
Abstract
Purpose of Review
The application of advanced genomic testing to develop tumor-specific molecular profiles is essential to facilitating precision medicine pharmacotherapy. These approaches are highly relevant in colorectal cancer, where tumors frequently contain druggable molecular mutations, as well as the potential to respond to immunotherapy. Here we review the literature characterizing biomarker-driven pharmacotherapy for colorectal cancer, and highlight the pivotal ongoing trials that will help inform future treatment of this disease.
Recent Findings
Both prospective and retrospective studies have confirmed that the benefit from adding anti-epidermal growth factor receptor therapy is limited to patients with stage IV disease, RAS wild-type tumors, and left-sided primary tumors. Furthermore, patients with BRAF-mutated tumors derive significantly less benefit from the addition anti-epidermal growth factor receptor therapy. The use of BRAF inhibitors in the second-line setting is associated with a relatively high response rate, and regimens incorporating first-line treatment with BRAF inhibitors may soon become standard of care for patients with BRAF-mutated tumors. In the relapsed setting, the use of targeted agents and immunotherapy should be prioritized for patients with respective tumor profiles.
Summary
There has been significant advancement in the understanding of how to utilize molecular profiling and tumor biomarkers to tailor pharmacotherapy in colorectal cancer. Future studies should continue to incorporate these tests at enrollment to further define patient cohorts deriving the greatest benefit from precision medicine, characterize ideal sequence of therapy, and advance understanding of drug resistance mechanisms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
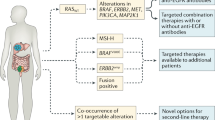
Colorectal cancer (CRC) is the second most common cause of cancer-related death in the USA, with an estimated incidence of 53,000 deaths annually [1]. Advances in the molecular testing of CRC tumors have enabled a more detailed understanding of mutational drivers of tumor biology and resistance to therapies. Increased availability of molecular profiling in CRC has facilitated incorporation of precision medicine through the development of targeted therapies with activity against specific tumor molecular characteristics [2]. Currently, biomarkers relevant to treatment of CRC include mutations in RAS (KRAS/NRAS) and BRAF, human epidermal growth factor 2 (HER2) amplifications and overexpression, microsatellite instability (MSI) and mismatch repair (MMR) status, as well as neurotrophic tyrosine kinase (NRTK) fusions (Fig. 1). In this review, we describe the evolution of molecular targets in CRC and highlight the most relevant recent literature describing the use of molecular profiling and biomarkers for its treatment.

Overview of molecular biomarkers in metastatic colorectal cancer. WT, wild type; MT, mutant, MSI-H, microsatellite instability high, NTRK, neurotrophic receptor tyrosine kinase; HER-2, human epidermal growth factor receptor 2. Co-existence of RAS and BRAF mutations occurs rarely and most commonly includes non-V600E BRAF mutations and/or atypical RAS mutations
Epidermal Growth Factor Receptor (EGFR)
Epidermal growth factor receptors (EGFRs) represent one form of transmembrane ERBB receptor tyrosine kinases (RTK) and are known to be upregulated in a variety of different tumor types. Upon ligand binding, EGFR dimerization leads to activation of a complicated downstream signaling pathway which facilitates tumor development and resistance via cellular proliferation, enhanced cell motility, increased protein secretion, and avoidance of apoptosis [3, 4]. These downstream signaling pathways are known to play an important role in the biology of CRC and include intracellular targets such as phosphoinositide 3-kinase (PI3K), the mitogen-activated protein kinase (MAPK), and RAS-RAF-mediated pathways [5,6,7].
RAS (KRAS/NRAS) Testing in Colorectal Cancer
In contrast to other disease entities like non-small cell lung cancer, there is no established role for testing for EGFR mutations in CRC as they are not predictive of response and therefore not routinely performed [8]. However, in patients being considered for anti-EGFR therapy, it is imperative to genotype for mutations in downstream targets, most notably RAS [2]. This is because RAS mutations in exons 2, 3, or 4 confer constitutive activation of signaling pathways downstream of EGFR, which renders anti-EGFR therapy ineffective. Thus, anti-EGFR therapies are only recommended for those patients with CRC harboring wild-type (WT) RAS genes [9, 10]. These activating mutations leading to therapy resistance occur more commonly in KRAS (up to 50%) compared to NRAS (up to 8%) mutated CRC [11]. Determination of RAS status is performed using DNA-based tests to identify specific gene mutations, or next-generation sequencing (NGS) panels which may also identify additional rare actionable mutations in tumor specimens such a BRAF [12, 13]. RAS testing should be performed at a CLIA-certified laboratory and may utilize specimens archived from primary tumors that have progressed or de novo metastatic sites [14].
Anti-EGFR Therapy for CRC
Cetuximab and panitumumab are monoclonal antibodies which bind to the extracellular domain of EGFR, thereby blocking the binding of endogenous ligands and suppressing EGFR signaling [7, 15]. The initial efficacy of anti-EGFR therapy was demonstrated in the third-line setting in previously treated stage IV metastatic CRC (mCRC) where cetuximab demonstrated improvement in overall response rates (ORR) (8.0% vs. 0%) and median overall survival (mOS) (6.1 vs. 4.6 months) but not median progression free survival (mPFS) (1.8 vs. 1.9 months) [16, 17]. Similarly, panitumumab was found to have activity compared to best supportive care in patients previously treated with two lines of chemotherapy with improvements in ORR (10% vs. 0%), mPFS (8.0 vs. 7.3 weeks), but no improvement in OS, conceivably because of crossover (76% crossed over from BSC to panitumumab) [18]. As evidenced by the relatively small numerical increases in these outcome measures, the clinical benefit of both panitumumab and cetuximab in this yet unselected patient population was modest. However, a subsequent retrospective analysis comparing outcomes of patients with and without KRAS mutations treated with panitumumab found that the clinical benefit of panitumumab was exclusive to WT KRAS patients (PFS 12.3 vs. 7.3 weeks) compared to KRAS-mutated patients (PFS 7.4 vs. 7.3 weeks) [19]. This study was the first to characterize the impact of KRAS status as a predictive biomarker of response to anti-EGFR therapy. A similar retrospective analysis was performed for cetuximab and also concluded that patients harboring KRAS mutations did not derive any clinical benefit from cetuximab [10]. These data were instrumental in identifying the importance of KRAS mutations in CRC, and KRAS mutational status would later be incorporated into the eligibility criteria for clinical studies investigating the activity of anti-EGFR therapy on CRC.
Subsequent investigations of both cetuximab and panitumumab evaluated their efficacy in the second-line setting when combined with standard chemotherapy backbones such FOLFOX and FOLFIRI. Similar to studies performed in the third-line setting, these studies (EPIC and 20050181) enrolled patients with and without KRAS mutations and demonstrated the addition of anti-EGFR therapy to standard chemotherapy provided modest benefits in terms of ORR and mPFS, and minimal or no benefit on mOS [20,21,22]. In these trials, when KRAS mutational testing was performed, patients harboring KRAS mutations had no improvement in outcome measures compared to chemotherapy alone [20, 22]. Around this same time, a similar open-label trial of chemotherapy with or without panitumumab for second-line treatment of mCRC (PICCOLO) amended its protocol during enrollment to include only patients with KRAS WT tumors [23]. Interestingly, despite excluding patients with KRAS mutations and restricting within-protocol crossover, no mOS benefit was noted in this trial (10.9 vs. 10.4 months) [23].
In the frontline setting, several phase II studies demonstrated the potential benefit of anti-EGFR therapy, which supported the development of larger phase III studies [24,25,26,27]. Subsequently there have been seven prospective phase III studies (CRYSTAL, PRIME, COIN, NORDIC, FIRE-3, TAILOR, CALGB/SWOG 80405) evaluating the efficacy of anti-EGFR therapy added to standard chemotherapy for the frontline treatment of CRC detailed in Table 1 [28,29,30,31,32,33,34]. Some of these earlier frontline studies of anti-EGFR therapy did not exclude patients harboring KRAS mutations, and similar to previous studies in the second and third line, the benefit of anti-EGFR therapy was consistently limited to KRAS WT patients. Furthermore, prior to the PRIME study, the initial use of KRAS mutations as a biomarker for response included mutations only in KRAS exon 2 codons 12 or 13. However, investigators in the PRIME study established the concept of extended RAS analysis, which included any KRAS mutation in exons 2–4 and NRAS mutations in exons 2–4 [29, 35]. Therefore, with the incorporation of extended RAS mutations, outcomes in the extended RAS WT populations have demonstrated more favorable outcomes for anti-EGFR therapy. The most informative of these trials are the FIRE-3, which showed improvement in mOS with the addition of cetuximab to chemotherapy vs. the addition of bevacizumab to chemotherapy, and CALGB/SWOG 80405, which showed no difference in mOS between these two treatments in the overall patient population [32, 33]. Based on these data, it would appear that either anti-VEGF therapy or anti-EGFR therapy are treatment options for first-line treatment of EGFR WT mCRC. However, subsequent analyses of these trials have identified the importance of primary tumor sidedness (PTS), discussed below, which can further refine the subgroup of patients deriving most benefit from frontline anti-EGFR therapy.
Lastly, anti-EGFR therapy is currently only indicated for patients with stage IV CRC. Two phase III studies have shown a lack of benefit in the adjuvant setting when added to an oxaliplatin-based regimen in stage III colon cancer [36, 37]. Additionally, several phase II studies as well as a meta-analysis suggested the potential benefit of anti-EGFR therapy for liver-limited metastases, improving resectability [38,39,40]. However, the recent results of a phase III trial (New EPOC) found that the addition of cetuximab to perioperative chemotherapy for resectable colorectal liver metastases (CRLM) leads to significant decreased mOS (55.4 vs. 81.0 months, HR 1.45) [41]. Based on this, it is recommended against using perioperative anti-EGFR therapy for resectable disease, and with caution in patients with unresectable disease when the goal is conversion to resectable status [42].
BRAF Mutations as Biomarker for Anti-EGFR Therapy
BRAF mutations in CRC as well as anti-BRAF therapy CRC are described more thoroughly below. However, it is important to know that these also play a role as a biomarker for response to anti-EGFR therapy in treatment of CRC. A multitude of retrospective studies have identified that patients harboring BRAF V600E mutations derive little or no clinical benefit from anti-EGFR therapy [9, 23, 43, 44]. Interpretation of these data is challenging as BRAF mutants occur most commonly in right-sided tumors. Taken together, for patients with BRAF V600E-mutated mCRC, clinicians should strongly consider alternative to anti-EGFR monotherapy or with chemotherapy unless the regimen also includes a BRAF inhibitor.
Summary of Biomarkers for Anti-EGFR Therapy
In summary, in either the first-line treatment of CRC or to downsize liver-limited unresectable disease, anti-EGFR therapy should only be offered to patients who have RAS WT, BRAF WT, and left-sided tumors only. There is no current role for adjuvant anti-EGFR therapy in treatment of CRC.
BRAF Testing in CRC
BRAF is a protein downstream of both EGFR and RAS in the MAPK kinase signaling pathway. Therefore, mutations in BRAF can lead to constitutive activation of the MAPK pathway independent of RAS or EGFR signaling, which triggers proliferation, differentiation, and cell survival [45]. Similar to RAS, testing for BRAF mutations is typically performed using a DNA-based test with polymerase chain reaction (PCR) or NGS methodologies [46]. Mutations in BRAF occur in 5–10% of CRC cases, are mutually exclusive with RAS mutations, and can be grouped in 3 classes: class 1-V600E (RAS independent signaling as monomer), class 2-codons 597/601 (RAS independent signaling as dimers) and class 3-codons 594/596 (RAS-dependent with impaired kinase activity) [47, 48]. Patients with class 1 V600E-mutated BRAF tend to be older age, female, right-sided primary, poor differentiation and poor prognosis, and frequently associated with MSI-H tumors. Patients with class 2 BRAF mutations share similar clinical and pathologic features as class 1. However, patients with class 3 BRAF-mutated metastatic CRCs are found to be more frequent in left-sided tumor and without peritoneal metastases [48]. The prognosis of patient with class 3 BRAF mutations is better than even for BRAF WT cancers [49].
BRAF Inhibitors for CRC
There are currently several oral tyrosine kinase inhibitors which have been developed to selectively target the mutant BRAF protein. These include vemurafenib, encorafenib, and dabrafenib, all of which were originally shown to be active in BRAF V600-mutated metastatic melanoma [50]. In contrast to their single-agent activity in metastatic melanoma, BRAF inhibitor monotherapy has shown very limited clinical activity for CRC [51, 52]. This is thought to be due to a feedback loop whereby BRAF inhibition triggers rapid activation of EGFR signaling, which permits continued cell growth and survival despite independent BRAF signaling. Subsequent combination strategies evaluated the potential efficacy of BRAF inhibition when combined with cytotoxic chemotherapy and/or anti-EGFR therapy [53, 54]. Based on this preliminary evidence, the SWOG 1406 trial was performed which was a randomized prospectively phase II study of irinotecan and cetuximab with or without vemurafenib in 106 BRAF-mutated patients with mCRC. The addition of vemurafenib increased PFS (4.4 vs. 2.0 months, HR 0.42) and these results informed future combination strategies with BRAF inhibitors [55].
The BEACON study demonstrated the efficacy of BRAF plus EGFR inhibition in the second- and third-line settings. This was a phase III open-label study of 655 patients with BRAF V600E-positive mCRC with progression after 1–2 prior treatment lines who were randomized to one of three cohorts: triplet biologic therapy (encorafenib + binimetinib (MEK inhibitor) + cetuximab), doublet biologics (encorafenib + cetuximab), or control (cetuximab + either irinotecan or FOLFIRI) [56]. As mentioned previously, encorafenib is an oral tyrosine kinase inhibitor targeting BRAF, and binimetinib is an oral tyrosine kinase inhibitor targeting MEK that has shown to be efficacious for BRAF-mutated metastatic melanoma [50]. The authors found that patients receiving doublet therapy demonstrated improved mOS of 9.3 months compared to 5.9 months in the control arm, and similar mOS in the triplet arm of 9.3 months [56, 57]. Given the incremental benefit with triplet therapy which was not statistically significant, but greater toxicity in this arm, in April 2020 the Food and Drug Administration (FDA) approved the combination of encorafenib and cetuximab for mCRC with BRAF V600E who progressed after one or two prior regimens [58]. Based on these data, the combination of cetuximab with encorafenib supports this combination as the new standard of care for second-line treatment of BRAFV600E-mutated mCRC [59]. More recently, the ANCHOR-CRC trial was the first study to investigate BRAF inhibitor therapy in the first-line setting for BRAFV600E-mutant mCRC. This was an open-label, single-arm, phase 2, two-stage study of 41 patients treated with the combination of encorafenib, binimetinib, and cetuximab. Results of the first stage demonstrated an ORR of 50% and mPFS 4.9 months in stage I of the study, and notably based on the number of responses in this high risk feature population this study will proceed with stage two to enroll an additional 54 patients. The second stage has finished accruing, and results are still anticipated [60]. In addition to this study, the phase III trial entitled BREAKWATER will evaluate the combination of encorafenib, cetuximab with or without chemotherapy in the first-line setting in patients with BRAF V600E-mutated mCRC [61].
Primary Tumor Sidedness as Biomarker for Anti-EGFR Therapy
In addition to mutations in RAS and BRAF, discussed below, PTS has been shown to be an important predictive biomarker for anti-EGFR efficacy [62]. Primary tumor locations refer to either right-sided or left-sided tumors, with right-sided tumors being defined by the proximal colon from cecum to two-thirds of the transverse colon which originated from the embryonic midgut. Right-sided tumors carry a negative prognostic effect, and PTS is thought to be a surrogate marker for oncogenic alterations such as BRAF, PIK3CA, AKT1, RNF43, and SMAD4 mutations which occur more commonly in right-sided primary tumors [47]. This effect has been shown in large aggregated retrospective studies combining first- and second-line use of anti-EGFR therapy for CRC (CRYSTAL, PRIME, PEAK, FIRE-3, CALGB 80403, 20050181). This meta-analysis revealed that the OS benefit of chemotherapy with anti-EGFR therapy was greatest in left-sided tumors (HR=0.75), and there was no significant OS benefit for the addition of anti-EGFR therapy to chemotherapy for right-sided tumors (HR=1.12) [63]. This effect has also been replicated in meta-analyses specific to first-line anti-EGFR therapy [64]. In addition to these combined analyses, the impact of PTS was also investigated in patients treated in the capstone CALGB 80405 trial. This analysis demonstrated that in KRAS WT patients treated with cetuximab, the mOS benefit was greater than twice as long for left-sided tumors compared to right-sided tumors (37.5 vs. 16.4 months). Additionally, patients with right-sided tumors treated with bevacizumab had improved mOS (24.5 months) compared to cetuximab [65]. Based on these data, the effect of PTS is most firmly established in the first-line setting use of anti-EGFR therapy, but the body of evidence suggests that this is likely predictive in subsequent lines of therapies as well. Interestingly, even when adjusted for all molecular alterations affecting response to EGFR antibody therapy, sidedness was still identified as an independent predictive factor.
Deficient Mismatch Repair (dMMR) and Microsatellite Instability High (MSI-H) Testing in CRC
Mismatch repair proteins, which include MLH1, MSH2, MSH6, and PMS2, repair insertions or deletions that appear in DNA replication. When this system is defective, mutations accumulate, and microsatellite instability emerges. This is known as deficient mismatch repair (dMMR), detected via immunohistochemistry, or MSI-H, detected via PCR or NGS [66,67,68]. These dMMR/MSI-H tumors are seen in a number of cancers, including gastrointestinal, uterine, ovarian, and prostate malignancies, but CRC has one of the highest prevalence of dMMR/MSI-H, in which it can range from 5 to 15% in a stage-dependent manner [69,70,71]. Importantly, these biomarkers can aid in the diagnosis of Lynch syndrome, and therefore universal MMR or MSI testing is recommended for all patients with CRC [2, 72]. dMMR/MSI-H tumors are associated with poor response to chemotherapy, but due to the high expression of neoantigens, they are considered good targets for immunotherapy approaches with single-agent or combined immune checkpoint inhibitors (ICI) [70, 73,74,75].
Immunotherapy for Treatment of dMMR and MSI-H CRC
In early stage disease, dMMR/MSI-H CRCs are thought to carry a favorable prognosis. This is based on several retrospective analyses which have demonstrated that this subtype is less likely to metastasize, is associated with improved outcomes, and typically does not derive the same benefit from fluoropyrimidine-based adjuvant chemotherapy compared to patients with MMR-proficient tumors [76,77,78,79,80,81]. Based on this, adjuvant chemotherapy should typically not be offered to patients with stage II dMMR/MSI-H cancers without other high risk features.
Initial efficacy of ICIs in dMMR/MSI-H mCRC was demonstrated in a phase II trial of pembrolizumab in previously treated patients with and without dMMR in a variety of tumor types [73]. This was the first study to establish the role of dMMR as a biomarker for response to checkpoint inhibition. In both CRC and non-CRC patients, dMMR tumors had an improved ORR when compared to those with MMR-proficient cancers. Neither OS nor PFS were not reached by patients in the dMMR CRC group at the time of publication [73]. Based on these findings, as well as several other phase I–II trials, in 2017 the FDA approved the use of pembrolizumab in the chemotherapy refractory setting for any solid tumor with dMMR/MSI-H [82]. More recently, data from the KEYNOTE-164 study further supported the efficacy of PD-1 inhibition in this patient population. KEYNOTE-164 was a phase II, open-label, single-arm study of pembrolizumab in 124 patients with previously treated dMMR/MSI-H CRC. Results demonstrated that in patients who previously received >2 prior lines of therapy, mOS was 31.4 months, and those with <1 prior line of therapy, mOS was not yet reached [83].
Combination ICI therapy has also been evaluated for patients in this setting. CheckMate-142 was a phase II single-arm trial evaluating the combination of nivolumab and ipilimumab in 45 patients with dMMR/MSI-H mCRC which were refractory or intolerant to chemotherapy [84, 85]. A 2020 update with median follow-up of 2 years revealed investigator-assessed ORR increased to 69% (95% CI 53–82), and PFS, OS, and median duration of response were not reached. Rates of PFS and OS at 2 years were 74% and 79%, respectively, and overall therapy was well tolerated with only 22% of subjects experiencing grade 3–4 AEs [85]. Ongoing studies are currently evaluating the efficacy of this combination in first-line treatment of dMMR/MSI-H mCRC [86]. Based on these results, the FDA approved nivolumab and ipilimumab in treatment-refractory dMMR/MSI-H mCRC [87].
ICI has also been investigated in the frontline setting in this patient population. KEYNOTE-177 was a phase III, randomized clinical trial evaluating pembrolizumab versus standard of care chemotherapy in 307 patients with untreated dMMR/MSI-H mCRC [88•]. In the most recent updated results from this trial, authors reported improved mPFS with pembrolizumab compared to chemotherapy (16.5 months vs. 8.2 months), and OS analysis is still ongoing. Additionally, despite a high rate of 59% crossover from the chemotherapy arm to immunotherapy, PFS2, defined by the time from the randomization to the second progression with the subsequent line of therapy, for pembrolizumab was not reached compared with the chemotherapy arm of 23.5 months [89]. These data support pembrolizumab as new standard of care first-line therapy in patients with MSI-high mCRC, and the FDA approved pembrolizumab for this indication in 2020 [90]. However, it is important to note that there is the higher rate of progression as best response with pembrolizumab (29.4% vs 12.6%) compared to chemotherapy. The identification of the subset of patient population that may not benefit from pembrolizumab is essential, and combination of chemotherapy and immunotherapy or doublet immunotherapies should be tested in this population. Additionally, further analysis of retrospective biospecimen data and PFS of subsequent therapy progression would help determine the primary and secondary resistant mechanism of MSI high tumor to pembrolizumab therapy.
Given the success of immunotherapy dMMR/MSI-H tumors, several studies are ongoing to evaluate its role in adjuvant and neoadjuvant setting in these patients. The ongoing NICHE study investigates the utility of ipilimumab and nivolumab in patients with early-stage CRC [91]. The results of 35 evaluable patients who had early-stage CRC were recently published and were very favorable. These patients each received one dose of ipilimumab and two doses of nivolumab prior to surgery. There was a 100% pathological response rate in the dMMR group: 12 patients had pathological complete responses, and 19 patients had major pathological responses (defined as ≤10% residual viable tumor). In the pMMR group, only 27% of patients had major pathological responses, and none of these were pathological complete responses [91]. These findings indicate that nivolumab and ipilimumab may have a role for a select group of early-stage colon cancer patients with dMMR/MSI-H, but larger studies are still needed to confirm this. Currently, ATOMIC and COMMIT trials are evaluating the benefit of addition of anti PD-L1 antibody (atezolizumab) to chemotherapy FOLFOX in patients with dMMR/MSI-H in adjuvant stage III and first-line stage IV colon cancer, respectively [92, 93].
Human Epidermal Growth Factor Receptor 2 (HER-2)
HER2, or ERBB2, is an extracellular tyrosine kinase that when overexpressed leads to activation of PI3K, AKT, and MAPK pathways [94]. The HER2 gene amplification and/or overexpression is observed in 2–6% of CRC patients, most commonly in left-sided and KRAS WT mCRC. Identification of HER2 can be done using either IHC, fluorescence in situ hybridization (FISH), or NGS [94, 95]. Various studies have investigated the role of anti-HER2 therapy in HER2-amplified mCRC, as these tumors are thought to be resistant to anti-EGFR therapies [96, 97]. Anti-HER2 agents are standard of care in breast and gastric cancer and include monoclonal antibodies trastuzumab and pertuzumab, the tyrosine kinase inhibitors lapatinib and tucatinib, and the antibody-drug conjugate fam-trastuzumab deruxtecan-nxki (T-DXd) [98].
The phase II HERACLES A trial evaluated the combination of trastuzumab and lapatinib in 27 patients with KRAS WT and HER2-amplified mCRC tumors refractory to standard of care therapy [99]. The authors reported an ORR of 30% (95% CI 14–50) and median OS of 46 weeks (95% CI 33–68). No drug-related serious adverse events were reported, and this served as proof of concept for HER2 targeting in CRC [99]. Another single-arm phase II study, MyPathway, evaluated the combination of trastuzumab and pertuzumab in 37 heavily pretreated patients with HER2-amplified mCRC with an ORR of 32% (95% CI 20–45) and mPFS and mOS of 2.9 months and 11.3 months, respectively [100]. These findings were replicated recently in the TAPUR study with a reported ORR of 14% (95% CI 4–33), mPFS of 17.2 weeks (95% CI 11.1–27.4), and 1 year OS rate of 58% (95% CI 37–75) [101]. A fourth phase II study, Destiny-CRC01, evaluated the use of T-DXd in HER2-expressing, RAS/BRAF WT, mCRC patients who had previously received two or more prior regimens [102]. Investigators split patients into three cohorts based on their level of HER2 expression and reported that in the highest HER2 expression cohort (IHC3+ or IHC2+/ISH+), ORR was 45.3%, mPFS was 6.9 months, and mOS was not reached. Cohort B (HER2 IHC 2+) and Cohort C (HER IHC 1+) did not have any confirmed response [102]. Importantly, T-DXd induced responses even in patients pretreated with anti-HER2 therapies.
Interpretation of these data to determine proper sequencing of anti-HER2 therapy in mCRC is challenging given the single-arm nature of these trials and the small numbers of patients evaluated. Additionally, it is important to consider the toxicity profile of these regimens, in particular the concern for interstitial lung disease with T-DXd which, although rare, can be fatal. All three of these regimens are options for previously treated mCRC patients with HER2 amplifications and may be reasonable for untreated patients who are not appropriate for intensive therapy. Importantly, when used for HER2-amplified mCRC, these anti-HER2 regimens are only indicated for RAS and BRAF WT disease, as patients with mutations in RAF/BRAF were excluded from these investigations. The ongoing SWOG 1613 study is comparing the efficacy of this combination versus cetuximab and irinotecan in HER2-amplified mCRC in 2nd or 3rd line setting [103]. Furthermore the MOUNTAINEER trial included mCRC patients with RAS WT, HER2 amplification by NGS, FISH, or IHC who were naïve to anti HER2 therapy [104]. Patients received the oral HER2 tyrosine kinase inhibitor tucatinib combined with trastuzumab. Preliminary results in 26 patients demonstrated an ORR of 52% and mPFS of 8.1 months and mOS of 18.7 months. Currently the trial is expanded to include additional patients randomized to two cohorts: one with trastuzumab and tucatinib and one with tucatinib only as part of a registration strategy for the combination [104].
Neurotrophic Receptor Tyrosine Kinase (NRTK)
The proteins TRKA (encoded by NTRK1), TRKB (encoded by NTRK2), and TRKC (encoded by NTRK3) comprise the family of NRTK and are vital in neural deployment [105, 106]. NTRKs consist of an extracellular ligand-binding domain, a transmembrane region, and an intracellular kinase domain, which together allow for downstream signal activation using the Ras–Raf–MAPK, PI3K–Akt–mTOR, and PLCc–PKC pathways. In 0.2–1% of solid tumors, fusion occurs due to rearrangement of NTRK genes where TRK fusion proteins initiate cell transformation, growth, and proliferation [105, 107, 108]. NTRK fusion can be detected via IHC, FISH, RT-PCR, and both RNA-based (preferred) and DNA-based NGS [109]. Their most common presentation is in females with RAS/BRAF WT disease that is primarily right-sided, and 50–70% of NTRK fusions are associated with the dMMR/MSI high phenotype [105, 110].
Given the rarity of these mutations, development of TRK inhibitors has been guided by basket studies enrolling patients with relapsed NTRK fusion-positive cancers across multiple tumor types. In a combination of three phase I–II trials of 55 patients with TRK fusion-positive cancers, the TRK inhibitor larotrectinib was shown to be associated with a high response rate (ORR 75%, 95% CI 61–85) with 55% of patients progression free at 1 year [111, 112]. Follow-up analysis of the 14 patients with TRK fusion gastro-intestinal cancer demonstrated a median PFS of 5.3 months (95% CI 2.2–9.0) and median OS of 33.4 months (95% CI 2.8–36.5) [113]. Based on these data, the FDA recently granted larotrectinib approval for use on TRK fusion cancers after reviewing the LOXO-TRK-14001, NAVIGATE, and SCOUT single-arm trials [114]. In a similar pooled analysis of phase I–II studies of patients with TRK fusion cancers, a second TRK inhibitor entrectinib demonstrated a high response rates (ORR 57%, 95% CI 43–71) and a median duration of response of 10 months. The trial only included 4 subjects with mCRC and of those only 1 (25%) had a treatment response [115]. Similarly, the FDA approved entrectinib for the treatment of adult and pediatric patients with NTRK fusion based on three single-arm trials (ALKA, STARTRK-1 (NCT02097810), and STARTRK-2) [116]. Together these data suggest that for the rare patient with relapse mCRC and NTRK fusion, either entrectinib or larotrectinib are potential treatment options
Tumor Mutational Burden (TMB)
Tumor mutational burden, which is defined by the number of somatic mutations per DNA megabase, has been shown to be one of the predictive markers for ICIs [117]. Currently, TMB calculations are obtained from targeted cancer gene panels from tissue biopsies or blood. The retrospective analysis of the phase III SWOG 80405 study showed that patients with a TMB greater than 8 derived greater benefit from chemotherapy [118]. The seminal paper of ICI therapy in MSI high cancers showed that higher TMB was correlated with longer PFS. A retrospective study of MSI high mCRC treated with ICI also confirmed that TMB is an important biomarker even in the MSI subset of patients. The cut-point TMB between 37 and 42 by Foundation Medicine (FMI) was correlated with higher response rate and longer PFS than lower TMB group [118].
In addition to the MSI high population, additional patients (3%) with high TMB were found with MSS mCRC who derived benefit from ICI therapies in case reports [119, 120]. The mutations in POLE and POLD1 lead to an ultra-mutated phenotype, and these patients were found to have durable response from ICI therapies [121, 122]. Recently the phase 2 KEYNOTE-158 trial showed that patients with a TMB of more than 10 by FMI have an objective response rate of 30% with pembrolizumab in multiple tumor types. However, it is important to note that there were no patients with mCRC in this analysis. This led to FDA’s tumor agnostic approval of ICI in the TMB >10 setting [123]. However, a recent correspondence showed that among 137 patients with mCRC who were treated with ICIs, the difference in OS between those with high TMB vs low TMB disappeared after stratification by MMR deficiency or pathogenic mutations in POLE and POLD1 [124]. A retrospective analysis of CCTG CO.26 trial of durvalumab and tremelimumab versus best supportive care in mCRC showed that high plasma TMB (>28) is correlated with improved outcomes in the ICI treated group, but the same is not true for tissue TMB [125]. Currently, the benefit of ICI on TMB is complex and may depend on histology. Further studies are required to establish a definitive benefit in CRC with high TMB without MMR deficiency or POLE or POLD mutation.
Conclusion
The integration of molecular profiling and biomarker testing has revolutionized treatment strategies for mCRC. Importantly, retrospective studies comparing outcomes of patients with different molecular tumor types have been critical to our understanding of tumor biology and resistance mechanisms, thereby informing current pharmacologic treatment algorithms for mCRC. Currently, there are a multitude of ongoing clinical trials seeking for further define subgroups of patients which benefit from different classes of agents, as well as optimizing sequence of therapy. Some of the notable trials that are anticipated to impact practice are listed in Table 2. Moving forward, future research is needed to further advance molecular and biomarker classification of mCRC, such as parallel biomarker testing of ctDNA, combination of targeted therapies in multiple pathways as well as appropriate sequencing of chemotherapy, immunotherapy, and targeted therapies, and these may enhance the use of these precision medicine techniques to optimize patient outcomes.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7–33. https://doi.org/10.3322/caac.21654.
Sepulveda AR, Hamilton SR, Allegra CJ, Grody W, Cushman-Vokoun AM, Funkhouser WK, et al. Molecular biomarkers for the evaluation of colorectal cancer: guideline from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and American Society of Clinical Oncology. J Mol Diagnostics. 2017;19:187–225. This is a Consortium guideline for biomarker testing in Colorectal Cancer which was developed by the American Society of Clinical Pathology (ASCP), College of American Pathologists (CAP), Association for Molecular Pathology (AMP), and American Society for Clinical Oncology (ASCO).
Wells A. EGF receptor. Int J Biochem Cell Biol. 1999;31:637–43. https://doi.org/10.1016/S1357-2725(99)00015-1.
Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:15–31.
Yarden Y. The EGFR family and its ligands in human cancer: signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37:3–8. https://doi.org/10.1016/s0959-8049(01)00230-1.
Molina JR, Adjei AA. The Ras/Raf/MAPK Pathway. J Thorac Oncol. 2006;1:7–9. https://doi.org/10.1016/s1556-0864(15)31506-9.
El Zouhairi M, Charabaty A, Pishvaian MJ. Molecularly targeted therapy for metastatic colon cancer: proven treatments and promising new agents. Gastrointest Cancer Res. 2011;4:15–21.
Krasinskas AM. EGFR signaling in colorectal carcinoma. Pathol Res Int. 2011;2011:1–6. https://doi.org/10.4061/2011/932932.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62. https://doi.org/10.1016/S1470-2045(10)70130-3.
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras Mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. https://doi.org/10.1056/nejmoa0804385.
Cercek A, Braghiroli MI, Chou JF, Hechtman JF, Kemeny N, Saltz L, et al. Clinical features and outcomes of patients with colorectal cancers harboring NRAS mutations. Clin Cancer Res. 2017;23:4753–60. https://doi.org/10.1158/1078-0432.CCR-17-0400.
Del Vecchio F, Mastroiaco V, Di Marco A, Compagnoni C, Capece D, Zazzeroni F, et al. Next-generation sequencing: recent applications to the analysis of colorectal cancer. J Transl Med. 2017;15:246. https://doi.org/10.1186/s12967-017-1353-y.
Semrad TJ, Kim EJ. Molecular testing to optimize therapeutic decision making in advanced colorectal cancer. J Gastrointest Oncol. 2016;7:S11–20.
Monzon FA, Ogino S, Hammond MEH, Halling KC, Bloom KJ, Nikiforova MN. The role of KRAS mutation testing in the management of patients with metastatic colorectal cancer. Arch Pathol Lab Med. 2009;133:1600–6.
García-Foncillas J, Sunakawa Y, Aderka D, Wainberg Z, Ronga P, Witzler P, et al. Distinguishing features of cetuximab and panitumumab in colorectal cancer and other solid tumors. Front Oncol. 2019;9:849.
Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au H-J, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8. https://doi.org/10.1056/NEJMoa071834.
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45. https://doi.org/10.1056/nejmoa033025.
Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy- refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–64. https://doi.org/10.1200/JCO.2006.08.1620.
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34. https://doi.org/10.1200/JCO.2007.14.7116. This was the first study to report the relationship between KRAS mutations and anti-EGFR therapy.
André T, Blons H, Mabro M, Chibaudel B, Bachet JB, Tournigand C, et al. Panitumumab combined with irinotecan for patients with KRAS wild-type metastatic colorectal cancer refractory to standard chemotherapy: a GERCOR efficacy, tolerance, and translational molecular study. Ann Oncol. 2013;24:412–9. https://doi.org/10.1093/annonc/mds465.
Sobrero AF, Maurel J, Fehrenbacher L, Scheithauer W, Abubakr YA, Lutz MP, et al. EPIC: Phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:2311–9. https://doi.org/10.1200/JCO.2007.13.1193.
Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28:4706–13. https://doi.org/10.1200/JCO.2009.27.6055.
Seymour MT, Brown SR, Middleton G, Maughan T, Richman S, Gwyther S, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013;14:749–59. https://doi.org/10.1016/S1470-2045(13)70163-3.
Cartwright T, Kuefler P, Cohn A, Hyman W, Berger M, Richards D, et al. Results of a phase II trial of cetuximab plus capecitabine/irinotecan as first-line therapy for patients with advanced and/or metastatic colorectal cancer. Clin Colorectal Cancer. 2008;7:390–7. https://doi.org/10.3816/CCC.2008.n.052.
Berlin J, Posey J, Tchekmedyian S, Hu E, Chan D, Malik I, et al. Panitumumab with irinotecan/leucovorin/5-fluorouracil for first-line treatment of metastatic colorectal cancer. Clin Colorectal Cancer. 2007;6:427–32. https://doi.org/10.3816/CCC.2007.n.011.
Köhne CH, Hofheinz R, Mineur L, Letocha H, Greil R, Thaler J, et al. First-line panitumumab plus irinotecan/5-Xuorouracil/leucovorin treatment in patients with metastatic colorectal cancer. J Cancer Res Clin Oncol. 2012;138:65–72. https://doi.org/10.1007/s00432-011-1061-6.
Bokemeyer C, Bondarenko I, Makhson A, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–671. https://doi.org/10.1200/JCO.2008.20.8397.
Van Cutsem E, Köhne C-H, Hitre E, Zaluski J, Chang Chien C-R, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–17. https://doi.org/10.1056/nejmoa0805019.
Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Randomized, Phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28:4697–705. https://doi.org/10.1200/JCO.2009.27.4860. This was the first study to report establishing role for extended RAS mutations.
Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377:2103–14. https://doi.org/10.1016/S0140-6736(11)60613-2.
Tveit KM, Guren T, Glimelius B, Pfeiffer P, Sorbye H, Pyrhonen S, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol. 2012;30:1755–62. https://doi.org/10.1200/JCO.2011.38.0915.
Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran S-E, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(10):1065–1075. https://doi.org/10.1016/S1470-2045(14)70330-4.
Venook AP, Niedzwiecki D, Lenz HJ, Innocenti F, Fruth B, Meyerhardt JA, et al. Effect of first-line chemotherapy combined with cetuximab or bevacizumab on overall survival in patients with KRAS wild-type advanced or metastatic colorectal cancer a randomized clinical trial. JAMA - J. Am Med Assoc. 2017;317:2392–401. https://doi.org/10.1001/jama.2017.7105.
Qin S, Li J, Wang L, Xu J, Cheng Y, Bai Y, et al. Efficacy and tolerability of first-line cetuximab plus leucovorin, fluorouracil, and oxaliplatin (FOLFOX-4) versus FOLFOX-4 in patientswith RASwild-typemetastatic colorectal cancer: The open-label, randomized, phase III TAILOR trial. J Clin Oncol. 2018;36:3031–9. https://doi.org/10.1200/JCO.2018.78.3183.
Al-Shamsi HO, Alhazzani W, Wolff RA. Extended RAS testing in metastatic colorectal cancer-refining the predictive molecular biomarkers. J Gastrointest Oncol. 2015;6:314–21.
Alberts SR, Sargent DJ, Nair S, Mahoney MR, Mooney M, Thibodeau SN, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA - J Am Med Assoc. 2012;307:1383–93. https://doi.org/10.1001/jama.2012.385.
Taieb J, Balogoun R, Le Malicot K, Tabernero J, Mini E, Folprecht G, et al. Adjuvant FOLFOX +/- cetuximab in full RAS and BRAF wildtype stage III colon cancer patients. Ann Oncol. 2017;28:824–30. https://doi.org/10.1093/annonc/mdw687.
Folprecht G, Gruenberger T, Bechstein W, Raab HR, Weitz J, Lordick F, et al. Survival of patients with initially unresectable colorectal liver metastases treated with FOLFOX/cetuximab or FOLFIRI/cetuximab in a multidisciplinary concept (CELIM study). Ann Oncol. 2014;25:1018–25. https://doi.org/10.1093/annonc/mdu088.
Modest DP, Martens UM, Riera-Knorrenschild J, Greeve J, Florschütz A, Wessendorf S, et al. FOLFOXIRI plus panitumumab as first-line treatment of RAS wild-type metastatic colorectal cancer: the randomized, open-label, phase II Volfi study (AIO KRK0109). J Clin Oncol. 2019;37:3401–11. https://doi.org/10.1200/JCO.19.01340.
Petrelli F, Barni S. Resectability and outcome with anti-EGFR agents in patients with KRAS wild-type colorectal liver-limited metastases: a meta-analysis. Int J Color Dis. 2012;27:997–1004.
Bridgewater JA, Pugh SA, Maishman T, Eminton Z, Mellor J, Whitehead A, et al. Systemic chemotherapy with or without cetuximab in patients with resectable colorectal liver metastasis (New EPOC): long-term results of a multicentre, randomised, controlled, phase 3 trial. Lancet Oncol. 2020;21:398–411. https://doi.org/10.1016/S1470-2045(19)30798-3.
Gholami S, Grothey A. EGFR antibodies in resectable metastatic colorectal liver metastasis: more harm than benefit? Lancet Oncol. 2020;21:324–6.
Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48:1466–75. https://doi.org/10.1016/j.ejca.2012.02.057.
Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–12. https://doi.org/10.1200/JCO.2008.18.0786.
Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–90.
Chen W, Frankel WL. A practical guide to biomarkers for the evaluation of colorectal cancer. Mod Pathol. 2019;32:1–15.
Yaeger R, Chatila WK, Lipsyc MD, Hechtman JF, Cercek A, Sanchez-Vega F, et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell. 2018;33:125–136.e3. https://doi.org/10.1016/j.ccell.2017.12.004.
Schirripa M, Biason P, Lonardi S, Pella N, Simona Pino M, Urbano F, et al. Class 1, 2, and 3 BRAF-mutated metastatic colorectal cancer: a detailed clinical, pathologic, and molecular characterization. Clin Cancer Res. 2019;25:3954–61. https://doi.org/10.1158/1078-0432.CCR-19-0311.
Jones JC, Renfro LA, Al-Shamsi HO, Schrock AB, Rankin A, Zhang BY, et al. Non-V600BRAF mutations define a clinically distinct molecular subtype of metastatic colorectal cancer. In Proceedings of the Journal of Clinical Oncology. Proc Am Soc Clin Oncol. 2017;35:2624–30.
Sanchez JN, Wang T, Cohen MS. BRAF and MEK inhibitors: use and resistance in BRAF-mutated cancers. Drugs. 2018;78:549–66.
Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379:1893–901. https://doi.org/10.1016/S0140-6736(12)60398-5.
Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33:4032–8. https://doi.org/10.1200/JCO.2015.63.2497.
Yaeger R, Cercek A, O’Reilly EM, Reidy DL, Kemeny N, Wolinsky T, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21:1313–20. https://doi.org/10.1158/1078-0432.CCR-14-2779.
Hong DS, Morris VK, El Osta B, Sorokin AV, Janku F, Fu S, et al. Phase IB study of vemurafenib in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with BRAFV600E mutation. Cancer Discov. 2016;6:1352–65. https://doi.org/10.1158/2159-8290.CD-16-0050.
Kopetz S, McDonough SL, Morris VK, Lenz H-J, Magliocco AM, Atreya CE, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF -mutant metastatic colorectal cancer (SWOG 1406). J Clin Oncol. 2017;35:520–0. https://doi.org/10.1200/jco.2017.35.4_suppl.520.
Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E–mutated colorectal cancer. N Engl J Med. 2019;381:1632–43. https://doi.org/10.1056/NEJMoa1908075.
Kopetz S, Grothey A, Van Cutsem E, Yaeger R, Wasan HS, Yoshino T, et al. Encorafenib plus cetuximab with or without binimetinib for BRAF V600E metastatic colorectal cancer: updated survival results from a randomized, three-arm, phase III study versus choice of either irinotecan or FOLFIRI plus cetuximab (BEACON CRC). J Clin Oncol. 2020;38:4001–1. https://doi.org/10.1200/jco.2020.38.15_suppl.4001.
FDA approves encorafenib in combination with cetuximab for metastatic colorectal cancer with a BRAF V600E mutation | FDA Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-encorafenib-combination-cetuximab-metastatic-colorectal-cancer-braf-v600e-mutation (accessed on Mar 11, 2021).
Grothey, A.; Fakih, M.; Tabernero, J. Management of BRAF-mutant metastatic colorectal cancer: a review of treatment options and evidence-based guidelines. Ann Oncol 2021, 0, https://doi.org/10.1016/j.annonc.2021.03.206.
Grothey A, Tabernero J, Taieb J, Yaeger R, Yoshino T, Maiello E, et al. LBA-5 ANCHOR CRC: a single-arm, phase 2 study of encorafenib, binimetinib plus cetuximab in previously untreated BRAF V600E-mutant metastatic colorectal cancer. Ann Oncol. 2020;31:S242–3. https://doi.org/10.1016/j.annonc.2020.04.080.
BRAF V600E-mutant Colorectal Cancer Study of Encorafenib Taken With Cetuximab Plus or Minus Chemotherapy (BREAKWATER) - Full Text View - ClinicalTrials.gov Available online: https://clinicaltrials.gov/ct2/show/NCT04607421 (accessed on Apr 11, 2021).
Moretto R, Cremolini C, Rossini D, Pietrantonio F, Battaglin F, Mennitto A, et al. Location of primary tumor and benefit from anti-epidermal growth factor receptor monoclonal antibodies in patients with RAS and BRAF wild-type metastatic colorectal cancer. Oncologist. 2016;21:988–94. https://doi.org/10.1634/theoncologist.2016-0084.
Arnold D, Lueza B, Douillard JY, Peeters M, Lenz HJ, Venook A, et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol. 2017;28:1713–29. https://doi.org/10.1093/annonc/mdx175.
Holch JW, Ricard I, Stintzing S, Modest DP, Heinemann V. The relevance of primary tumour location in patients with metastatic colorectal cancer: a meta-analysis of first-line clinical trials. Eur J Cancer. 2017;70:87–98. https://doi.org/10.1016/j.ejca.2016.10.007.
Venook AP, Niedzwiecki D, Innocenti F, Fruth B, Greene C, O’Neil BH, et al. Impact of primary (1o) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol. 2016;34:3504–4. https://doi.org/10.1200/jco.2016.34.15_suppl.3504.
Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. https://doi.org/10.1126/science.8484121.
Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–61. https://doi.org/10.1038/363558a0.
Koopman M, Kortman GAM, Mekenkamp L, Ligtenberg MJL, Hoogerbrugge N, Antonini NF, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer. 2009;100:266–73. https://doi.org/10.1038/sj.bjc.6604867.
Shukla N, Abrha A, Longacre TA, Koff R, Ford JM, Fisher GA, et al. Prevalence and molecular etiology of mismatch repair deficiency among gastrointestinal cancers. J Clin Oncol. 2019;37:215–5. https://doi.org/10.1200/jco.2019.37.4_suppl.215.
Oliveira AF, Bretes L, Furtado I. Review of PD-1/PD-L1 inhibitors in metastatic dMMR/MSI-H colorectal cancer. Front Oncol. 2019;9. https://doi.org/10.3389/fonc.2019.00396.
Venderbosch S, Nagtegaal ID, Maughan TS, Smith CG, Cheadle JP, Fisher D, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res. 2014;20:5322–30. https://doi.org/10.1158/1078-0432.CCR-14-0332.
Rubenstein JH, Enns R, Heidelbaugh J, Barkun A, Adams MA, Dorn SD, et al. American Gastroenterological Association Institute guideline on the diagnosis and management of Lynch syndrome. Gastroenterology. 2015;149:777–82. https://doi.org/10.1053/j.gastro.2015.07.036.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20. https://doi.org/10.1056/NEJMoa1500596.
Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51. https://doi.org/10.1158/2159-8290.CD-14-0863.
Shulman K, Barnett-Griness O, Friedman V, Greenson JK, Gruber SB, Lejbkowicz F, et al. Outcomes of chemotherapy for microsatellite instable–high metastatic colorectal cancers. JCO Precis Oncol. 2018:1–10. https://doi.org/10.1200/PO.17.00253.
Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28:466–74. https://doi.org/10.1200/JCO.2009.23.3452.
Bertagnolli MM, Redston M, Compton CC, Niedzwiecki D, Mayer RJ, Goldberg RM, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: pospective evaluation of biomarkers for stages II and III colon cancer - a study of CALGB 9581 and 89803. J Clin Oncol. 2011;29:3153–62. https://doi.org/10.1200/JCO.2010.33.0092.
Hutchins G, Southward K, Handley K, Magill L, Beaumont C, Stahlschmidt J, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol. 2011;29:1261–70. https://doi.org/10.1200/JCO.2010.30.1366.
Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219–26. https://doi.org/10.1200/JCO.2009.27.1825.
Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–57. https://doi.org/10.1056/nejmoa022289.
Klingbiel D, Saridaki Z, Roth AD, Bosman FT, Delorenzi M, Tejpar S. Prognosis of stage II and III colon cancer treated with adjuvant 5-fluorouracil or FOLFIRI in relation to microsatellite status: results of the PETACC-3 trial. Ann Oncol. 2015;26:126–32. https://doi.org/10.1093/annonc/mdu499.
FDA approves first cancer treatment for any solid tumor with a specific genetic feature | FDA Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-cancer-treatment-any-solid-tumor-specific-genetic-feature (accessed on Apr 11, 2021).
Le DT, Kim TW, van Cutsem E, Geva R, Jäger D, Hara H, et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability–high/mismatch repair–deficient metastatic colorectal cancer: KEYNOTE-164. In Proceedings of the Journal of Clinical Oncology; American Society of Clinical Oncology. 2020;38:11–9. This was the first study to report of dMMR predictive marker of response to immune checkpoint inhibition.
Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–9. https://doi.org/10.1200/JCO.2017.76.9901.
Lenz H-J, Lonardi S, Zagonel V, Van Cutsem E, Limon ML, Wong M, et al. Nivolumab (NIVO) + low-dose ipilimumab (IPI) as first-line (1L) therapy in microsatellite instability-high/mismatch repair-deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC): two-year clinical update. J Clin Oncol. 2020;38:4040–0. https://doi.org/10.1200/jco.2020.38.15_suppl.4040.
A Study of Nivolumab, Nivolumab Plus Ipilimumab, or Investigator’s Choice Chemotherapy for the Treatment of Participants With Deficient Mismatch Repair (dMMR)/Microsatellite Instability High (MSI-H) Metastatic Colorectal Cancer (mCRC) - Full Text View - ClinicalTrials.gov Available online: https://www.clinicaltrials.gov/ct2/show/NCT04008030 (accessed on Apr 11, 2021).
FDA grants accelerated approval to ipilimumab for MSI-H or dMMR metastatic colorectal cancer | FDA Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-ipilimumab-msi-h-or-dmmr-metastatic-colorectal-cancer (accessed on Apr 11, 2021).
• André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-instability–high advanced colorectal cancer. N Engl J Med. 2020;383:2207–18. https://doi.org/10.1056/NEJMoa2017699. This study established the standard of care for dMMR/MSI-H mCRC with pembrolizumab ove chemotherapy.
Shiu K-K, Andre T, Kim TW, Jensen BV, Jensen LH, Punt CJA, et al. KEYNOTE-177: Phase III randomized study of pembrolizumab versus chemotherapy for microsatellite instability-high advanced colorectal cancer. J Clin Oncol. 2021;39:6–6. https://doi.org/10.1200/jco.2021.39.3_suppl.6.
FDA approves pembrolizumab for first-line treatment of MSI-H/dMMR colorectal cancer | FDA Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-first-line-treatment-msi-hdmmr-colorectal-cancer (accessed on Apr 20, 2021).
Chalabi M, Fanchi LF, Dijkstra KK, Van den Berg JG, Aalbers AG, Sikorska K, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med. 2020;26:566–76. https://doi.org/10.1038/s41591-020-0805-8.
Combination Chemotherapy With or Without Atezolizumab in Treating Patients With Stage III Colon Cancer and Deficient DNA Mismatch Repair - Full Text View - ClinicalTrials.gov Available online: https://www.clinicaltrials.gov/ct2/show/NCT02912559 (accessed on Apr 11, 2021).
Combination Chemotherapy, Bevacizumab, and/or Atezolizumab in Treating Patients With Deficient DNA Mismatch Repair Metastatic Colorectal Cancer, the COMMIT Study - Full Text View - ClinicalTrials.gov Available online: https://clinicaltrials.gov/ct2/show/NCT02997228 (accessed on Apr 21, 2021).
Cenaj O, Ligon AH, Hornick JL, Sholl LM. Detection of ERBB2 amplification by next-generation sequencing predicts HER2 expression in colorectal carcinoma. Am J Clin Pathol. 2019;152:97–108. https://doi.org/10.1093/ajcp/aqz031.
Valtorta E, Martino C, Sartore-Bianchi A, Penaullt-Llorca F, Viale G, Risio M, et al. Assessment of a HER2 scoring system for colorectal cancer: results from a validation study. Mod Pathol. 2015;28:1481–91. https://doi.org/10.1038/modpathol.2015.98.
Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra86. https://doi.org/10.1126/scitranslmed.3002442.
Raghav KPS, Overman MJ, Yu R, Meric-Bernstam F, Menter D, Kee BK, et al. HER2 amplification as a negative predictive biomarker for anti-epidermal growth factor receptor antibody therapy in metastatic colorectal cancer. J Clin Oncol. 2016;34:3517–7. https://doi.org/10.1200/jco.2016.34.15_suppl.3517.
La Salvia A, Lopez-Gomez V, Garcia-Carbonero R. HER2-targeted therapy: an emerging strategy in advanced colorectal cancer. Expert Opin Investig Drugs. 2019;28:29–38.
Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:738–46. https://doi.org/10.1016/S1470-2045(16)00150-9.
Meric-Bernstam F, Hurwitz H, Raghav KPS, McWilliams RR, Fakih M, VanderWalde A, et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019;20:518–30. https://doi.org/10.1016/S1470-2045(18)30904-5.
Gupta R, Garrett-Mayer E, Halabi S, Mangat PK, D’Andre SD, Meiri E, et al. Pertuzumab plus trastuzumab (P+T) in patients (Pts) with colorectal cancer (CRC) with ERBB2 amplification or overexpression: results from the TAPUR Study. J Clin Oncol. 2020;38:132–2. https://doi.org/10.1200/jco.2020.38.4_suppl.132.
Siena S, Di Bartolomeo M, Raghav KPS, Masuishi T, Loupakis F, Kawakami H, et al. A phase II, multicenter, open-label study of trastuzumab deruxtecan (T-DXd; DS-8201) in patients (pts) with HER2-expressing metastatic colorectal cancer (mCRC): DESTINY-CRC01. J Clin Oncol. 2020;38:4000–0. https://doi.org/10.1200/jco.2020.38.15_suppl.4000.
S1613, Trastuzumab and Pertuzumab or Cetuximab and Irinotecan Hydrochloride in Treating Patients With Locally Advanced or Metastatic HER2/Neu Amplified Colorectal Cancer That Cannot Be Removed by Surgery - Full Text View - ClinicalTrials.gov Available online: https://clinicaltrials.gov/ct2/show/NCT03365882 (accessed on Apr 12, 2021).
Strickler JH, Ng K, Cercek A, Fountzilas C, Sanchez FA, Hubbard JM, et al. MOUNTAINEER:open-label, phase II study of tucatinib combined with trastuzumab for HER2-positive metastatic colorectal cancer (SGNTUC-017, trial in progress). J Clin Oncol. 2021;39:TPS153–3. https://doi.org/10.1200/jco.2021.39.3_suppl.tps153.
Pietrantonio F, Di Nicolantonio F, Schrock AB, Lee J, Tejpar S, Sartore-Bianchi A, et al. ALK, ROS1, and NTRK rearrangements in metastatic colorectal cancer. JNCI J Natl Cancer Inst. 2017:109. https://doi.org/10.1093/jnci/djx089.
Pagani F, Randon G, Guarini V, Raimondi A, Prisciandaro M, Lobefaro R, et al. The landscape of actionable gene fusions in colorectal cancer. Int J Mol Sci. 2019:20.
Okamura R, Boichard A, Kato S, Sicklick JK, Bazhenova L, Kurzrock R. Analysis of NTRK alterations in pan-cancer adult and pediatric malignancies: implications for NTRK-targeted therapeutics. JCO Precis Oncol. 2018;2018:1–20. https://doi.org/10.1200/po.18.00183.
Gatalica Z, Xiu J, Swensen J, Vranic S. Molecular characterization of cancers with NTRK gene fusions. Mod Pathol. 2019;32:147–53. https://doi.org/10.1038/s41379-018-0118-3.
Solomon JP, Benayed R, Hechtman JF, Ladanyi M. Identifying patients with NTRK fusion cancer. Ann Oncol Off J Eur Soc Med Oncol. 2019;30(Suppl 8):viii16–22. https://doi.org/10.1093/annonc/mdz384.
Cocco E, Benhamida J, Middha S, Zehir A, Mullaney K, Shia J, et al. Colorectal carcinomas containing hypermethylated MLH1 promoter and wild-type BRAF/KRAS are enriched for targetable kinase fusions. Cancer Res. 2019;79:1047–53. https://doi.org/10.1158/0008-5472.CAN-18-3126.
Hong DS, DuBois SG, Kummar S, Farago AF, Albert CM, Rohrberg KS, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020;21:531–40. https://doi.org/10.1016/S1470-2045(19)30856-3.
Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N Engl J Med. 2018;378:731–9. https://doi.org/10.1056/nejmoa1714448.
Berlin J, Hong DS, Deeken JF, Boni V, Oh D-Y, Patel JD, et al. Efficacy and safety of larotrectinib in patients with TRK fusion gastrointestinal cancer. J Clin Oncol. 2020;38:824–4. https://doi.org/10.1200/jco.2020.38.4_suppl.824.
FDA approves larotrectinib for solid tumors with NTRK gene fusions | FDA Available online: https://www.fda.gov/drugs/fda-approves-larotrectinib-solid-tumors-ntrk-gene-fusions (accessed on Apr 12, 2021).
Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21:271–82. https://doi.org/10.1016/S1470-2045(19)30691-6.
FDA approves entrectinib for NTRK solid tumors and ROS-1 NSCLC | FDA Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-entrectinib-ntrk-solid-tumors-and-ros-1-nsclc (accessed on Apr 12, 2021).
Cohen R, Colle R, Pudlarz T, Heran M, Duval A, Svrcek M, et al. Immune checkpoint inhibition in metastatic colorectal cancer harboring microsatellite instability or mismatch repair deficiency. Cancers (Basel). 2021;13:1–10.
Schrock AB, Ouyang C, Sandhu J, Sokol E, Jin D, Ross JS, et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann Oncol. 2019;30:1096–103. https://doi.org/10.1093/annonc/mdz134.
Gong J, Robertson MD, Kim E, Fakih M, Schrock AB, Tam KW, et al. Efficacy of PD-1 blockade in refractory microsatellite-stable colorectal cancer with high tumor mutation burden. Clin Colorectal Cancer. 2019;18:307–9. https://doi.org/10.1016/j.clcc.2019.08.001.
Fabrizio DA, George TJ, Dunne RF, Frampton G, Sun J, Gowen K, et al. Beyond microsatellite testing: assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J Gastrointest Oncol. 2018;9:610–7. https://doi.org/10.21037/jgo.2018.05.06.
Bellido F, Pineda M, Aiza G, Valdés-Mas R, Navarro M, Puente DA, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med. 2016;18:325–32. https://doi.org/10.1038/gim.2015.75.
Wang F, Zhao Q, Wang YN, Jin Y, He MM, Liu ZX, et al. Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol. 2019;5:1504–6.
Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353–65. https://doi.org/10.1016/S1470-2045(20)30445-9.
Marabelle A, Jin F, Norwood K, Aurora-Garg D. Tumour mutational burden in treatment-resistant tumours – Authors’ reply. Lancet Oncol. 2020;21:e552.
Loree JM, Topham JT, Kennecke HF, Feilotter H, Keshavarz-Rahaghi F, Lee YS, et al. Tissue and plasma tumor mutation burden (TMB) as predictive biomarkers in the CO.26 trial of durvalumab + tremelimumab (D+T) versus best supportive care (BSC) in metastatic colorectal cancer (mCRC). J Clin Oncol. 2021;39:61–1. https://doi.org/10.1200/jco.2021.39.3_suppl.61.
Chibaudel B, Bonnetain F, Tournigand C, de Larauze MH, de Gramont A, Laurent-Puig P, et al. STRATEGIC-1: a multiple-lines, randomized, open-label GERCOR phase III study in patients with unresectable wild-type RAS metastatic colorectal cancer. BMC Cancer. 2015;15. https://doi.org/10.1186/s12885-015-1503-7.
Borelli B, Moretto R, Lonardi S, Bonetti A, Antoniotti C, Pietrantonio F, et al. TRIPLETE: A randomised phase III study of modified FOLFOXIRI plus panitumumab versus mFOLFOX6 plus panitumumab as initial therapy for patients with unresectable RAS and BRAF wild-type metastatic colorectal cancer. ESMO Open. 2018;3. https://doi.org/10.1136/esmoopen-2018-000403.
Rechallenge With Panitumumab Driven by RAS Dynamic of Resistance - Full Text View - ClinicalTrials.gov Available online: https://clinicaltrials.gov/ct2/show/NCT03227926 (accessed on Apr 21, 2021).
Author information
Authors and Affiliations
Contributions
Conceptualization, MC and AG; original draft preparation, MC and RJB; review and editing, MC, RJB, SG, and AG; and supervision, MC and AG. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Ryan Beechinor and Sepideh Gholami each declare no potential conflicts of interest. May Cho has received personal fees from Amgen, Pfizer, AstraZeneca, BMS, and Seagen. Axel Grothey has received honoraria payments and grants from Bayer, Genentech, Natera, CARIS, Daiichi, Pfizer, and Merck.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Surgery and Surgical Innovations in Colorectal Cancer
Rights and permissions
About this article

Cite this article
Cho, M., Beechinor, R., Gholami, S. et al. Precision Medicine for the Treatment of Colorectal Cancer: the Evolution and Status of Molecular Profiling and Biomarkers. Curr Colorectal Cancer Rep 17, 55–68 (2021). https://doi.org/10.1007/s11888-021-00466-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11888-021-00466-7