s10620-015-3834-2.
2018 Patient Registry Annual Data Report. Cystic Fibrosis Foundation. 2019. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2018-Patient-Registry-Annual-Data-Report.pdf. Accessed 8 May 2021.
Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, et al. Altered chloride ion channel kinetics associated with the ΔF508 cystic fibrosis mutation. Nature. 1991;354:526–8. https://doi.org/10.1038/354526a0.
Lukacs GL, Chang XB, Bear C, Kartner N, Mohamed A, Riordan JR, et al. The ΔF508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1993;268:21592–8.
Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for phe508del CFTR. N Engl J Med. 2015;373:220–31. https://doi.org/10.1056/NEJMoa1409547.
Konstan MW, McKone EF, Moss RB, Marigowda G, Tian S, Waltz D, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir Med. 2017;5:107–18. https://doi.org/10.1016/S2213-2600(16)30427-1.
Hubert D, Chiron R, Camara B, Grenet D, Prévotat A, Bassinet L, et al. Real-life initiation of lumacaftor/ivacaftor combination in adults with cystic fibrosis homozygous for the Phe508del CFTR mutation and severe lung disease. J Cyst Fibros. 2017;16:388–91. https://doi.org/10.1016/j.jcf.2017.03.003.
Labaste A, Ohlmann C, Mainguy C, Jubin V, Perceval M, Coutier L, et al. Real-life acute lung function changes after lumacaftor/ivacaftor first administration in pediatric patients with cystic fibrosis. J Cyst Fibros. 2017;16:709–12. https://doi.org/10.1016/j.jcf.2017.05.002.
Jennings MT, Dezube R, Paranjape S, West NE, Hong G, Braun A, et al. An observational study of outcomes and tolerances in patients with cystic fibrosis initiated on lumacaftor/ivacaftor. Ann Am Thorac Soc. 2017;14:1662–6. https://doi.org/10.1513/AnnalsATS.201701-058OC.
Schneider EK. Cytochrome P450 3A4 induction: lumacaftor versus ivacaftor potentially resulting in significantly reduced plasma concentration of ivacaftor. Drug Metab Lett. 2018;12:71–4. https://doi.org/10.2174/1872312812666180328105259.
Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E, et al. Tezacaftor–ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med. 2017;377:2024–35. https://doi.org/10.1056/NEJMoa1709847.
Taylor-Cousar JL, Munck A, McKone EF, Van Der Ent CK, Moeller A, Simard C, et al. Tezacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med. 2017;377:2013–23. https://doi.org/10.1056/NEJMoa1709846.
Lommatzsch ST, Taylor-Cousar JL. The combination of tezacaftor and ivacaftor in the treatment of patients with cystic fibrosis: clinical evidence and future prospects in cystic fibrosis therapy. Ther Adv Respir Dis. 2019;13:1753466619844424. https://doi.org/10.1177/1753466619844424.
SYMDEKO® (tezacaftor/ivacaftor) Package Insert. Vertex Pharmaceuticals Incorporated. 2019. https://pi.vrtx.com/files/uspi_tezacaftor_ivacaftor.pdf. Accessed 8 May 2021.
Research Milestones. Cystic Fibrosis Foundation. Available from: https://www.cff.org/Research/About-Our-Research/Research-Milestones/ Accessed 27 Sep 2020.
Ridley K, Condren M. Elexacaftor-tezacaftor-ivacaftor: The first triple-combination cystic fibrosis transmembrane conductance regulator modulating therapy. J Pediatr Pharmacol Ther. 2020;25:192–7. https://doi.org/10.5863/1551-6776-25.3.192.
Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 2019;394:1940–8. https://doi.org/10.1016/S0140-6736(19)32597-8. This trial demonstrated efficacy of elexacaftor-tezacaftor-ivacaftor in patients with cystic fibrosis with a homozygous F508del mutation.
Zemanick ET, Taylor-Cousar JL, Davies J, Gibson RL, Mall MA, McKone EF, et al. A phase 3 open-label study of ELX/TEZ/IVA in children 6 through 11 years of age with cf and at least one F508del Allele. Am J Respir Crit Care Med. 2021. https://doi.org/10.1164/rccm.202102-0509oc.
Trikafta [package insert]. Vertex Pharmaceuticals Incorporated. 2020. https://pi.vrtx.com/files/uspi_elexacaftor_tezacaftor_ivacaftor.pdf. Accessed 8 May 2021.
Cystic Fibrosis Agents. In: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. National Institute of Diabetes and Digestive and Kidney Diseases. 2018. https://www.ncbi.nlm.nih.gov/books/NBK547889/ Accessed 27 Sep 2020.
Bessonova L, Volkova N, Higgins M, Bengtsson L, Tian S, Simard C, et al. Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax. 2018;73:731–40. https://doi.org/10.1136/thoraxjnl-2017-210394.
Sergeev V, Chou FY, Lam GY, Hamilton CM, Wilcox PG, Quon BS. The extrapulmonary effects of cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. Ann Am Thorac Soc. 2020;17:147–54. https://doi.org/10.1513/AnnalsATS.201909-671CME.
Kutney K, Donnola SB, Flask CA, Gubitosi-Klug R, O’Riordan M, Mcbennett K, et al. Lumacaftor/ivacaftor therapy is associated with reduced hepatic steatosis in cystic fibrosis patients. World J Hepatol. 2019;11:761–72. https://doi.org/10.4254/wjh.v11.i12.761.
van de Peppel IP, Doktorova M, Berkers G, de Jonge HR, Houwen RHJ, Verkade HJ, et al. IVACAFTOR restores FGF19 regulated bile acid homeostasis in cystic fibrosis patients with an S1251N or a G551D gating mutation. J Cyst Fibros. 2019;18:286–93. https://doi.org/10.1016/j.jcf.2018.09.001.
Carrion A, Borowitz DS, Freedman SD, Siracusa CM, Goralski JL, Hadjiliadis D, et al. Reduction of recurrence risk of pancreatitis in cystic fibrosis with ivacaftor: case series. J Pediatr Gastroenterol Nutr. 2018;66:451–4. https://doi.org/10.1097/MPG.0000000000001788.
Bellin MD, Laguna T, Leschyshyn J, Regelmann W, Dunitz J, Billings J, et al. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study. Pediatr Diabetes. 2013;14:417–21. https://doi.org/10.1111/pedi.12026.
Christian F, Thierman A, Shirley E, Allen K, Cross C, Jones K. Sustained glycemic control with ivacaftor in cystic fibrosis–related diabetes. J Investig Med High Impact Case Rep. 2019. https://doi.org/10.1177/2324709619842898.
Hayes D, McCoy KS, Sheikh SI. Resolution of cystic fibrosis-related diabetes with ivacaftor therapy. Am J Respir Crit Care Med. 2014;190:590–1. https://doi.org/10.1164/rccm.201405-0882LE.
Tsabari R, Elyashar HI, Cymberknowh MC, Breuer O, Armoni S, Livnat G, et al. CFTR potentiator therapy ameliorates impaired insulin secretion in CF patients with a gating mutation. J Cyst Fibros. 2016;15:e25–7. https://doi.org/10.1016/j.jcf.2015.10.012.
Mutyam V, Libby EF, Peng N, Hadjiliadis D, Bonk M, Solomon GM, et al. Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. J Cyst Fibros. 2017;16:24–9. https://doi.org/10.1016/j.jcf.2016.09.005.
McNamara JJ, McColley SA, Marigowda G, Liu F, Tian S, Owen CA, et al. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2–5 years with cystic fibrosis homozygous for F508del-CFTR: an open-label phase 3 study. Lancet Respir Med. 2019;7:325–35. https://doi.org/10.1016/S2213-2600(18)30460-0.
Ratjen F, Hug C, Marigowda G, Tian S, Huang X, Stanojevic S, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2017;5:557–67. https://doi.org/10.1016/S2213-2600(17)30215-1.
Ooi CY, Syed SA, Rossi L, Garg M, Needham B, Avolio J, et al. Impact of CFTR modulation with Ivacaftor on Gut Microbiota and Intestinal Inflammation. Sci Rep. 2018;8:17834. https://doi.org/10.1038/s41598-018-36364-6. This study demonstrated associations between ivacaftor therapy and changes in the gut microbiome as well as decreased intestinal inflammation.
Zeybel GL, Pearson JP, Krishnan A, Bourke SJ, Doe S, Anderson A, et al. Ivacaftor and symptoms of extra-oesophageal reflux in patients with cystic fibrosis and G551D mutation. J Cyst Fibros. 2017;16:124–31. https://doi.org/10.1016/j.jcf.2016.07.004.
Hjelm M, Shaikhkhalil AK. Celiac Disease in Patients With Cystic Fibrosis on Ivacaftor: A Case Series. J Pediatr Gastroenterol Nutr. 2020;71:257–60. https://doi.org/10.1097/MPG.0000000000002736
Gelfond D, Heltshe S, Ma C, Rowe SM, Frederick C, Uluer A, et al. Impact of CFTR modulation on intestinal pH, motility, and clinical outcomes in patients with cystic fibrosis and the G551D mutation. Clin Transl Gastroenterol. 2017;8(3): e81. https://doi.org/10.1038/ctg.2017.10.
Acknowledgements
We thank Sarah Kathryn Stevens for her work as medical illustrator for Figure 1 and the University Hospitals Cleveland Medical Center Department of Surgery for her salary support.
Author information
Authors and Affiliations
Contributions
Conceptualization (DBK, LCC); Writing—Original Draft Preparation (DBK, LCC); Writing—Editing and Reviewing (LCC).
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Small Intestine
Rights and permissions
About this article

Cite this article
Karb, D.B., Cummings, L.C. The Intestinal Microbiome and Cystic Fibrosis Transmembrane Conductance Regulator Modulators: Emerging Themes in the Management of Gastrointestinal Manifestations of Cystic Fibrosis. Curr Gastroenterol Rep 23, 17 (2021). https://doi.org/10.1007/s11894-021-00817-2
Accepted:
Published:
DOI: https://doi.org/10.1007/s11894-021-00817-2