
Abstract
Purpose of review
The discovery of Helicobacter pylori and other organisms colonizing the stomach and the intestines has shed some light on the importance of microbiome in maintaining overall health and developing pathological conditions when alterations in biodiversity are present. The gastric acidity plays a crucial role in filtering out bacteria and preventing development of enteric infections. In this article, we discuss the physiology of gastric acid secretion and bacterial contribution to the composition of gastric and intestinal barriers and review the current literature on the role of proton pump inhibitors (PPIs) in the microbial biodiversity of the gastrointestinal tract.
Recent findings
Culture-independent techniques, such as 16S rRNA sequencing, have revolutionized our understanding of the microbial biodiversity in the gastrointestinal tract. Luminal and mucosa-associated microbial populations are not identical. Streptococcus is overrepresented in the biopsies of patients with antral gastritis and may also be responsible for the development of peptic ulcer disease. The use of PPIs favors relative streptococcal abundance irrespective of H. pylori status and may explain the persistence of dyspeptic symptoms in patients on PPI therapy. Increased risk of enteric infections has also been seen in patients taking PPIs. The overuse of PPIs leads to significant shift of the gastrointestinal microbiome towards a less healthy state.
Summary
With the advent of PPIs, many studies have demonstrated the significant changes in the microbial composition of both gastric and intestinal microbiota. Although they are considered relatively safe over-the-counter medications, PPIs in many cases are over- and even inappropriately used. Future studies assessing the safety of PPIs and their role in the development of microbiome changes should be encouraged.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Physiology of Gastric Acid Secretion
The regulation of gastric acid secretion has been traditionally divided into three phases: (1) cephalic, (2) gastric, and (3) intestinal. [1] The cephalic phase is initiated before the food enters the stomach. Stimuli such as smell, taste, sight, and even thought of food are sent to the nervous system where they are processed. The release of the neurotransmitter acetylcholine by vagal efferent fibers exerts a stimulatory effect on gastric acid secretion via muscarinic M3 receptors found on the parietal cells of the oxyntic glands. Histamine-producing enterochromaffin-like (ECL) cells can also be directly upregulated by pituitary adenylate cyclase-activating polypeptide (PACAP) that binds to its receptor, PAC1, found on ECL cells. [2] The gastric phase of acid secretion is regulated centrally and peripherally. The chemical composition of food directly stimulates gastric acid secretion. For instance, amino acids stimulate, while fats and carbohydrates inhibit, the release of gastrin by G cells. Interestingly, it has been revealed that two active compounds (A and B) found in beer can directly stimulate the muscarinic M3 receptors on parietal cells. [3] Gastric distention can produce either stimulatory or inhibitory effects on gastric acid secretion depending on the extent of the distention. Low-grade distention activates vasoactive intestinal peptide (VIP) neurons which in turn stimulate somatostatin secretion, leading to decreased gastrin production by G cells. Alternatively, higher-grade distention results in the recruitment of cholinergic neurons and therefore leads to increased gastric acid secretion. [4] The intestinal phase of gastric acid secretion occurs when chyme reaches the duodenum. Its contribution to the regulation of gastric acid secretion is minimal and elicited mainly via the enterogastric reflex. It is not known whether the microbiome or metabolome is involved in this process. The result of this reflex activation is the inhibition of gastric acid secretion. [5]
Different pathways are involved in the regulation of gastric acid secretion: (1) neural (vagal), (2) hormonal (gastrin), and (3) paracrine (histamine, somatostatin). [6, 7] The main stimulatory effect of the parasympathetic nervous system is due to binding to M3 receptors on parietal cells. [8] Recently, it has been shown that the activation of M4 receptors found on D cells (responsible for somatostatin secretion) can indirectly enhance gastric acid secretion by inhibiting the release of somatostatin. [9] Gastrin is the main hormonal regulator of gastric acid secretion. It is secreted by G cells of the stomach that are localized to the antrum. [10] Gastrin can stimulate gastric acid production by two distinctive mechanisms. Its major action (indirect mechanism) is via binding to cholecystokinin-2 (CCK-2) receptors found on ECL cells. Also, gastrin can directly stimulate gastric acid secretion binding to CCK-2 receptors on parietal cells. [11] The main paracrine stimulatory substance for gastric acid secretion is histamine. It is released from ECL cells that are located in the oxyntic or body mucosa. The main activating signal to ECL comes from gastrin as demonstrated in Fig. 1. Other noteworthy mechanism that was described earlier is the binding of PACAP to PAC1 receptors on ECL cells. As a potent gastric secretagogue, histamine binds to H2 receptors expressed on parietal cells. Moreover, histamine has been shown to inhibit somatostatin secretion via binding to H3 receptors. [12] Somatostatin released from D cells of the stomach exerts inhibitory effects on the secretion of histamine and gastrin by ECL cells and D cells, respectively. [13] Its direct negative impact on gastric acid secretion is of lesser importance. Somatostatin synthesis is increased by gastrin and gastric acid, as well as the activation of VIP. The parasympathetic nervous system decreases the release of somatostatin from D cells. [9] Other substances that can either stimulate (gastrin-releasing peptide (GRP), ghrelin) or inhibit (ghrelin, secretin, cholecystokinin (CCK), and leptin) gastric acid secretion have also been described in the literature (Fig. 1). [14,15,16,17,18]

Physiology of gastric acid secretion
These diverse inputs are integrated by parietal cells to regulate recruitment of the H+K+-ATPase (proton pump) to the cell surface. It is a heterodimeric protein containing α (catalytic) and β subunits. The latter one is responsible for N-glycosylation which involves the assembly, maturation, and sorting of the enzyme. [19] Its function is to exchange luminal K+ ions for cytoplasmic H+ ions in a 1:1 ratio. [20] This is an energy-dependent transport driven by ATP hydrolysis. In the resting state, the H+K+-ATPases are located in the intracellular tubulovesicles. [21] Upon stimulation, these tubulovesicles fuse with the apical membrane containing numerous canaliculi. This significantly increases the surface area of the plasma membrane and therefore the number of proton pumps capable of ion exchange. Chloride ions are also an integral part of hydrochloric acid. They can cross the apical membrane of the parietal cells due to their intracytoplasmic excess which, in turn, is the result of intracytoplasmic exchange of HCO3 − for serum Cl− via chloride/bicarbonate exchangers. [22]
Pharmacology of PPI Action
The discovery of the proton pump and the recognition of its paramount role in gastric acid secretion have led to the development of the class of drugs known as proton pump inhibitors (PPIs). Structurally, almost all of them are substituted benzimidazoles. [23] The only exception is the relatively novel PPI tenatoprazole which molecule contains an imidazopyridine backbone. [24] This difference in the chemical structure contributes to the prolonged plasma half-life of tenatoprazole compared to other PPIs. [25] The PPIs are prodrugs that can be activated only in the acidic environment where they transform into their protonated active forms: sulfonamide and sulfenic acid. These thiophilic compounds can bind to the SH group of cysteine-containing portions of the active H+K+-ATPases. This interaction results in a relatively stable covalent disulfide bond. [26] Although different PPIs bind to their specific SH groups at different sites of the enzyme, they all have been found to react with one common site which is Cys813. [27] Since the PPIs can only covalently inhibit active pumps, it takes several days in order for them to reach steady-state inhibition of acid secretion. The inhibitory activity of the PPIs directly correlates with the stability of the PPI binding. It has been shown that the enzymatic activity of the proton pumps can be restored by a reducing agent such as glutathione. Another way of obtaining the recovery of gastric acid secretion is via de novo synthesis of proton pumps. [28] The inhibitory effect of the PPIs is dose-dependent and results in diminished secretion of both basal and stimulated gastric acid secretion. Additionally, area under the curve (AUC) has also been found to correlate with the activity of the PPIs. [29] Conversely, plasma level of the drug has no relationship with its inhibitory potential. [26] All PPIs are recommended to be taken once daily, usually in the morning before breakfast. [30] The presence of H. pylori infection predominantly affecting the gastric corpus can significantly affect the activity of PPI. It has been revealed that H. pylori-positive patients may have enhanced intragastric pH control which is mainly seen overnight. Therefore, patients with H. pylori infection are less likely to develop nocturnal gastric acid breakthrough. [31]
The metabolism of the PPIs that takes place in the liver is regulated by two enzymes of the cytochrome P450 (cytP450) family: CYP2C19 and CYP3A4. [32] Omeprazole is a racemic molecule that can exist in two enantiomers: R-omeprazole and S-omeprazole. The latter one has been revealed to have lower sensitivity to the action of CYP2C19. This property is responsible for the increased plasma concentration as well as higher AUC of esomeprazole (S-omeprazole). [33, 34] Genetic variation in CYP2C19 contributes to differences in the inhibitory activity of the PPIs. For instance, inactivating mutations of CYP2C19 have been described in Asian populations. [35] Fifteen to twenty percent of Asians and 2–6% of Caucasians are known to be “slow metabolizers.” [36]
Gastric Microbiome
Up until the discovery of H. pylori in 1982 by Marshall and Warren, the acidic environment of the stomach was considered sterile. [37] In fact, that discovery not only helped treat patients with various conditions where H. pylori is considered a causative agent but also shed some light on the possibility that other bacteria reside in the stomach. The combination of both (1) H. pylori and (2) other commensal organisms comprises the gastric microbiome.
Helicobacter pylori
Nearly half of the world population are carriers of H. pylori. [38] In recent decades, a decline in prevalence has been observed in many countries which can be attributed to improved socioeconomic conditions. [39] In the USA, McJunkin et al. observed a dramatic decline in the prevalence of H. pylori in an endoscopy-referral population over a 10-year period (from 65.8 down to 6.8%). [40] However, in developing countries, H. pylori is still very common with its prevalence hitting 80% in some countries. [41] The most likely mode of transmission that has been repeatedly mentioned in the literature is person-to-person contact. [42, 43] Despite the stomach representing an extremely hostile environment for most of the bacteria, H. pylori is able to persist in the gastric niche. Interestingly, the stomach is the only known habitat for H. pylori. After H. pylori colonizes the stomach mucosa, it becomes the predominant species of the gastric microbiome. [44] H. pylori is a Gram-negative, spiral-shaped, microaerophilic, and flagellated bacterium. [45] The mechanism behind the unique adaptation of H. pylori to survive and proliferate in the gastric environment has been widely described: (1) urease activity, (2) ability of H. pylori to penetrate through the gastric mucus layer (spiral shape and the presence of flagella, mucolytic enzymes, and the production of host mucin), and (3) binding of bacterial surface adhesins to the corresponding receptors of the gastric epithelial cells. [46, 47] The unique interaction with gastric receptors may explain why H. pylori exclusively colonizes the gastric epithelium. [48] Every H. pylori carrier will ultimately develop chronic gastritis. Of note, in the majority of cases, the course of that inflammation is asymptomatic and affected individuals will never seek medical attention. Only 15% of infected patients will develop H. pylori-related symptoms. [49] Depending on the location of the affected part of the stomach, different changes in the gastric acid secretion can be expected. Antral predominant gastritis is characterized by the deficiency of antral somatostatin production which leads to gastrin-induced increased gastric acid secretion. Conversely, patients with pangastritis or corpus-predominant gastritis develop extensive gastric atrophy resulting in hypo- and achlorhydria and, therefore, decreased gastric acid secretion. [50] Different conditions are known to be related to the presence of H. pylori in the gastric mucosa: acute and chronic gastritis, peptic ulcer disease, non-ulcer dyspepsia, atrophic gastritis, intestinal metaplasia and gastric adenocarcinoma, and gastric mucosa-associated lymphoid tissue (MALT) lymphoma. [51,52,53,54] Several extra-gastroduodenal disorders have also been linked to H. pylori and include iron-deficiency anemia, scleroderma, rosacea, atherosclerosis, and autoimmune thyroid disease. [55] It is worth mentioning that H. pylori has been described to have a protective effect against gastroesophageal reflux disorder (GERD) and esophageal cancer. For instance, it has been reported that more virulent cagA-positive strains and pro-inflammatory genotypes of the IL-1β gene lead to gastric atrophy that, in turn, results in hypochlorhydria contributing to the protection of the esophageal mucosa. [56] The protective properties of H. pylori on asthma and allergy have also been well established. [57] Both non-invasive and invasive methods of detecting H. pylori are currently available. The former include the urea breath test, fecal antigen test, and H. pylori-specific IgG test. Endoscopy with subsequent biopsy of multiple sites of the stomach (two from the lesser curvature, two from the greater curvature, and one from the incisura angularis) is also used in selected group of patients. [58] Although culture techniques are considered a gold standard for the diagnosis of H. pylori, they have several significant limitations: (1) low sensitivity, (2) high expense, and (3) slow growth in culture (H. pylori is a fastidious organism). [59] In fact, PCR of biopsy samples, saliva, and feces is widely used with sensitivity and specificity approaching 100%. [60] However, due to extensive polymorphism of H. pylori genes, the applicability of PCR is sometimes considered questionable. It has been revealed that H. pylori-specific region of the 16S ribosomal RNA sequence is unique for most H. pylori strains and therefore can be used for qualitative and quantitative assessment of the presence of H. pylori in biological specimens. [61]
Other Commensal Organisms
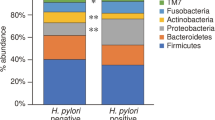
Although the discovery of H. pylori in 1982 ended the debate over the possibility of bacterial colonization of the stomach, it was still challenging to reveal other potential gastric microflora. Undoubtedly, many bacterial strains (Streptococcus, Neisseria, Lactobacillus, and others) have been repeatedly detected in the gastric fluid. However, whether these bacteria are simply “guests” in the stomach (transient microbiome) or permanent colonizers has been difficult to establish. In fact, bacteria can travel down to the stomach from the oral cavity. [62] To determine whether these bacteria can colonize in the gastric milieu, studies have shifted from studying gastric fluid to bacterial strains in the gastric mucosa. These compartments have distinct microbial compositions, as Firmicutes, Bacteroidetes, and Actinobacteria are common inhabitants of the gastric fluid while the gastric mucosa is predominantly colonized by Firmicutes and Proteobacteria. [44] In recent years, 16S rRNA sequencing has been used to characterize the composition of the gastric microbiome in a culture-independent manner which avoids the limitations of earlier studies which could only detect bacteria that can be cultured with current techniques. [63] In their study, Bik et al. identified 128 phylotypes from 23 subjects and showed that the most common bacteria of the stomach mucosa belong to the following five phyla: Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria (includes H. pylori), and Fusobacteria. It is noteworthy that although Proteobacteria was the predominant phylum in H. pylori-positive subjects (around 96% of all bacteria), the remaining four phyla were also consistently detected regardless of H. pylori status. [44] In another study, Li et al. analyzed both body and antrum biopsies from five normal individuals and five patients with non-H. pylori and non-NSAID (NHNN) gastritis. [64] They determined that the most common bacterial genera are the following: Streptococcus (phylum Firmicutes), Prevotella and Porphyromonas (Bacteroidetes), and Neisseria and Haemophilus (Proteobacteria). In this small cohort, the Firmicutes phylum (primarily the Streptococcus genus) was overrepresented in the biopsies from antral gastritis subjects. Of note, Streptococcus and Prevotella were also found to be the most common non-Helicobacter genera in the previously mentioned study by Bik et al. Recently, the presence of Streptococcus has been associated with peptic ulcer disease. [65•] Aviles-Jimenez et al. found evidence of a gradual change in gastric microbial diversity in patients from non-atrophic gastritis (NAG) to intestinal metaplasia (IM) to gastric cancer (GC). The authors concluded that with the progression of neoplastic changes, the gastric environment becomes less favorable for bacterial colonization. [66] The findings from these and other studies suggest that the overall composition of the gastric microbiome is quite similar among individuals from different ethnic and geographic populations. [67, 68]
Relationship Between H. pylori and Other Commensal Organisms
Intuitively, one can assume that the gastric microbiome population should have an impact on the course of diseases caused by H. pylori. For instance, it was shown that identical mouse strains from different vendors developed different response after being infected with H. pylori. [69••] In the study with the Mongolian gerbils, it was demonstrated that three Lactobacillus species (L. reuteri, L. johnsonii, and L. murinus) exerted an inhibitory effect on the growth of H. pylori. Similarly, in humans, two strains of L. reuteri have been revealed to have profound antimicrobial effect against H. pylori as well as strong antioxidative properties. [70] Additionally, Streptococcus mitis has also been found to inhibit H. pylori growth, resulting in conversion of the latter one to coccoid cells. [71] The effect of H. pylori on the composition of the gastric microbiome has been assessed in multiple studies. Some of them concluded that there is a higher diversity of bacteria in H. pylori-negative patients. [72] In contrast, other studies did not find any changes in the gastric bacterial complexity regardless of H. pylori status. [65•] Recently, Brawner et al. characterized the gastric microbiota of 86 children and adults. The data from this study suggest that H. pylori status was only associated with significant differences in gastric bacterial composition in children. Also, H. pylori-positive children were found to have more diverse gastric microbiota, larger abundance of non-Helicobacter Proteobacteria, and smaller abundance of Firmicutes than H. pylori-positive adults. [73] Llorca et al. characterized the gastric microbiota in H. pylori-positive and H. pylori-negative children. They reported higher microbial diversity and richness in H. pylori-negative subjects. [74] Several factors have been suggested that may account for shifts in the gastric microbiome with H. pylori infection: [1] increased gastric pH secondary to long-term H. pylori infection favors the colonization of transient bacteria, [2] ammonia and bicarbonate produced as a result of urease activity may be used as substrates for other bacteria, and [3] H. pylori-induced decreased gastric motility. [75]
PPI Use and Gastric Microbiome
PPIs are among the top 10 most commonly used medications worldwide. They are widely prescribed for the H. pylori eradication. Their actions against H. pylori have been extensively described and include the following possible effects: (1) direct bacteriostatic effect due to inhibition of the bacterial P-type ATPase and (2) inhibition of bacterial urease activity. [76, 77] It should be mentioned that the reduction of urease activity was achieved only when high doses of PPI (omeprazole 80 mg/day) were administered.
There are at least two known mechanisms by which PPIs can affect the gastric bacterial composition: (1) directly targeting bacterial and fungal proton pumps and (2) disrupting the normal gastric microenvironment by increasing gastric pH. [78] It has been shown that oropharyngeal-like and fecal-like bacteria are more widely represented in the gastric microflora after PPI use. [79] Sterbini et al. revealed a significant increase in the relative abundance of Streptococcus in patients taking PPIs irrespective of H. pylori status. They concluded that Streptococcus may serve as an independent indicator of the gastric microbiome changes in dyspeptic patients secondary to the use of PPIs. That observation may explain the exacerbation or persistence of dyspepsia in patients on PPI therapy. [80••]
Intestinal Microbiome
There are trillions of microorganisms residing in the human gut. The colon alone is the home of approximately 70% of human bacteria. [81] Soon after birth, the infant gut is colonized by maternal microorganisms which may be influenced by whether the infant breastfeeds. Similarities between maternal vaginal microflora and infant microbiota have been shown. [82] However, in infants born via cesarean delivery, there are significant changes in microbial composition of the gut compared to those born via vaginal delivery. [83] It is suggested that several factors can influence intestinal microbiota during the first year of life: (1) the transfer of the maternal gut microbiota to the newborn, (2) diet, (3) environmental exposures, (4) antimicrobial medications, and (5) genetic factors. Over time (by 3 years of age), the number and diversity of the gut microorganisms increase and the gut microbiota of children stabilizes and resembles that of an adult. [84] Of note, the changes in the number and diversity of microorganisms can be appreciated when traveling down the gastrointestinal tract. While the number of bacteria in the stomach is 101 cells/g, it exponentially increases when moving distally (103 in the small intestine) and reaches 1012 cells/g in the colon. [85] Interestingly, in terms of the source of the cellular respiration, bacteria in the different parts of the digestive tract differ as well: Gram-positive aerobes in the duodenum but Gram-negative and Gram-positive anaerobes and facultative anaerobes in the terminal ileum and the predominance of obligate anaerobes (mainly genus Bacteroides) in the colon. [86, 87]
The balance (both quantitative and qualitative) of the microorganisms in the different parts of the gastrointestinal tract is exceptionally important for maintaining proper digestive functioning. For instance, small intestinal bacterial overgrowth (SIBO), the condition when the number of small intestinal bacteria exceeds 105−106 organisms/mL, results mainly from diminished gastric acid secretion (H. pylori-induced, related to aging, secondary to the use of histamine type 2 receptor (H2) blockers or PPIs) and small intestinal dysmotility. [88]
Additionally, one should take into account that the composition of the luminal microbiome is not the same as the one attached to the mucosa or adjacent to the epithelial cells. In other words, not all bacteria can reach mucosal and epithelial “spaces” of the gut. [89] Two dominant phyla of the gut microbiota are Firmicutes and Bacteroidetes. Among other less common phyla are Proteobacteria, Actinobacteria, Fusobacteria, Cyanobacteria, and Verrucomicrobia. [90] Obviously, the role of intestinal microbiota in maintaining human health is well established. [91] Referred to as “a forgotten organ,” the gut flora exerts numerous crucial functions in the host. [85] It is involved in (1) the establishment of both the intestinal mucosal and systemic immune systems, (2) protecting the host against pathogens by being an important part of the physical barrier, (3) promoting structural and functional maturation of the digestive tract, and (4) regulating various metabolic processes including those related to drug metabolisms. [92] Apart from influences on the intestinal barrier, it was suggested that the gut microbiota may have role in regulating the blood-brain (BBB) and blood-testis (BTB) barriers. [93] Recently, it was discovered that the gut microbiome composition may play a role in aging progression. [94]
The number of diseases directly or indirectly linked to the alteration of the gut microbiota composition has significantly increased over the last several years. They include but are not limited to gastrointestinal disorders (irritable bowel syndrome (IBS), inflammatory bowel disease (IBD), SIBO, celiac disease, and colorectal cancer), metabolic impairments (obesity and type 2 diabetes), allergy, asthma, and cardiovascular and neuropsychiatric conditions. [95,96,97,98,99,100,101]
The Gut Microbiota and the Gut Barrier
As mentioned above, the gut microbiome has been proven to have an enormous impact on functional and structural maturation of the gut. Specifically, Bacteroides thetaiotaomicron is known for its ability to induce expression of sppr2a protein required for the maintenance of desmosomes at the epithelial villi. [102] Additionally, Toll-like receptor 2 (TLR2)-mediated signaling which is stimulated by the microbial cell wall peptidoglycan maintains tight junctions and downregulates apoptosis. [103] The development of intestinal microvasculature has been associated with the microbiota-induced stimulation of transcription factor angiotensin-3. [104] Also, several observations show that germ-free (GF) mice have meaningful structural and functional changes of their intestines: (1) greatly enlarged ceca, (2) reduced intestinal surface area, (3) altered peristaltic movements, (4) diminished regenerative capacity of the intestinal epithelium resulting in thin villi, and (5) prolonged cell cycle time. [105,106,107,108] Some commensal organisms are able to modulate mucosal glycosylation patterns since carbohydrate moieties serve as microbial attachment sites. This process takes place both at the cellular and subcellular levels. It has been shown that a signaling molecule secreted by Bacteroides thetaiotaomicron induces the expression of fucose on the cell surface glucoconjugates. [109]
The Gut Microbiota and the Gastric Barrier
The role of the stomach in digestion and metabolism of different substances including the breakdown of proteins by means of HCl-induced pepsinogen activation is well known. However, Beasley et al. suggested that the evolution of stomach acidity plays an important role in maintaining balanced composition of gut microbiome in humans. [110] For example, it has been shown that one of the main factors determining gastric acidity is the diet habit of the host. Vultures which are considered obligate scavengers have one of the most hostile gastric environment (pH close to 0) which prevents colonization by many microbial strains. [111] Generally speaking, scavengers and carnivores have higher gastric acidity when compared to herbivores. There have been several proposed explanations why the stomach pH values in humans are similar to those of carrions: (1) carrion feeding in our direct ancestors and (2) natural selection favoring high gastric acidity aimed at prevention of infections caused by numerous fecal-oral pathogens. [110] Although increased gastric acidity prevents colonization by many pathogenic bacteria, the other side of the coin is that it also “filters out” many mutualistic microbes, thus preventing their colonization in certain patients (for example, after antibiotic use). [112]
Conditions resulting in decreased acidity of the gastric lumen have been associated with the increased risk of Clostridium difficile infection. [113] It is a well-known fact that the gastric pH in elderly patients is lower compared to young individuals. [114] This explains why advanced age is one of the most notable risk factors (along with systemic antibiotics and acid suppression medications) for developing C. difficile infection. [115]
PPI Use and Intestinal Microbiome
Undoubtedly, the best way to evaluate the impact of long-term PPI use on intestinal microbiome would be to conduct a randomized, double-blinded, placebo-controlled trial. However, due to ethical reasons, this approach is not feasible in humans. [116] In rat models, long-term use of lansoprazole (50 weeks) was associated with significant shift in terminal ileum bacterial composition: a predominance of Proteobacteria (93.9%, mainly Escherichia and Pasteurella genera) in the controls and the abundance of Firmicutes (66.9%, mainly Clostridium and Lactobacillus genera) in the treatment group. [117]
A number of observational studies and clinical trials have been conducted on humans to determine the impact of PPI use on gut microbial diversity. Leonard et al. conducted a systematic review of the risk of enteric infections in patients taking gastric acid suppression medications. They concluded that there was an association between taking antisecretory medications (higher risk with PPI use) and the development of Salmonella, Campylobacter, and other enteric infections. [118] Dial reported similar findings regarding the association of PPI use and enteric infections. [119] In 2015, Jackson et al. published the results of the study in which they collected fecal samples of 1827 healthy twins and using 16S rRNA amplification evaluated the impact of PPI use on the composition of the gut microbiota. They revealed that PPI users had significantly lower numbers of commensal organisms as well as lower bacterial diversity. Additionally, they recognized an association between PPI use and a significantly higher abundance of pharyngeal commensals in the gut. Altogether, 10 families were more common among PPI users versus non-users (mainly, Streptococcaceae and Micrococcaceae). Interestingly, a higher abundance of Streptococcaceae was observed in PPI users in monozygotic twins discordant for PPI use. [120••] In an open-label crossover study, Freedberg et al. investigated whether PPIs could alter the gastrointestinal microbiome. They did not reveal any significant impact of PPI use on within-individual difference in microbial diversity. However, significant changes in the abundance of taxa associated with C. difficile infection (increased Streptococcaceae and Enterococcaceae and decreased Clostridiales) and SIBO (increased Staphylococcaceae and Micrococcaceae) were noted in the subjects after PPI use. [121] Imhann et al. assessed the gut microbiome composition of 1815 individuals (healthy subjects and patients with IBD and IBS) in the Netherlands. They concluded that PPI use (211 participants) was associated with the abundance of families Streptococcaceae and Micrococcaceae in all cohorts. Also, they observed significantly increased amount of oral cavity bacteria (genera Rothia, Scardovia, and Actinomyces) in the gut microbiome of PPI users. Of note, the potentially pathogenic strains of Escherichia coli were also increased in PPI users. [122••] Lo and Chan reported a meta-analysis of 11 studies (3134 subjects) which showed that there was a positive correlation between PPI use and the development of SIBO. Interestingly, such correlation was noted only when the diagnosis of SIBO was made by an invasive test (duodenal or jejunal aspirate culture). [123] It has been suggested that PPIs can exacerbate non-steroidal anti-inflammatory drug (NSAID)-induced enteropathy. It is believed this effect is the result of intestinal “dysbiosis” secondary to PPI use. [124] Moreover, it has been shown that concomitant use of PPIs and NSAIDs was associated with an increased risk of lower gastrointestinal bleeding (LGB). [125]
Conclusion
In recent years, numerous studies have consistently demonstrated the paramount role of the gastrointestinal microbiome in overall human health. Maintaining the unique balance of the microorganisms in the gut has become a challenging task due in large part to medications. The overuse of PPIs has been reported to significantly shift the gastrointestinal microbiome towards less healthy state. It is worth mentioning, however, that many of the abovementioned studies have used fecal samples for analyzing the gut microbiome. Hence, they do not necessarily represent the changes in the biodiversity of mucosa-associated microbiota. More studies need to be conducted to detect any confounders that may have impact on the role of PPIs in the composition of the gastrointestinal microbiome. Regardless of the availability of future studies, we appreciate the clinical significance of PPIs. Therefore, searching for new therapies addressing risks and complications secondary to PPI use is of significant importance. For instance, it has been shown that modulation of microbiota with probiotic supplementation can reverse pathological changes in patients with inflammatory intestinal conditions. [126] Overutilization of PPIs needs more attention. Undoubtedly, patient education and counseling about the risks associated with PPI use and role of the gut microbiome should not be overlooked by healthcare providers.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ramsay PT, Carr A. Gastric acid and digestive physiology. Surg Clin North Am. 2011;91(5):977–82.
Zeng N, Athmann C, Kang T, et al. PACAP type I receptor activation regulates ECL cells and gastric acid secretion. J Clin Invest. 1999;104(10):1383–91.
Yamaji N, Yokoo Y, Iwashita T, et al. Structural determination of two active compounds that bind to the muscarinic M3 receptor in beer. Alcohol Clin Exp Res. 2007;31:S9–S14.
Schubert ML, Makhlouf GM. Gastrin secretion induced by distention is mediated by gastric cholinergic and vasoactive intestinal peptide neurons in rats. Gastroenterology. 1993;104(3):834–9.
Orloff SL, Bunnett NW, Wong H, Walsh JH, Debas HT. Neural and hormonal mechanisms mediate the enterogastric reflex: a study in intestinal transplants in rats. Gastroenterology. 1991;101(3):734–42.
Phan J, Benhammou JN, Pisegna JR. Gastric hypersecretory states: investigation and management. Curr Treat Options Gastroenterol. 2015;13(4):386–97.
Schubert ML. Gastric acid secretion. Curr Opin Gastroenterol. 2016;32(6):452–60.
Schubert ML, Peura DA. Control of gastric acid secretion in health and disease. Gastroenterology. 2008;134(7):1842–60.
Takeuchi K, Endoh T, Hayashi S, Aihara T. Activation of muscarinic acetylcholine receptor subtype 4 is essential for cholinergic stimulation of gastric acid secretion: relation to D cell/somatostatin. Front Pharmacol. 2016;7:278.
Frick C, Rettenberger AT, Lunz ML, Breer H. Complex morphology of gastrin-releasing G-cells in the antral region of the mouse stomach. Cell Tissue Res. 2016;366(2):301–10.
Bitziou E, Patel BA. Simultaneous detection of gastric acid and histamine release to unravel the regulation of acid secretion from the guinea pig stomach. Am J Physiol Gastrointest Liver Physiol. 2012;303(3):G396–403.
Vuyyuru L, Schubert ML. Histamine, acting via H3 receptors, inhibits somatostatin and stimulates acid secretion in isolated mouse stomach. Gastroenterology. 1997;113(5):1545–52.
Schubert ML, Hightower J, Makhlouf GM. Linkage between somatostatin and acid secretion: evidence from use of pertussis toxin. Am J Phys. 1989;256(2 Pt 1):G418–22.
Beales ILP. Regulation of gastric function by gastrin releasing peptide. Gut. 2002;50(6):897–8.
Yakabi K, Kawashima J, Kato S. Ghrelin and gastric acid secretion. World J Gastroenterol. 2008;14(41):6334–8.
Shimizu K, Li P, Lee KY, Chang TM, Chey WY. The mechanism of inhibitory action of secretin on gastric acid secretion in conscious rats. J Physiol. 1995;488(Pt 2):501–8.
Schmidt WE, Schenk S, Nustede R, Holst JJ, Folsch UR, Creutzfeldt W. Cholecystokinin is a negative regulator of gastric acid secretion and postprandial release of gastrin in humans. Gastroenterology. 1994;107(6):1610–20.
Konturek JW, Konturek SJ, Kwiecien N, et al. Leptin in the control of gastric secretion and gut hormones in humans infected with Helicobacter pylori. Scand J Gastroenterol. 2001;36(11):1148–54.
Shin JM, Munson K, Vagin O, Sachs G. The gastric HK-ATPase: structure, function, and inhibition. Pflugers Archiv. 2009;457(3):609–22.
Caplan MJ. The future of the pump. J Clin Gastroenterol. 2007;41(Suppl 2):S217–22.
Hersey SJ, Sachs G. Gastric acid secretion. Physiol Rev. 1995;75(1):155–89.
Fujii T, Fujita K, Takeguchi N, Sakai H. Function of K(+)-Cl(−) cotransporters in the acid secretory mechanism of gastric parietal cells. Biol Pharm Bull. 2011;34(6):810–2.
Fellenius E, Berglindh T, Sachs G, et al. Substituted benzimidazoles inhibit gastric acid secretion by blocking (H+ + K+) ATPase. Nature. 1981;290(5802):159–61.
Li H, Meng L, Liu F, Wei JF, Wang YQ. H+/K+-ATPase inhibitors: a patent review. Expert Opin Ther Pat. 2013;23(1):99–111.
Galmiche JP, Bruley Des Varannes S, Ducrotte P, et al. Tenatoprazole, a novel proton pump inhibitor with a prolonged plasma half-life: effects on intragastric pH and comparison with esomeprazole in healthy volunteers. Aliment Pharmacol Ther. 2004;19(6):655–62.
Shin JM, Kim N. Pharmacokinetics and pharmacodynamics of the proton pump inhibitors. J Neurogastroenterol Motil. 2013;19(1):25–35.
Sachs G, Shin JM. The basis of differentiation of PPIs. Drugs Today (Barc). 2004;40(Suppl A):9–14.
Shin JM, Sachs G. Restoration of acid secretion following treatment with proton pump inhibitors. Gastroenterology. 2002;123(5):1588–97.
Stedman CA, Barclay ML. Review article: comparison of the pharmacokinetics, acid suppression and efficacy of proton pump inhibitors. Aliment Pharmacol Ther. 2000;14(8):963–78.
Hatlebakk JG, Katz PO, Camacho-Lobato L, Castell DO. Proton pump inhibitors: better acid suppression when taken before a meal than without a meal. Aliment Pharmacol Ther. 2000;14(10):1267–72.
Katsube T, Adachi K, Kawamura A, et al. Helicobacter pylori infection influences nocturnal gastric acid breakthrough. Aliment Pharmacol Ther. 2000;14(8):1049–56.
Ishizaki T, Horai Y. Review article: cytochrome P450 and the metabolism of proton pump inhibitors—emphasis on rabeprazole. Aliment Pharmacol Ther. 1999;13(Suppl 3):27–36.
Abelo A, Andersson TB, Antonsson M, Naudot AK, Skanberg I, Weidolf L. Stereoselective metabolism of omeprazole by human cytochrome P450 enzymes. Drug Metab Dispos. 2000;28(8):966–72.
Andersson T, Hassan-Alin M, Hasselgren G, Rohss K. Drug interaction studies with esomeprazole, the (S)-isomer of omeprazole. Clin Pharmacokinet. 2001;40(7):523–37.
Furuta T, Ohashi K, Kamata T, et al. Effect of genetic differences in omeprazole metabolism on cure rates for Helicobacter pylori infection and peptic ulcer. Ann Intern Med. 1998;129(12):1027–30.
Tang HL, Li Y, Hu YF, Xie HG, Zhai SD. Effects of CYP2C19 loss-of-function variants on the eradication of H. pylori infection in patients treated with proton pump inhibitor-based triple therapy regimens: a meta-analysis of randomized clinical trials. PloS One. 2013;8(4):e62162.
Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1(8390):1311–5.
Brown LM. Helicobacter pylori: epidemiology and routes of transmission. Epidemiol Rev. 2000;22(2):283–97.
Eusebi LH, Zagari RM, Bazzoli F. Epidemiology of Helicobacter pylori infection. Helicobacter. 2014;19:1–5.
McJunkin B, Sissoko M, Levien J, Upchurch J, Ahmed A. Dramatic decline in prevalence of Helicobacter pylori and peptic ulcer disease in an endoscopy-referral population. Am J Med. 124(3):260–4.
Segal I, Ally R, Mitchell H. Helicobacter pylori—an African perspective. QJM: Int J Med. 2001;94(10):561–5.
Cave DR. How is Helicobacter pylori transmitted? Gastroenterology. 1997;113(6 Suppl):S9–14.
van Duynhoven YT, de Jonge R. Transmission of Helicobacter pylori: a role for food? Bull World Health Organ. 2001;79(5):455–60.
Bik EM, Eckburg PB, Gill SR, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103(3):732–7.
Blaser MJ. Helicobacter pylori: microbiology of a ‘slow’ bacterial infection. Trends Microbiol. 1993;1(7):255–60.
Stingl K, Altendorf K, Bakker EP. Acid survival of Helicobacter pylori: how does urease activity trigger cytoplasmic pH homeostasis? Trends Microbiol. 2002;10(2):70–4.
Kao C-Y, Sheu B-S, Wu J-J. Helicobacter pylori infection: an overview of bacterial virulence factors and pathogenesis. Biom J. 2016;39(1):14–23.
Logan RP. Adherence of Helicobacter pylori. Aliment Pharmacol Ther. 1996;10(Suppl 1):3–15.
Bytzer P, Dahlerup JF, Eriksen JR, Jarbol DE, Rosenstock S, Wildt S. Diagnosis and treatment of Helicobacter pylori infection. Dan Med Bull. 2011;58(4):C4271.
McColl KE, El-Omar E, Gillen D. Interactions between H. pylori infection, gastric acid secretion and anti-secretory therapy. Br Med Bull. 1998;54(1):121–38.
Chen XY, Liu WZ, Shi Y, Zhang DZ, Xiao SD, Tytgat GNJ. Helicobacter pylori associated gastric diseases and lymphoid tissue hyperplasia in gastric antral mucosa. J Clin Pathol. 2002;55(2):133–7.
Peterson WL. Helicobacter pylori and peptic ulcer disease. N Engl J Med. 1991;324(15):1043–8.
Jaakkimainen RL, Boyle E, Tudiver F. Is Helicobacter pylori associated with non-ulcer dyspepsia and will eradication improve symptoms? A meta-analysis. BMJ. 1999;319(7216):1040–4.
Kim SS, Ruiz VE, Carroll JD, Moss SF. Helicobacter pylori in the pathogenesis of gastric cancer and gastric lymphoma. Cancer Lett. 2011;305(2):228–38.
Gasbarrini A, Carloni E, Gasbarrini G, Chisholm SA. Helicobacter pylori and extragastric diseases—other Helicobacters. Helicobacter. 2004;9:57–66.
Shahabi S, Rasmi Y, Jazani NH, Hassan ZM. Protective effects of Helicobacter pylori against gastroesophageal reflux disease may be due to a neuroimmunological anti-inflammatory mechanism. Immunol Cell Biol. 2007;86(2):175–8.
Amedei A, Codolo G, Del Prete G, de Bernard M, D’Elios MM. The effect of Helicobacter pylori on asthma and allergy. J Asthma Allergy. 2010;3:139–47.
Lee JY, Kim N. Diagnosis of Helicobacter pylori by invasive test: histology. Ann Transl Med. 2015;3(1):10.
Testerman TL, Morris J. Beyond the stomach: an updated view of Helicobacter pylori pathogenesis, diagnosis, and treatment. World J Gastroenterol: WJG. 2014;20(36):12781–808.
Li C, Ha T, Ferguson DA Jr, et al. A newly developed PCR assay of H. pylori in gastric biopsy, saliva, and feces. Evidence of high prevalence of H. pylori in saliva supports oral transmission. Dig Dis Sci. 1996;41(11):2142–9.
Liu H, Rahman A, Semino-Mora C, Doi SQ, Dubois A. Specific and sensitive detection of H. pylori in biological specimens by real-time RT-PCR and in situ hybridization. PloS One. 2008;3(7):e2689.
Bassis CM, Erb-Downward JR, Dickson RP, et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. mBio. 2015;6(2):e00037.
Jovel J, Patterson J, Wang W, et al. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front Microbiol. 2016;7:459.
Li X-X, Wong GL-H, To K-F, et al. Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non-steroidal anti-inflammatory drug use. PLoS One. 2009;4(11):e7985.
• Khosravi Y, Dieye Y, Poh BH, et al. Culturable bacterial microbiota of the stomach of Helicobacter pylori positive and negative gastric disease patients. Sci World J. 2014;2014:610421. This study showed the correlation between Streptococcus and peptic ulcer disease. Burkholderia pseudomallei was also isolated from the gastric samples, suggesting geographical variations in the biodiversity of gastric microbiome.
Aviles-Jimenez F, Vazquez-Jimenez F, Medrano-Guzman R, Mantilla A, Torres J. Stomach microbiota composition varies between patients with non-atrophic gastritis and patients with intestinal type of gastric cancer. Sci Rep. 2014;4:4202.
Engstrand L, Lindberg M. Helicobacter pylori and the gastric microbiota. Best Pract Res Clin Gastroenterol. 2013;27(1):39–45.
Delgado S, Cabrera-Rubio R, Mira A, Suarez A, Mayo B. Microbiological survey of the human gastric ecosystem using culturing and pyrosequencing methods. Microb Ecol. 2013;65(3):763–72.
•• Rolig AS, Cech C, Ahler E, Carter JE, Ottemann KM. The degree of Helicobacter pylori-triggered inflammation is manipulated by preinfection host microbiota. Infect Immun. 2013;81(5):1382–9. The authors revealed that the clinical course and outcome of H. pylori disease are influenced by the host gastric bacterial composition. They suggested that gastric microbiome could be used as a diagnostic marker to predict the outcome of H. pylori infection.
Delgado S, Leite AM, Ruas-Madiedo P, Mayo B. Probiotic and technological properties of Lactobacillus spp. strains from the human stomach in the search for potential candidates against gastric microbial dysbiosis. Front Microbiol. 2014;5:766.
Khosravi Y, Dieye Y, Loke MF, Goh KL, Vadivelu J. Streptococcus mitis induces conversion of Helicobacter pylori to coccoid cells during co-culture in vitro. PLoS One. 2014;9(11):e112214.
Andersson AF, Lindberg M, Jakobsson H, Backhed F, Nyren P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One. 2008;3(7):e2836.
Brawner KM, Kumar R, Serrano CA, et al. Helicobacter pylori infection is associated with an altered gastric microbiota in children. Mucosal Immunol. 2017;
Llorca L, Perez-Perez G, Urruzuno P, et al. Characterization of the gastric microbiota in a pediatric population according to Helicobacter pylori status. Pediatr Infect Dis J. 2017;36(2):173–8.
Nardone G, Compare D. The human gastric microbiota: is it time to rethink the pathogenesis of stomach diseases? United European Gastroenterol J. 2015;3(3):255–60.
Mauch F, Bode G, Malfertheiner P. Identification and characterization of an ATPase system of Helicobacter pylori and the effect of proton pump inhibitors. Am J Gastroenterol. 1993;88(10):1801–2.
Stoschus B, Dominguez-Munoz JE, Kalhori N, Sauerbruch T, Malfertheiner P. Effect of omeprazole on Helicobacter pylori urease activity in vivo. Eur J Gastroenterol Hepatol. 1996;8(8):811–3.
Vesper BJ, Jawdi A, Altman KW, Haines GK 3rd, Tao L, Radosevich JA. The effect of proton pump inhibitors on the human microbiota. Curr Drug Metab. 2009;10(1):84–9.
Sanduleanu S, Jonkers D, De Bruine A, Hameeteman W, Stockbrugger RW. Non-Helicobacter pylori bacterial flora during acid-suppressive therapy: differential findings in gastric juice and gastric mucosa. Aliment Pharmacol Ther. 2001;15(3):379–88.
•• Paroni Sterbini F, Palladini A, Masucci L, et al. Effects of proton pump inhibitors on the gastric mucosa-associated microbiota in dyspeptic patients. Appl Environm Microbiol. 2016;82(22):6633–44. Firmicutes , particularly the Streptococcus genus, were found in a relative abundance in patients taking PPIs. The authors suggested that it may explain the exacerbation and persistence of dyspeptic symptoms in patients on PPI therapy.
Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124(4):837–48.
Mandar R, Mikelsaar M. Transmission of mother’s microflora to the newborn at birth. Biol Neonate. 1996;69(1):30–5.
Neu J, Rushing J. Cesarean versus vaginal delivery: long term infant outcomes and the hygiene hypothesis. Clin Perinatol. 2011;38(2):321–31.
Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–7.
O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7(7):688–93.
Simrén M, Barbara G, Flint HJ, et al. Intestinal microbiota in functional bowel disorders: a Rome foundation report. Gut. 2013;62(1):159–76.
Canny GO, McCormick BA. Bacteria in the intestine, helpful residents or enemies from within? Infect Immun. 2008;76(8):3360–73.
Dukowicz AC, Lacy BE, Levine GM. Small intestinal bacterial overgrowth: a comprehensive review. Gastroenterol Hepatol. 2007;3(2):112–22.
Swidsinski A, Loening-Baucke V, Lochs H, Hale LP. Spatial organization of bacterial flora in normal and inflamed intestine: a fluorescence in situ hybridization study in mice. World J Gastroenterol. 2005;11(8):1131–40.
Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–8.
Kahrstrom CT, Pariente N, Weiss U. Intestinal microbiota in health and disease. Nature. 2016;535(7610):47–7.
Sekirov I, Russell SL, Antunes LCM, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90(3):859–904.
Al-Asmakh M, Hedin L. Microbiota and the control of blood-tissue barriers. Tissue Barriers. 2015;3(3):e1039691.
Smith P, Willemsen D, Popkes ML, et al. Regulation of life span by the gut microbiota in the short-lived African turquoise killifish. bioRxiv. 2017;
Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis. 2015;26 doi:10.3402/mehd.v3426.26191.
Salonen A, de Vos WM, Palva A. Gastrointestinal microbiota in irritable bowel syndrome: present state and perspectives. Microbiology. 2010;156(Pt 11):3205–15.
Vogtmann E, Hua X, Zeller G, et al. Colorectal cancer and the human gut microbiome: reproducibility with whole-genome shotgun sequencing. PLoS One. 2016;11(5):e0155362.
De Palma G, Nadal I, Medina M, et al. Intestinal dysbiosis and reduced immunoglobulin-coated bacteria associated with coeliac disease in children. BMC Microbiol. 2010;10:63.
Tang WHW, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J Clin Invest. 2014;124(10):4204–11.
Fujimura KE, Lynch SV. Microbiota in allergy and asthma and the emerging relationship with the gut microbiome. Cell Host Microbe. 2015;17(5):592–602.
Petra AI, Panagiotidou S, Hatziagelaki E, Stewart JM, Conti P, Theoharides TC. Gut-microbiota-brain axis and effect on neuropsychiatric disorders with suspected immune dysregulation. Clin Ther. 2015;37(5):984–95.
Lutgendorff F, Akkermans LM, Soderholm JD. The role of microbiota and probiotics in stress-induced gastro-intestinal damage. Curr Mol Med. 2008;8(4):282–98.
Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology. 2007;132(4):1359–74.
Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci U S A. 2002;99(24):15451–5.
Gordon HA, Bruckner-Kardoss E. Effect of normal microbial flora on intestinal surface area. Am J Phys. 1961;201:175–8.
Wostmann B, Bruckner-Kardoss E. Development of cecal distention in germ-free baby rats. Am J Phys. 1959;197:1345–6.
Husebye E, Hellstrom PM, Midtvedt T. Intestinal microflora stimulates myoelectric activity of rat small intestine by promoting cyclic initiation and aboral propagation of migrating myoelectric complex. Dig Dis Sci. 1994;39(5):946–56.
Alam M, Midtvedt T, Uribe A. Differential cell kinetics in the ileum and colon of germfree rats. Scand J Gastroenterol. 1994;29(5):445–51.
Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292(5519):1115–8.
Beasley DE, Koltz AM, Lambert JE, Fierer N, Dunn RR. The evolution of stomach acidity and its relevance to the human microbiome. PLoS One. 2015;10(7):e0134116.
Roggenbuck M, Baerholm Schnell I, Blom N, et al. The microbiome of new world vultures. Nat Commun. 2014;5:5498.
Lange K, Buerger M, Stallmach A, Bruns T. Effects of antibiotics on gut microbiota. Dig Dis. 2016;34(3):260–8.
Jump RLP. Clostridium difficile infection in older adults. Aging Health. 2013;9(4):403–14.
Russell TL, Berardi RR, Barnett JL, et al. Upper gastrointestinal pH in seventy-nine healthy, elderly, North American men and women. Pharm Res. 1993;10(2):187–96.
Loo VG, Bourgault AM, Poirier L, et al. Host and pathogen factors for Clostridium difficile infection and colonization. N Engl J Med. 2011;365(18):1693–703.
Lee C, Hong SN. Does long-term proton pump inhibitor therapy affect the health of gut microbiota? Gut Liver. 2016;10(6):865–6.
Shin CM, Kim N, Kim YS, et al. Impact of long-term proton pump inhibitor therapy on gut microbiota in F344 rats: pilot study. Gut Liver. 2016;10(6):896–901.
Leonard J, Marshall JK, Moayyedi P. Systematic review of the risk of enteric infection in patients taking acid suppression. Am J Gastroenterol. 2007;102(9):2047–56. quiz 2057
Dial MS. Proton pump inhibitor use and enteric infections. Am J Gastroenterol. 2009;104(Suppl 2):S10–6.
•• Jackson MA, Goodrich JK, Maxan M-E, et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut. 2016;65(5):749–56. It is a large study (1827 healthy twins) that showed negative correlation between PPI use and microbial diversity of the gut. Also, overrepresentation of the upper gastrointestinal tract bacteria in the lower parts of the gut was detected.
Freedberg DE, Toussaint NC, Chen SP, et al. Proton pump inhibitors alter specific taxa in the human gastrointestinal microbiome: a crossover trial. Gastroenterology. 2015;149(4):883–5. e889
•• Imhann F, Bonder MJ, Vich Vila A, et al. Proton pump inhibitors affect the gut microbiome. Gut. 2016;65(5):740–8. The findings of this European study involving both healthy subjects and patients with GI disease (1815 participants) supported the idea that PPI use is associated with the development of less healthy gut microbiome. Of note, a significant increase in pathogenic bacteria has been revealed.
Lo WK, Chan WW. Proton pump inhibitor use and the risk of small intestinal bacterial overgrowth: a meta-analysis. Clin Gastroenterol Hepatol. 2013;11(5):483–90.
Fujimori S. What are the effects of proton pump inhibitors on the small intestine? World J Gastroenterol: WJG. 2015;21(22):6817–9.
Lué A, Lanas A. Protons pump inhibitor treatment and lower gastrointestinal bleeding: balancing risks and benefits. World J Gastroenterol. 2016;22(48):10477–81.
Gallo A, Passaro G, Gasbarrini A, Landolfi R, Montalto M. Modulation of microbiota as treatment for intestinal inflammatory disorders: an uptodate. World J Gastroenterol. 2016;22(32):7186–202.
Acknowledgements
This work received grant support from the Department of Veterans Affairs RR&D Merit Review (JRP) I01 RX000194; Human Studies CORE through CURE: Digestive Diseases Research Center supported by NIH grant P30DK41301.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Stomach and Duodenum
Rights and permissions
About this article

Cite this article
Minalyan, A., Gabrielyan, L., Scott, D. et al. The Gastric and Intestinal Microbiome: Role of Proton Pump Inhibitors. Curr Gastroenterol Rep 19, 42 (2017). https://doi.org/10.1007/s11894-017-0577-6
Published:
DOI: https://doi.org/10.1007/s11894-017-0577-6