
Abstract
Progressive kidney disease is a common companion to both type 1 and type 2 diabetes. However, the majority of people with diabetes do not develop diabetic kidney disease. This may in part be explained by good control of glucose, blood pressure, obesity and other risk factors for kidney disease. It may also be partly due to their genetic makeup or ethnicity. However, the vast majority of the variability in incident nephropathy remains unaccounted for by conventional risk factors or genetics. Epigenetics has recently emerged as an increasingly powerful paradigm to understand and potentially explain complex non-Mendelian conditions—including diabetic kidney disease. Persistent epigenetic changes can be acquired during development or as adaptations to environmental exposure, including metabolic fluctuations associated with diabetes. These epigenetic modifications—including DNA methylation, histone modifications, non-coding RNAs and other changes in chromatin structure and function—individually and co-operatively act to register, store, retain and recall past experiences in a way to shape the transcription of specific genes and, therefore, cellular functions. This review will explore the emerging evidence for the role of epigenetic modifications in programming the legacy of hyperglycaemia for kidney disease in diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The burden of diabetic kidney disease (DKD) is unequally shared. At least half of all patients with type 2 diabetes and one third of those with type 1 diabetes develop overt kidney disease owing to their diabetes and/or other comorbidity, such as vascular disease, hypertension and heart failure [1•]. At the same time, the majority of individuals with diabetes remain free of kidney disease, sometimes, despite long periods of persistent hyperglycaemia, dyslipidaemia or hypertension. Indeed, the long-term survival of some of the very first patients Banting and Best treated with insulin without the advantages or intensity of modern treatment regimens stand as a testament to the fact that some individuals appear to be ‘protected’ despite many decades of marked hyperglycaemia and the prolonged absence of effective treatment. The mechanism(s) by which protection occurs in some diabetic individuals, or through which some individuals with diabetes seem predisposed to progressive chronic kidney disease (CKD), remains to be established.
Most patients and their practitioners believe that diabetic complications, like kidney disease, are largely the cumulative result of inadequate metabolic control over many years of diabetes. Indeed, the very fact that diabetic nephropathy is not observed in the absence of hyperglycaemia, confirms that excess glucose flux is required for its pathogenesis. But while patients with the highest glucose levels have a greater risk for developing progressive kidney disease than those with good control, it is clear that even with intensive intervention and dedicated compliance, renal complications may still occur. Indeed, we have shown in the FinnDiane cohort of patients with type 1 diabetes that the majority of patients who develop albuminuria have very good glucose control [1•]. At the same time, many patients do surprisingly well and do not develop diabetic complications, despite their poor metabolic control. Consequently, the vast majority of the variability in incident nephropathy remains unaccounted for by conventional risk factors.
It is also assumed that coding differences in specific genes then explain why some individuals seem programmed for an inordinate burden of complications, while others remain unaffected despite similar or worse metabolic control [2]. Indeed, the idea of genetic determinism still pervades much of diabetes practice. Many patients will now ask if there is a genetic test to determine their risks, as well as their subsequent preventive health actions. An inherited predisposition for DKD is certainly, evident [3]. DKD is common in some families, but absent from others despite a shared predisposition for diabetes. For example, in the Diabetes Control and Complications Trial (DCCT), there was an increased risk of DKD in relatives of patients with DKD when compared to relatives of subjects with diabetes but without DKD (odds ratio = 5.4; P < 0.001) [3]. Some ethnic groups also have a high incidence of kidney disease associated with diabetes (e.g. Native American, Pacific Islanders, First Nation Australian (Aboriginal) [1•], and this difference may be partly genetically determined. A number of potential loci have been reproducibly associated with DKD. However, only three meet stringent criteria for significance, the ACE I/D polymorphism (ACE rs179975), the lipoprotein polymorphisms (APOE E2/3/4) and the polyol pathway polymorphism (AKRB1 CA repeat Z−2) [2]. Comprehensive sequencing across the whole genome has also identified additional targets, mostly in non-coding or other regulatory regions of the genome, such as enhancers [4]. However, any role for these polymorphisms, alone or in combination, in the molecular pathobiology of DKD remains to be established. With the exception of rare monogenetic disorders, current evidence suggests that the genetic code explains only a fraction of why some individuals develop DKD and some do not [2].
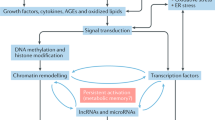
Although any genetic programming for DKD remains elusive, it is now clear that risk can be imprinted through other means. In particular, epigenetics has emerged as an increasingly powerful paradigm to understand and potentially explain complex non-Mendelian conditions—including DKD [5•]. Persistent epigenetic changes can be acquired during development or as structural adaptations following environmental exposure (the so-called environmental footprint) including metabolic fluctuations associated with diabetes. These epigenetic modifications—including DNA methylation, histone modifications, non-coding RNAs (ncRNAs) and other changes in chromatin structure and function—individually and co-operatively act to register, store, retain and recall past experiences in a way to shape the transcription of specific genes and, therefore, cellular functions. Indeed, epigenetics may be considered the language through which environmental factors and our genes interact to create and adapt a kind of cellular memory. And in much the same way that memories in our brain steer us away from future problems and/or steel us against them, epigenetic adaptations are supposed to be there to programme our safety. Indeed, epigenetic memories may underlie (programming) adaptations in important homeostatic pathways, including those associated with hyperglycaemia in diabetes. The problem comes when these responses become maladaptive, out of context or out of place. This review will explore the emerging evidence for the role of epigenetic modifications in programming for kidney disease in diabetes.
Epigenetics—Same Genes, But Different Outcomes
Epigenetic modifications refer to dynamic changes written on and erased in and around our genes by specialised enzymes, which do not alter the DNA nucleotide sequence itself, but instead modify how it is transcribed. DNA does not exist naked within a eukaryotic cell, but is dynamically packaged as a DNA–protein complex called chromatin, that facilitates the packaging of extraordinary lengths of DNA into the tight confines of the cell nucleus. When a gene product is needed, chromatin is selectively unwound and made ‘open’ to allow access to transcription factors (known as euchromatin). Potentially more so than the DNA sequence itself, changes to the structure and accessibility of chromatin significantly influence the regulation of gene expression, both between different cells and within an individual cell over its lifetime. These changes are partly determined by epigenetics. This means that the same genes can result in different phenotypes without changes in the DNA sequence. For example, every cell in the body is genetically identical and has a same gene for insulin, but only the β-cells of the pancreas have permissive epigenetic changes allowing open chromatin and insulin gene transcription. Elsewhere insulin expression is silenced by repressive epigenetic changes leading to chromatin condensation (known as heterochromatin). In the same way, genetically identical twins can become progressively more different as they age through accumulating epigenetic changes, even though their genetic similarity never changes.
The term ‘epigenetic’ is catch-all phrase encompassing many different and dynamic changes in and around our genes. The most well-known and best characterised is DNA methylation, which converts the DNA nucleotide, cytosine, into 5-methyl-cytosine, in essence creating a ‘fifth base’ mostly in the body of genes or adjacent to CpG ‘islands’ typically found in gene promoters. The regulatory effects of DNA methylation are complex and highly dependent of the genomic context. DNA methylation may serve a variety of roles including gene activation or silencing, modulation of transposable elements, regulation of transcription elongation and alternative splicing. This may be partly achieved by altering the binding of transcription factors and/or triggering the recruitment of transcriptional co-repressors to keep genes silenced. DNA methylation serves a critical regulatory function: to prevent genes being activated in the wrong setting, or from being silenced when they need to be transcribed. Indeed, it is now recognised that inopportune epigenetic silencing of tumour-suppressor genes or regulatory microRNA by hyper-methylation of their CpG islands is a key step in the origin of many cancers [6]. At the same time, there is also often progressive loss of loss-of-imprinting and epigenetic remodelling of repeat elements in cancers, particularly loss of (suppressive) methylation at satellite DNA and oncogene promoters which potentially contributes to chromosomal instability (leading to increased rates of mutation) and inopportune reactivation of silenced gene elements [6].
Another important epigenetic mark is the modification of histone proteins around which a little less than two turns of DNA (147 base pairs) is wound to form the nucleosome. Specifically, enzyme-mediated post-translational modification of the unstructured amino-terminal tails of histones (by acetylation, methylation, ubiquitination citrullination, phosphorylation and/or sumoylation) or the removal of these modifications are able to facilitate or repress gene transcription through altering the electrostatic interaction of histones with adjacent chromosomal DNA to regulate chromatin accessibility to transcription factors and RNA polymerase II. The specific site, type, extent and diversity of post-translational histone modifications are independently associated with distinguishable gene expression patterns, in what is referred to as the histone code (Fig. 1) [7]. For example, methylation of histone H3 lysine 4 (H3K4) H3K36 and H3K79 are activating signatures, associated with increased gene expression. Histone acetylation (such as acetylation at H3K9, H3K14 and H4K5) at gene promoters also correlates with transcriptional activation. By contrast, di- or tri-methylation at H3K9, H3K27 and H4K20 generally function as repressive marks [8].

The histone code. The specific site, type, extent and diversity of post-translational modifications histone proteins leads to specific signalling effects, including the repression (red signal) or activation (green signal) of gene expression
While the majority of the genome is transcribed into RNA, only a fraction codes for protein. The remainder is non-coding single-strand RNA. The best studied examples include microRNA (usually 20–25 nucleotides in length; miRNA) and long non-coding RNA (>200 nucleotides; lncRNA) [9]. Non-coding RNA is thought to confer an additional level of regulation of gene expression both at the level of transcription as well as translation. For example, non-coding RNA is able altering chromatin structure both directly or indirectly by recruiting chromatin-modifying complexes to target gene loci [10••] and forming a docking platform for recruiting epigenetic modifiers [9]. NcRNAs are also able to regulate RNA stability, the processing of (pre)-mRNAs and mRNA decay. MiRNA bind to 6–8 nucleotide sequences (known as called ‘seeds’) located in the 3′ untranslated region of protein-coding mRNA to regulate translational gene expression by promoting mRNA degradation and translational repression [11].
DNA Methylation and Diabetic Kidney Disease
Epigenetic imprinting is thought to be important for determining the predisposition for chronic and latent diseases, like DKD [5•]. We have previously shown that exposure of microvascular endothelial cells to hyperglycaemia is able to induce changes in DNA methylation on genome wide ChIP-Seq, leading to changes in gene expression, including activation of pro-inflammatory pathways implicated in diabetic complications such as DKD [5•, 12, 13•]. Studies in the zebrafish also demonstrate that hyperglycaemia-induced DNA methylation changes. Diabetes is also induces aberrant DNA methylation in the proximal tubules of the kidney, including key targets implicated in glucose metabolism and transport, leading to a resistance to the effects of pioglitazone [14]. However, an elevated glucose level is not the only factor that leads to maladaptive epigenetic modifications in diabetes. DNA methylation can also be influenced by reactive oxygen species, both directly through oxidative modification DNA preventing methylation and indirectly through its effects on methylation writing/erasing enzymes [15]. Many other factors including hypoxia, inflammation, cytokines and growth factors, drugs, nutrition and even physical activity can modify epigenetic profiles [16, 17]; the sum of which and their interactions being the key determinant of the resulting phenotype.
The potential importance of these experimental findings to clinical DKD is unclear. Certainly, skin cells cultured from individuals with DKD express different genes to those that do not have DKD [18], potentially reflecting their epigenetic programming. However, the application of high-throughput sequencing technologies beyond merely DNA sequencing, to include epigenetic modifications on a genome-wide scale, represents a new course towards the understanding of any predisposition to DKD. As an early taste of future epigenetic fingerprinting in DKD, researchers at Temple University Medical School have recently identified 187 gene targets that were differentially methylated in patients with type 2 diabetes and end-stage kidney disease when compared to diabetic patients without nephropathy [19]. Interestingly, 21 % of these genes had been previously implicated with CKD in genome association studies. Similarly, in a study of patients with type 2 diabetes, when compared to individuals without DKD, genomic DNA from patients with nephropathy relative demonstrated clear differential methylation at several genes, including UNC13B, which has previously been linked to DKD [20]. DNA methylation profiles in microdissected tubules obtained from patients with DKD also demonstrated differentially methylated genes targets implicated in fibrogenesis [21]. Finally, in a case control study of individuals with type 1 diabetes with and without DKD, differential methylation has been observed, including changes in genes that influence mitochondrial function associated with kidney disease [22]. Taken together, these and other data suggest that research may have identified the right gene targets for DKD all along, but that we failed to appreciate that variability in their expression was only more significantly determined by epigenetic programming than genetic polymorphism. For example, as stated above, polymorphisms in the ACE gene are associated with incident nephropathy in patients with diabetes [2], as well as changes in ACE expression. Somatic ACE contains two CpG islands in its proximal promoter region. When this DNA is de-methylated and associated histones are acetylated, ACE expression is markedly upregulated. But when these sites are methylated, ACE expression is silenced [23].
Histone Modifications and Diabetic Kidney Disease
Post-translational modification of nucleosomal histones are among the best characterised of epigenetic modifications with respect to diabetes and are clearly implicated in the induction in the expression of genes implicated in DKD [8, 24]. For example, following exposure to glucose there is persistent transcriptional upregulation of expression of the pro-inflammatory mediator NF-κB (p65; Rel (A)) in vitro and in vivo. This is specifically associated with mono-methylation of H3K4 adjacent to the p65 proximal promoter, such that inhibition of Set7-dependent methylation at this site is able to prevent its induction without restoring euglycaemia [8, 24]. We have also recently reported the persistent induction of other pathogenic genes that may be mediated by H3K4m1 writing events, including the induction of IL-8 following exposure to transient hyperglycaemia [25••]. Exposure to hyperglycaemia also dynamically changes histone acetylation in cells exposed to hyperglycaemia [12, 13•] and diabetic patients. More recently, genome-wide increases in monocyte H3 acetylation were associated with conventional treatment compared with intensive treatment group subjects of the Diabetes Control and Complications Trial (DCCT), indicating a possible mechanism of metabolic memory in humans [26•]. However, overall transcriptional activity is more likely to be dependent on the sum of multiple histone marks, and their interaction with other epigenetic modifications (e.g. DNA methylation) rather than any individual changes [27]. For example, glomerulosclerosis in diabetic mice is associated with enrichment of H3 histones dimethylated at K4, acetylated at K9 and K27, and phosphorylated at S10.
Non-coding RNA and DKD
Aberrant expression of miRNAs has been implicated in numerous disease states, including diabetes and its complications. For example, we have shown that miR-29a/b/c, Let-7b and miR200a can exert translational repression on collagen expression and can antagonise the pro-fibrotic effects of TGF-β, a pro-fibrotic cytokine of particular relevance to renal scarring seen in DKD [28–30]. Similarly, miR-21 is known to modulate the expression and activities of key pro-fibrotic factors associated with diabetic kidney disease [31]. The so-called redoximiRs miR-146a and miR-25 downregulate the expression of NADPH oxidase 4 (NOX-4) [32], a key mediator of renal injury in diabetes as demonstrated by the protection from renal injury observed in diabetic NOX-4 KO mice [33]. Recent reports have also suggest a critical role for long non-coding RNA in DKD, partly by acting as a host gene for miRNA [5•]. Most miRNAs are associated with RNA binding-proteins or packaged into small microvesicular particles (known as exosomes) that provide protection from RNAse and aid in their intercellular trafficking [34]. This also means that they are intrinsically protected against nuclease degradation and withstand repetitive freezing/thawing, so can potentially be used as biomarkers of the epigenetic milieu and the resulting phenotype [35, 36].
Epigenetics as the Language of Metabolic Memories?
It is clear from clinical studies that periods of good glycaemic control can have long-lasting effects on the development of complications in diabetes, potentially even decades after that good control is lost. Equally periods of poor control can cast a long shadow with an increased risk of cardiovascular disease, renal disease, retinopathy and neuropathy, even if glucose control is subsequently restored. For example, patients with type 1 diabetes in the DCCT who received intensive treatment (HbA1c 7.3 vs. 9.0 %) benefitted with a slower incidence and progression of complications relative to those who were in the conventional treatment group. Moreover, long-term follow-up of this cohort in the Epidemiology of Diabetic Complications and Interventions (EDIC) study demonstrated a persistently reduced complications rate (for nephropathy, retinopathy, hypertension cardiovascular disease and mortality) in patients randomised to receive intensive treatment for over a decade after the original study, even though the large differences in HbA1C between the two treatment groups that existed during the study had dissipated within a year of the completion of the DCCT [37•]. Similar finding were observed in the United Kingdom Prospective Diabetes Study Post Trial (UKPDS post-trial study) [38••] and ADVANCE-ON [39].
This phenomenon has become known as ‘metabolic memory’, ‘the legacy effect’ or ‘metabolic karma’ [40] and has been used to explain many clinical observations surrounding diabetes and its management. For example, the key impediment to benefits in many short and intermediate-term trials of glucose lowering belatedly undertaken in high-risk patients with advanced diabetes may be due to the ‘bad metabolic karma’ from years of poor control prior to enrolment the study [41]. More recently, the idea of ‘good metabolic karma’ has also been used to promote the concept of early intensive diabetes management, as a means to establish a long-term legacy for good vascular health.
A number of different factors potentially contribute to ‘metabolic karma’, including increasing power from longer observation, lead-time effects, long-lasting post-translational modification, compositional changes and the irreversibility of cumulative damage (i.e. once broken, it cannot be mended). However, another key factor may be epigenetic programming induced by periods of hyperglycaemia, obesity or other components of the diabetic milieu which promote maladaptive activation or pathogenic pathways, even when such risk factors are belatedly treated. Certainly, transient exposure to glucose in vitro results in changes in gene expression, that persistent for many days after glucose exposure has returned to normal [8, 24]. That these persistent changes in gene expression are largely mediated by epigenetic modifications is demonstrated by the finding that inhibition of the histone methyltransferase enzyme, SET-7, is able to prevent these changes [24, 25••]. Furthermore, persistent epigenetic effects of transient exposure to hyperglycaemia have been demonstrated in animal models of DKD [42]. Moreover, in epigenetic screening of blood monocytes from DCCT participants persistent differences in histone modifications, and, in particular, those associated with pro-inflammatory signalling, were still detectable 10 years after its completion, with those developing complications having more marked changes compared to those who remained free of microvascular disease [26•]
Developmental Programming and Epigenetics
Epigenetic programming also occurs during gestation and early development and is determined by the intrauterine environment, as well a pre-conception nutrition and health of both the mother and father [43]. Cells are potentially more sensitive to ‘in utero environmental priming’ during development and differentiation, when gene regulatory regions are becoming established. Famously children prenatally exposed to famine during the Dutch Hunger Winter of 1944–1945 had both higher prevalence of type 2 diabetes and subsequently diabetic complications including DKD. Whether this was mediated through intrauterine growth retardation (i.e. compositional endowment—such as small kidneys with reduced nephron number) or persistent epigenetic programming is still unclear [44]. Another example of in utero programming may be found in the children of mothers who develop gestational diabetes, also have a higher prevalence of type 2 diabetes, and subsequently diabetic complications including DKD. Again, one component of this predisposition may be epigenetic in origin [45]. Similarly, maternal smoking in pregnancy is associated with an increased risk of nephropathy in offspring who later develop diabetes, presumably through dysfunction programmed during development.
Trans-generational Persistence of Epigenetic Adaptations
The importance of epigenetic modifications is not only that they stably alter gene expression but also that some epigenetic signatures may be transferred through mitosis and meiosis, to the next generation of cells or progeny. This is consistent with the evolutionary theories of Jean-Baptiste Lamarck who proposed that ‘if an organism changes during life in order to adapt to its environment, some of those changes may be passed onto its offspring’ (Fig. 2). It is now clear that the external environment’s effects upon our genes can influence disease, and some of these effects can be inherited. For example, in Överkalix, Sweden, it was shown that if a grandfather did not have enough food available to him, his grandsons were less likely to die from cardiovascular disease or diabetes [46••]. By contrast, food abundance was associated with a shortened life span of his grandchildren. These findings suggest that the environment can cause stable (epigenetic) changes to (imprinted) genes that are passed down through many generations and that these alterations can affect susceptibility to certain diseases, including diabetes and its complications. The best example of a clear epigenetic basis of this phenomenon is the agouti mouse. In this model, the ‘agouti viable yellow’ (Avy) gene codes for a phenotype associated with yellow fur, adult-onset obesity and diabetes. Expression of this gene can be silenced by DNA methylation, and this modification can be inherited for several generations [47]. Epigenetic programming has also been implicated in the effects of parents’ diet on obesity in offspring [48] and trans-generational immune priming in mice, whereby fathers provide immune protection to offspring [49].

The impacts of environmental exposure prior to reproduction may be passed onto its offspring through epigenetic programming in order to sustain adaptations to its environment
Epigenetics as a Therapeutic Target?
Although it is not possible to change our genes or our grandparents, the cumulative effects of environmental exposure are not irrevocable. Unlike the genetic code, the human epigenome is surprisingly plastic and responsive to new stimuli. Whether it is possible to completely erase ‘bad karma’ through austerity (i.e. metabolic control) is unclear, but at the very least there is evidence of partial responsiveness. Indeed, it is highly likely that changes in epigenetic profiles partly mediate the long-term benefits of multifactorial interventions, including exercise [16], blockade of the renin angiotensin system [27] and glucose control. At present, the best opportunity for our patients is to achieve and maintain control early in the course of diabetes and accumulate karma from dedicated control, rather than trying to belatedly or inexpertly expunge its negative effects down the track. Given the very specific nature of epigenetic modification, it is hoped that in the future, it may be possible to rewrite our epigenome in way to promote health. Already, a number of epigenetic modifiers and antagomirs (miRNA inhibitors) are in clinical development, mostly for the treatment of cancer [50]. But given the burden of diabetes, they will soon be explored in diabetes and its complications as well. For example, sodium butyrate is able to promote insulin sensitivity through epigenetic mechanisms [51]
Conclusions
Epigenetic modifications potential provide one explanation for why some patients seem programmed for an inordinate burden of disease, including diabetic complications like DKD. Much more remains to be understood before it will be possible to read an epigenome to predict future clinical outcomes or the utility of intervention. At the same time, the plasticity of the human epigenome means that it is theoretically possible to undo adverse programming instilled by poor metabolic control. The best means to achieve this remains to be established. But this is an important area of ongoing research that will likely influence the management of diabetes in decades to come.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Thomas MC, Brownlee M, Susztak K, et al. Diabetic kidney disease. Nature Reviews (Disease Primers). July 2015. doi: 10.1038/nrdp.2015.18. This collaborative paper provides an expert review of the pathogenesis, epidemiology, diagnosis, management and treatment of diabetic kidney disease.
Thomas MC, Groop PH, Tryggvason K. Towards understanding the inherited susceptibility for nephropathy in diabetes. Curr Opin Nephrol Hypertens. 2012;21(2):195–202.
Clustering of long-term complications in families with diabetes in the diabetes control and complications trial. The Diabetes Control and Complications Trial Research Group. Diabetes. 1997;46(11):1829-1839
Iyengar SK, Abboud HE, Goddard KA, et al. Genome-wide scans for diabetic nephropathy and albuminuria in multiethnic populations: the family investigation of nephropathy and diabetes (FIND). Diabetes. 2007;56(6):1577–85.
Reddy MA, Zhang E, Natarajan R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia. 2015;58(3):443–55. This paper provides a state-of-the-art review of evidence pertaining to the role of epigenetic modifications in diabetic complications.
Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–59.
Grace BS, Clayton P, McDonald SP. Increases in renal replacement therapy in Australia and New Zealand: understanding trends in diabetic nephropathy. Nephrology. 2012;17(1):76–84.
Brasacchio D, Okabe J, Tikellis C, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes. 2009;58(5):1229–36.
Kornfeld JW, Bruning JC. Regulation of metabolism by long, non-coding RNAs. Front Genet. 2014;5:57.
Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482(7385):339–46. This paper provides an excellent overview of mechanistic evidence pertaining to non-coding RNAs.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Pirola L, Balcerczyk A, Tothill RW, et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 2011;21(10):1601–15.
Cooper ME, El-Osta A. Epigenetics: mechanisms and implications for diabetic complications. Circ Res. 2010;107(12):1403–13. This paper also provides a review of evidence pertaining to the role of epigenetic modifications in diabetic complications.
Marumo T, Yagi S, Kawarazaki W, et al. Diabetes induces aberrant DNA methylation in the proximal tubules of the kidney. J Am Soc Nephrol. 2015.
Richter K, Konzack A, Pihlajaniemi T, Heljasvaara R, Kietzmann T. Redox-fibrosis: impact of TGFbeta1 on ROS generators, mediators and functional consequences. Redox biology. 2015;6:344–52.
Horsburgh S, Robson-Ansley P, Adams R, Smith C. Exercise and inflammation-related epigenetic modifications: focus on DNA methylation. Exerc Immunol Rev. 2015;21:26–41.
Milagro FI, Mansego ML, De Miguel C, Martinez JA. Dietary factors, epigenetic modifications and obesity outcomes: progresses and perspectives. Mol Aspects Med. 2013;34(4):782–812.
Caramori ML, Kim Y, Goldfine AB, et al. Differential gene expression in diabetic nephropathy in individuals with type 1 diabetes. J Clin Endocrinol Metab. 2015;100(6):E876–82.
Sapienza C, Lee J, Powell J, et al. DNA methylation profiling identifies epigenetic differences between diabetes patients with ESRD and diabetes patients without nephropathy. Epigenetics. 2011;6(1):20–8.
Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genomics. 2010;3:33.
Hasegawa K, Wakino S, Simic P, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. 2013;19(11):1496–504.
Swan EJ, Maxwell AP, McKnight AJ. Distinct methylation patterns in genes that affect mitochondrial function are associated with kidney disease in blood-derived DNA from individuals with type 1 diabetes. Diabet Med. 2015;32(8):1110–5.
Riviere G, Lienhard D, Andrieu T, Vieau D, Frey BM, Frey FJ. Epigenetic regulation of somatic angiotensin-converting enzyme by DNA methylation and histone acetylation. Epigenetics. 2011;6(4):478–89.
El-Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205(10):2409–17.
Okabe J, Orlowski C, Balcerczyk A, et al. Distinguishing hyperglycemic changes by Set7 in vascular endothelial cells. Circ Res. 2012;110(8):1067–76. This paper elaborates one key mechanism through which exposure to elevated glucose levels is able to imprint persistent epigenetic markers.
Miao F, Chen Z, Genuth S, et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes. 2014;63(5):1748–62. This paper documents the epigenetic changes induced by intensive control during the Diabetes Control and Complications Trial.
Reddy MA, Sumanth P, Lanting L, et al. Losartan reverses permissive epigenetic changes in renal glomeruli of diabetic db/db mice. Kidney Int. 2014;85(2):362–73.
Wang B, Jha JC, Hagiwara S, et al. Transforming growth factor-beta1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 2014;85(2):352–61.
Wang B, Komers R, Carew R, et al. Suppression of microRNA-29 expression by TGF-beta1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol. 2012;23(2):252–65.
Wang B, Koh P, Winbanks C, et al. miR-200a prevents renal fibrogenesis through repression of TGF-beta2 expression. Diabetes. 2011;60(1):280–7.
Zhong X, Chung AC, Chen HY, et al. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia. 2013;56(3):663–74.
Cheng X, Ku CH, Siow RC. Regulation of the Nrf2 antioxidant pathway by microRNAs: new players in micromanaging redox homeostasis. Free Radic Biol Med. 2013;64:4–11.
Thallas-Bonke V, Jandeleit-Dahm KA, Cooper ME. Nox-4 and progressive kidney disease. Curr Opin Nephrol Hypertens. 2015;24(1):74–80.
Mitchell PS, Parkin RK, Kroh EM, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105(30):10513–8.
Kantharidis P, Wang B, Carew RM, Lan HY. Diabetes complications: the microRNA perspective. Diabetes. 2011;60(7):1832–7.
McClelland A, Hagiwara S, Kantharidis P. Where are we in diabetic nephropathy: microRNAs and biomarkers? Curr Opin Nephrol Hypertens. 2014;23(1):80–6.
EDIC. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. JAMA. 2003;290(16):2159–67. This paper describes the persistent benefits achieved by intensive glucose control during the Diabetes Control and Complications Trial.
Chalmers J, Cooper ME. UKPDS and the legacy effect. N Engl J Med. 2008;359(15):1618–20. This editorial discusses the game-changing results of the UKPDS follow-up study and coins the now widely used term “the legacy effect”.
Zoungas S, Chalmers J, Neal B, et al. Follow-up of blood-pressure lowering and glucose control in type 2 diabetes. N Engl J Med. 2014;371(15):1392–406.
Thomas MC. Glycemic exposure, glycemic control, and metabolic karma in diabetic complications. Adv Chronic Kidney Dis. 2014;21(3):311–7.
Bianchi C, Del Prato S. Metabolic memory and individual treatment aims in type 2 diabetes—outcome-lessons learned from large clinical trials. Rev Diabet Stud. 2011;8(3):432–40.
Kowluru RA, Abbas SN, Odenbach S. Reversal of hyperglycemia and diabetic nephropathy: effect of reinstitution of good metabolic control on oxidative stress in the kidney of diabetic rats. J Diabetes Complications. 2004;18(5):282–8.
Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol. 2011;31(3):363–73.
Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105(44):17046–9.
Binder AM, LaRocca J, Lesseur C, Marsit CJ, Michels KB. Epigenome-wide and transcriptome-wide analyses reveal gestational diabetes is associated with alterations in the human leukocyte antigen complex. Clinical epigenetics. 2015;7(1):79.
Kaati G, Bygren LO, Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur J Hum Genet. 2002;10(11):682–8. This seminal paper documents the trans-generational effects of diet.
Waterland RA, Travisano M, Tahiliani KG. Diet-induced hypermethylation at agouti viable yellow is not inherited transgenerationally through the female. FASEB J. 2007;21(12):3380–5.
Slyvka Y, Zhang Y, Nowak FV. Epigenetic effects of paternal diet on offspring: emphasis on obesity. Endocrine. 2015;48(1):36–46.
Eggert H, Kurtz J, Diddens-de Buhr MF. Different effects of paternal trans-generational immune priming on survival and immunity in step and genetic offspring. Proceedings Biological sciences / The Royal Society. 2014;281:1797.
Smyth LJ, Duffy S, Maxwell AP, McKnight AJ. Genetic and epigenetic factors influencing chronic kidney disease. Am J Physiol Renal Physiol. 2014;307(7):F757–76.
Henagan TM, Stefanska B, Fang Z, et al. Sodium butyrate epigenetically modulates high-fat diet-induced skeletal muscle mitochondrial adaptation, obesity and insulin resistance through nucleosome positioning. Br J Pharmacol. 2015;172(11):2782–98.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Merlin C. Thomas declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Microvascular Complications—Nephropathy
Rights and permissions
About this article

Cite this article
Thomas, M.C. Epigenetic Mechanisms in Diabetic Kidney Disease. Curr Diab Rep 16, 31 (2016). https://doi.org/10.1007/s11892-016-0723-9
Published:
DOI: https://doi.org/10.1007/s11892-016-0723-9