
Abstract
In the past 25 years, treatment of metastatic colorectal cancer (mCRC) has undergone profound changes. The approval of newer chemotherapeutics such as irinotecan and oxaliplatin was followed in 2005 by the first targeted therapies, for example, monoclonal antibodies directed against the epidermal growth factor receptor (EGFR), as cetuximab and panitumumab, or the angiogenesis inhibitors bevacizumab, ramucirumab, and aflibercept. With the rapidly progressing molecular characterization of mCRC in the last 10 years and the classification of the disease in four consensus subtypes, further changes are emerging, which will promote, among other things, the introduction of protein-kinase inhibitors developed for specific molecular aberrations as well as immune checkpoint inhibitors into the treatment algorithm.
Thorough molecular pathologic testing is indispensable today for guideline-compliant treatment of mCRC patients. In addition to RAS testing as a precondition for the therapy decision with regard to cetuximab and panitumumab, BRAF testing is of considerable relevance to allow decision making with regard to the newly approved chemotherapy-free combination of the BRAF inhibitor encorafenib and cetuximab in cases where a BRAF-V600E mutation is detected. Additional diagnostic tests should also include genome instability (microsatellite instability). Overall, more and more molecular alterations need to be investigated simultaneously, so that the use of focused next-generation sequencing is increasingly recommended.
This overview describes the prognostic relevance of BRAF testing in the context of molecular pathologic diagnostics of mCRC, presents new treatment options for BRAF-mutated mCRC patients, and explains which modern DNA analytical and immunohistochemical methods are available to detect BRAF mutations in mCRC patients.
Zusammenfassung
Die Therapie des metastasierten kolorektalen Karzinoms (mKRK) hat in den letzten 25 Jahren tief greifende Veränderungen erfahren. Auf die Zulassung neuerer Chemotherapeutika folgten ab 2005 die ersten zielgerichteten Therapien, die sich gegen den epidermalen Wachstumsfaktorrezeptor (EGFR) bzw. gegen Rezeptoren vaskulärer endothelialer Wachstumsfaktoren (VEGFR) richteten. Mit der fortschreitenden molekularen Charakterisierung des mKRK in den letzten 10 Jahren und der Einteilung der Erkrankung in 4 Konsensus-Subtypen zeichnet sich weiterer Wandel ab, unter anderem durch Einführung speziell entwickelter Proteinkinaseinhibitoren wie auch Immuncheckpoint-Inhibitoren in den Therapiealgorithmus.
Eine angepasste molekularpathologische Testung ist heute für eine leitliniengerechte Behandlung von mKRK-Patienten unabdingbar. Neben der RAS-Testung als Voraussetzung für die Therapieentscheidung bezüglich Cetuximab und Panitumumab ist die BRAF-Testung äußerst relevant, um – im Falle des Nachweises einer BRAF-V600E-Mutation – eine Therapieentscheidung zugunsten der neu zugelassenen, chemotherapiefreien Kombination aus dem BRAF-Inhibitor Encorafenib und Cetuximab treffen zu können. Eine erweiterte Diagnostik sollte auch die Genominstabilität (Mikrosatelliten-Instabilität) einbeziehen. Insgesamt müssen immer mehr molekulare Alterationen simultan untersucht werden, sodass sich zunehmend die Verwendung des fokussierten Next Generation Sequencing empfiehlt.
Diese Übersichtsarbeit beschreibt die prognostische Relevanz der BRAF-Testung im Rahmen der molekularpathologischen Diagnostik des mKRK, stellt neue Therapieoptionen zur Behandlung BRAF-mutierter mKRK-Patienten vor und erläutert, welche modernen DNA-analytischen und immunohistochemischen Verfahren zur BRAF-Diagnostik von mKRK-Patienten zur Verfügung stehen.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In patients with colorectal cancer (CRC), BRAF testing together with RAS testing is an established component of molecular biological diagnostics before initiating first-line therapy according to guidelines. The aim of this review is to provide an up-to-date overview of the significance of BRAF as a prognostic and predictive biomarker, to show new therapeutic options for metastatic CRC (mCRC) patients with BRAF mutations, and to describe the currently available diagnostic methods for BRAF testing.
Despite significant advances in treatment, CRC continues to be one of the cancer entities with an unfavorable prognosis in Europe with about 250,000 deaths per year and an annual incidence of more than 500,000 new cases [1]. As the molecular characterization of metastatic CRC (mCRC) and the classification of CRC into molecular subtypes progresses [2], the number of options for the use of targeted therapies is also increasing, with molecular diagnostics becoming ever more important.
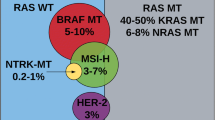
BRAF mutations are present in around 8–12% of patients with mCRC [3, 4]. More than 95% of all BRAF mutations are BRAF-V600 mutations, where valine (V) is mostly substituted by glutamic acid (E) at position 1799 in codon 600 (in exon 15) of the BRAF gene. Apart from this most frequent mutation, BRAFV600E, there are also less common mutations in codon 600, in which valine at position 1799 is substituted by lysine (BRAFV600K), aspartic acid (BRAFV600D), methionine (BRAFV600M), or arginine (BRAFV600R) [5]. Clinically, a comparison of the BRAF-V600 mutation with the significantly less common BRAF mutations in codons 594 and 596 shows that the former is more often found in right-sided and mucinous primary tumors with peritoneal metastasis, whereas BRAF594/596 tumors have a better prognosis [3]. Unless expressly described otherwise, all the statements made in the following sections of this paper refer to BRAFV600E mutations in mCRC.
B‑Raf is a key kinase in the Ras/RAF/MEK-mitogen-activated protein kinase (MAPK) signaling pathway, which is involved in the regulation of cell growth. The alteration of the BRAF gene due to mutation leads to the constitutive activation of this protein kinase, thus causing uncontrolled cell division and consecutively leading to (neo-)angiogenesis and metastasis [6]. Resulting from studies on the CRC transcriptome, mCRC has been classified into four consensus subtypes (consensus molecular subtypes, CMS). KRAS mutations predominantly occur in the epithelial, “metabolic” subtype CMS3, which is characterized by metabolic dysregulation and partly also by chromosomal and microsatellite instability (MSI) [2, 7]. BRAF mutations, however, are often seen in the “MSI-immune” subtype CMS1, which is dominated by somatic hypermutation and MSI.
It is extremely rare that BRAF mutations occur together with a mutation of the RAS gene. According to the current German S3 guideline, molecular testing for the presence of both mutations should be carried out before initiating first-line therapy, wherever possible. In this context, BRAF testing should best be done simultaneously with the RAS test or sequentially after exclusion of the RAS mutation [3].
Oncogenic properties of the BRAF mutation
BRAF, which is an oncogenic driver in mCRC patients, was already established as a therapeutic target structure in malignant melanoma many years ago [8]. Serrated adenomas of the intestine that are associated with a BRAF mutation show molecular, morphological, clinical, and epidemiological characteristics that differ from those of adenomas and which develop during a “classic adenoma-carcinoma sequence” based on mutations of the APC gene [9, 10]. The BRAF-driven form of sessile serrated adenomas (SSA) leads to impaired apoptosis of crypt epithelia followed by senescence with epigenetic promoter (CpG) methylation and decreased expression of various genes (e.g., hMLH1, MGMT, p16) [3, 9]. SSAs as a tumor pre-stage and precursor lesion are flat polyps that barely protrude from the mucosa, predominantly occur in the right-sided colon, and are difficult to detect even endoscopically [3, 9]. Patients with large SSA have a higher risk of developing colorectal cancer; women with SSA have a five-fold higher risk than men [3].
BRAF mutation: a clearly negative prognostic factor in CRC
In mCRC, advanced age is a negative prognostic marker, as is tumor location proximal to the left flexure [11]. As part of an investigation on further potential prognostic markers in this indication, the influence of BRAF mutations and MSI on metastatic spread and prognosis was analyzed within a large retrospective case series: BRAF-mutant tumors, particularly those harboring a V600E mutation, are associated with a significantly poorer overall survival (OS) than BRAF wild-type tumors (median 10.4 versus 34.7 months; hazard ratio [HR] = 10.66, p < 0.001), as well as with a higher rate of peritoneal and distant lymph node metastasis [12]. The prognostically highly unfavorable impact of BRAFV600 was also repeatedly reported in randomized controlled trials [4, 13, 14]; a detailed discussion of the prognostic impact of BRAF mutations and their connection with microsatellite stability and instability as a further biomarker can be found in two recent reviews [6, 15]. Apart from hereditary non-polyposis-associated colorectal carcinoma (HNPCC), MSI occurs in mCRC patients with an estimated frequency of only 4–8% [4]. If BRAF mutations and MSI occur simultaneously—the frequency is reciprocally about one third each—these alterations constitute sporadic defects of mismatch repair (dMMR) [3, 4]. MSI patients appear to have a better prognosis than patients with microsatellite stability (MSS) [16]; although the number of published cases is still limited, the available clinical evidence suggests that patients with a BRAFV600E mutation and MSS have a poorer outcome than those with BRAFV600E and high MSI status. In the metastatic setting, the combination of BRAFV600E and MSS seems to predominate, with the BRAF mutation determining the poor outcome [6, 13, 17, 18, 19, 20].
BRAF mutation: unclear predictive value with regard to former conventional therapies
The predictive relevance of BRAF mutations for the use of anti-epidermal growth factor receptor (EGFR) therapy, i.e., the two monoclonal antibodies cetuximab and panitumumab, is currently under debate due to the fact that BRAF and RAS mutations are almost mutually exclusive [21] and that RAS mutations are known negative predictive factors for the use of anti-EGFR therapy. In CRC, cetuximab and panitumumab are approved for use in RAS wild-type patients only: for their use in patients with a BRAF mutation, only limited data are available from subgroup analyses of larger confirmatory studies (Table 1), as well as from retrospective case series derived from clinical routine data [22, 23].
Two partly overlapping meta-analyses confirmed a clinical benefit for anti-EGFR antibody therapy in patients with wild-type RAS and wild-type BRAF; in RAS wild-type patients harboring a BRAF mutation, however, data showed only a limited, non-significant clinical benefit in terms of progression-free survival (PFS) as well as OS [24, 25]. The current body of evidence, on the other hand, does not justify the exclusion of anti-EGFR antibodies from the therapeutic repertoire for BRAFV600E-mutant patients either. Data from the German randomized phase-II study VOLFI comparing panitumumab plus chemotherapy vs. mono-chemotherapy in first-line treatment showed that the addition of panitumumab to chemotherapy tended to increase the overall response rate (ORR) in the 14 patients with BRAF-mutated tumors (odds ratio = 14.93, 95% confidence interval [CI] 1.03–200.00) [26].
Anti-VEGF therapy, which—as with anti-EGFR therapy—is given in combination with oxaliplatin-containing (mostly in first-line) or irinotecan-containing chemotherapy (mostly in second-line), is a clinically relevant routine treatment of mCRC and can be used independent of RAS status. However, since no or only indirect comparisons are available to date, the predictive value of BRAF testing with regard to this treatment regimen is still unclear. Results of a small phase-II study [27] and subgroup analyses of two large phase-III studies [28, 29] also do not allow a clear overall assessment of the intensified chemotherapy backbone (FOLFOXIRI) in BRAF-mutant patients. A meta-analysis of five randomized studies of quite differing case numbers [range: 70–679] recently found that in BRAF-mutated patients—with the total case numbers still being small—intensified combination therapy does not provide an additional benefit in the first-line setting [30]. A meta-analysis performed by the ARCAD study group, published in autumn 2020 and pooling data from two studies comparing chemotherapy plus-anti-EGFR with chemotherapy plus-anti-VEGFR therapy as first-line options for mCRC, could not demonstrate a significant difference in OS for the subgroup of BRAF-mutated patients (n = 138) that received either bevacizumab-based or cetuximab-based therapy (HR = 1.01 [95% CI 0.69–1.48]) [31]. The benefit of anti-VEGF therapy with bevacizumab per se and the predictive role of BRAF for initiating bevacizumab-based therapy still requires further investigation (Table 1).
Although targeted tyrosine kinase inhibitors have been used in clinical routine since 2011 with very good outcomes in BRAF V600-mutated melanoma, BRAF-mutated mCRC proved to be less sensitive to monotherapy [32, 33]. The reason behind this may be CRC-specific resistance mechanisms in the MAPK signaling cascade. In-vitro studies demonstrated suppression of the negative feedback loop between extracellular signal-regulated kinase (ERK) and the EGFR under BRAF monotherapy with overall high EGFR expression in CRC and possibly even stronger activation of the receptor by its ligands (Fig. 1; [33, 34]). This results in a reactivation of the EGFR pathway, e.g., by-passing the mutated BRAF protein via CRAF. Thus, it seems important to inhibit the EGFR pathway by simultaneously administering a therapy directed against EGFR in addition to BRAF blockade in order to block the multi-track resistance mechanisms within the MAPK signaling pathway [33, 35].

The MAPK signaling pathway (figure modified from Taieb et al. [15] CC BY licence). a MAPK pathway: signal enhancement in the presence of an activating BRAF mutation. b Inhibition of BRAF activated by mutation leading to suppression of the ERK-mediated negative feedback loop and reactivation of the MAPK signaling pathway via CRAF. c Counteracting resistance mechanisms: mechanism of action of combined BRAF and EGFR blockade. BRAF rapidly accelerated fibrosarcoma isoform B; CRAF rapidly accelerated fibrosarcoma isoform C; EGFR epidermal growth factor receptor; ERK extracellular signal-regulated kinase; MAPK mitogen-activated protein kinase; MEK MAPK/ERK kinase; RAS rat sarcoma protooncogene
Therapeutic options in BRAF-mutant mCRC
Until recently, first- and second-line treatment of mCRC has generally been based on the use of combination chemotherapies, mostly including—in the case of left-sided RAS wild-type tumors—EGFR antibody therapy, as described in the previous section [3, 4]Footnote 1. Current treatment recommendations for mCRC are drawing much attention to the general condition of the patients, which are typically of older age. Regarding patients fit enough for systemic treatment, a distinction is made between the therapeutic objectives of “cytoreduction,” i.e., reduction of the tumor mass, and “disease control,” i.e., delaying further progression.
First-line therapy in BRAF-mutant mCRC: a controversial treatment standard
The combination of an antimetabolite (5-fluorouracil, plus leucovorin as a folinic acid derivative) and a platinum compound interfering with DNA replication (oxaliplatin) together with a topoisomerase I inhibitor (irinotecan) and the angiogenesis inhibitor (anti-VEGF) bevacizumab represents the current European guideline standard for BRAF-mutant mCRC patients in good general health [4]. However, considering the evidence level in the BRAF-mutated subgroup, it has to be noted that this recommendation is based on a very small number of patients (N = 28) from the TRIBE study and is therefore associated with uncertainties [28]. In the BRAF subgroup of this phase-III study, the OS under FOLFOXIRI plus bevacizumab was 19 months with an ORR of 56%; however, there was no significant difference to the comparator group consisting of FOLFIRI plus bevacizumab (Table 1; [28]). This first-line standard is presently the subject of controversial discussion. A recently published meta-analysis based on five randomized studies on FOLFOXIRI plus bevacizumab vs. doublet chemotherapy plus bevacizumab showed a non-significant trend in BRAF-mutant tumors favoring the less intensive regime (n = 115; HR = 1.12 [95% CI 0.75–1.68]) [30].
Due to their poor prognosis, the concept of an ‘aggressive’ first-line treatment in BRAFV600E -mutated mCRC, using almost the entire arsenal of therapy modalities, is currently of clinical relevance with regard to the current recommendations for this mCRC patient collective—especially when cytoreduction is the therapeutic objective. On the other hand, it is unclear to what extent patients in Germany are treated with this intensified first-line therapy, which is associated with relevant adverse events (AE).
As mentioned above, the issue of using anti-EGFR-based therapy in BRAF-mutant tumors is currently the subject of controversial debates due to the inconclusive results of two meta-analyses [24, 25].
New chemotherapy-free, targeted option after systemic therapy
Due to the limited therapeutic options after completion of first-line treatment, no clear recommendations could be drawn to date for second- and third-line therapy of BRAF-mutant mCRC patients [3, 4]. The German S3 guideline stated in 2019 that “Individual (presently) not approved therapeutic approaches, e.g. with a BRAF inhibitor, MEK inhibitor and anti-EGFR antibody or, if possible, treatment within a clinical study (are) to be taken into consideration”; until recently, none of the therapeutic options mentioned here was approved for this indication.
In June 2020, however, the combination of the BRAF inhibitor encorafenib and the anti-EGFR antibody cetuximab was granted European Union (EU) approval, making such a chemotherapy-free, purely targeted dual blockade available for routine care. This combination is indicated for the treatment of adult patients with mCRC and a BRAFV600E mutation who have received prior systemic treatment [43].
The phase-III BEACON CRC trial, on which the approval was based, investigated the triple blockade with encorafenib and cetuximab plus the MEK inhibitor binimetinib and the dual blockade with encorafenib and cetuximab vs. control therapy consisting of irinotecan-based chemotherapy plus cetuximab in patients with BRAFV600E-mutant mCRC having previously received one or two palliative therapy lines (Fig. 2; [44]). Primary endpoints were ORR and OS of the triple blockade vs. the control. The study was powered on the key secondary endpoint: OS of dual blockade versus control. Using a test hierarchy, the secondary endpoints OS, ORR, and PFS of dual blockade vs. control and PFS of triple blockade vs. control were also alpha-controlled and thus confirmatory [44]. A total of 665 patients were randomized in a 1:1:1 ratio to the three therapy arms.

Design of the BEACONCRC phase-III study in patients with pretreated metastatic colorectal cancer (mCRC) and BRAFV600E mutation. BID twice daily; BM biomarker; FOLFIRI folic acid + fluorouracil + irinotecan; ORR overall response rate; OS overall survival; PD progressive disease; PFS progression-free survival; PK pharmacokinetics; Q1W weekly; QD once daily; R randomization; ° grade
The primary endpoints of the study were reached. In the primary analysis (median follow-up for OS: 7.8 months), the combination of encorafenib, cetuximab, and binimetinib showed an ORR of 26% [95%CI 18–35] vs. 2% [95%CI < 1–7] in the control arm (p < 0.001). Median OS of the triple blockade vs. control was 9.0 vs. 5.4 months (HR = 0.52 [95% CI 0.39–0.70]; p < 0.001) [44]. For the alpha-controlled key secondary endpoint—the confirmatory comparison of OS of the dual combination therapy vs. control—an extension of the median OS by 3 months (8.4 vs. 5.4 months; HR = 0.60 [95% CI 0.45–0.79], p < 0.001) could be demonstrated for encorafenib plus cetuximab vs. the control group; the ORR was 20% [95%CI 13–29] vs. 2% [95%CI < 1–7] (p < 0.001).
The tolerability of the dual blockade was slightly more favorable than that of the triple blockade and of the control group (Fig. 2). The safety profile of the combination of encorafenib and cetuximab was well manageable and showed, in terms of AE, the expected class effects. The most common AEs included: elevated creatinine level (50%), nausea (34%), diarrhea (33%), low hemoglobin level (32%), fatigue (30%), acneiform dermatitis (29%), and decreased appetite (27%) [44].
Due to the comparable efficacy results of triple versus dual blockade and the slightly more favorable tolerability of encorafenib plus cetuximab, the European Medicines Agency (EMA) approved the dual combination regimen in June 2020 [43].
A new update analysis after a median follow-up of 12.8 months confirmed the above-described results with a consistent tolerability profile [45]. Median OS was 9.3 months [95% CI 8.2–10.8] for the triplet blockade, 9.3 months [95% CI 8.0–11.3] for the EMA-approved double blockade of encorafenib and cetuximab, and 5.9 months [95% CI 5.1–7.1] for the control group.
The targeted triple blockade consisting of encorafenib, cetuximab, and binimetinib is currently being further investigated as a first-line therapy in the two-stage ANCHOR CRC phase-II study (Fig. 3).

Design of the ANCHOR phase-II study in patients with previously untreated mCRC and BRAFV600E mutation. ORR overall response rate; OS overall survival; PD progressive disease
BRAF diagnostics
As a consequence of the above-described clinical situation, diagnostic testing for the presence of RAS and BRAF mutations in mCRC is essential: for BRAF-mutated patients, encorafenib plus cetuximab can be taken into consideration as a newly available therapeutic option.
The guidelines recommend performing these tests either before initiating first-line therapy in mCRC, or already at the time of initial diagnosis of CRC, in order to exclude the presence of Lynch syndrome via additional dMMR testing [3, 4]. In the same way as BRAF-mutated tumors, such hereditary CRCs without polyposis (HNPCC) also constitute a biologically distinct subtype of CRC. If a BRAF mutation is present in a dMMR/MSI tumor, Lynch syndrome can be mostly excluded. Thus, the determination of BRAF mutation status is of diagnostic and therapeutic relevance and helps to differentiate somatic from genetic “mismatch” repair defects [46, 47, 48]. The diagnosis of a sporadic tumor, thus excluding HNPCC/Lynch syndrome, can be supported by analyzing MLH1 promoter methylation, since the presence of such methylation additionally corroborates the diagnosis of a sporadic, high MSI (Fig. 4). BRAF testing can be performed either simultaneously with RAS testing or stepwise after excluding a RAS mutation. Nowadays, however, the simultaneous approach is recommended applying gene panel diagnostics based on focused next-generation sequencing (NGS).

Diagnostic algorithm MSI/MSS—BRAF—MLH1 promoter methylation (at/after exclusion of a RAS mutation [3]; prognostic statements according to Lochhead et al. [17]). BRAF rapidly accelerated fibrosarcoma isoform B; MLH1, MSH2, MSH6, PMS2 DNA repair enzymes/complexes; MSI microsatellite instability; MSS microsatellite stability; wt wild-type
Sample preparation
In the course of the diagnostic workup, specimens are mostly obtained during colonoscopy or surgical removal of the primary tumor. After fixation via 10% neutral buffered formalin (4% formaldehyde) for 24–48 h and embedding in paraffin, the specimens are well suited for the tests indicated in Fig. 4. For molecular determination of MSI status, healthy tissue samples distant from the tumor should also be stored and analyzed [4]. If tissue samples contain a low percentage of tumor cells, a macro-dissection prior to DNA extraction is highly recommended for cancer cell enrichment.
Molecular pathological, DNA analytical methods
Various test methods with different specificity and sensitivity are available to determine BRAFV600E mutation status. With classical test methods (Table 2) such as Sanger sequencing, 99% of all mutations can be detected with a specificity of 100%. However, caution is required to ensure that the tumor cell proportion remains above 20–30%. Detection of the BRAFV600E mutation by means of high-resolution melting (HRM) analysis or pyrosequencing provides higher sensitivity than Sanger sequencing; here, a tumor cell proportion of about 10–15% (5% detection limit) is sufficient [49, 50]. Commercial tests such as the ThxID-BRAF-Kit® (V600E and V600K), the cobas 4800® BRAF mutation test (only V600E), or the Idylla BRAF mutation test are of similar sensitivity (Table 3; [51, 52, 53]).
In recent years, NGS methods have been increasingly used in molecular diagnostics, allowing the detection of selected genes/genomic regions relevant for diagnosis and therapy (targeted NGS) simultaneously with high sensitivity and specificity. Therefore, BRAF mutation testing in CRC is often no longer carried out as an isolated individual test but integrated into the parallel detection of other molecular alterations such as KRAS and NRAS. The sensitivity of NGS-based methods is generally very high (approximately 1% detection limit), but hampered by artifacts occurring during formalin fixation. In many laboratories, a threshold of 5% variant allele frequency (VAF) is therefore requested, which can however be undercut in specific situations [49, 50]. Today, ThermoFisher Scientific and Illumina are the most prominent NGS platforms available. Both platforms enable analysis of numerous commercial or in-house gene panels, based on either amplicon-based (multiplex polymerase chain reaction, PCR) or hybrid-capture methods for enriching the selected target regions. Numerous bioinformatic programs are available for the evaluation of NGS data. However, these should be used by scientists/physicians that have profound experience in molecular diagnostics.
In the BEACON CRC study, which was conducted in a total of 221 centers in 28 countries (111 of which were centers in Europe), evaluation of the procedures used for BRAF status determination from 510 samples revealed the following picture: in 48.8% of the analyses, single gene detection was still used for BRAF testing; protein-based methods (immunohistology) were used in 0.7% of the analyses. However, the majority of BRAF tests were performed together with the detection of other gene alterations (e.g., as focused, amplicon-based NGS) (50.5%). Discrepancies observed between local and central testing show the relevance of standardization of diagnostic procedures, especially in view of the increasing importance of targeted therapeutic approaches: clear confirmation of the locally detected BRAFV600E mutation was found in just 90.7% of central testing. Of note, this discrepancy was largely due to insufficient neoplastic tissue in the sample, most likely resulting from the fact that BRAF-mutant tumors are generally associated with mucinous adenocarcinoma that contain fewer tumor cells. In 1.6% of the central repeat tests, the local result was clearly negated. Taking into account this possibility of a discrepancy between local and central testing, the study protocol allowed the inclusion of patients based on local BRAFV600E detection in molecular prescreening, but additionally required central confirmation, as part of the inclusion criteria, within 30 days of initial receipt of study medication. Once the study had reached the pre-specified number of discrepant test results, the assay, which was developed and then approved in the USA as a “companion diagnostic,” became a prerequisite for the inclusion of all further patients.
The EU and US regulations on in-vitro diagnostics (IVD) are fundamentally different: in the US, such tests are subject to central approval (Premarket Approval) by the Food and Drug Administration (FDA), while in the EU manufacturers can chose an accredited “notified body” that evaluates the conformity of their test; once confirmed, the manufacturer is allowed to label its product with the so-called CE label (CE: Conformité Européene)Footnote 2. Furthermore, the FDA usually grants approval for targeted therapies only in conjunction with a defined and simultaneously approved companion diagnostic, which, at the time of drug approval, has an exclusivity of use: the usage of the respective companion diagnostic is therefore a prerequisite in the USA for the prescription of the drug by the physician [54].
The US legislation distinguishes such standardized, mostly commercially available tests from so-called laboratory-developed tests (LDT), which—like the classical test methods such as Sanger sequencing—are designed, validated, and applied by institutes for their own use. The German Accreditation Body (DAkkS) refers to such LDTs as “in-house tests.” They are normally not subject to formal approval or labeling requirements; however, a few years ago, the DAkkS issued a guideline for the validation of molecular pathological examination methods [55, 56]. In the US, a discussion paper was presented by the FDA in January 2017, advocating stronger prospective regulation of LDTs due to their increasing prognostic and predictive importance—this applies in particular to the role and growing importance of NGS [57, 58]. With the FoundationOne® CDx (F1CDx) test, an NGS method was approved as a companion diagnostic for the first time in the USA in late 2017 (Table 3; [59]).
Against this background, the diversity of competing classical and modern DNA analytical methods for BRAF mutation determination is easier to understand. Of the various commercial procedures using allele-specific PCR techniques, only the Qiagen therascreen® test is currently recommended for BRAF-mutated mCRC in the US; in Europe, this test is CE-labeled. It is to be expected that for the tests currently approved for melanoma only, appropriate adjustments will soon be made in the US with regard to CRC. In the BEACON CRC approval study, the only methods allowed by the study protocol were PCR and NGS based on local assays [44].
A comparison between commercially available (i.e., FDA-approved) tests and LDTs for EGFR, KRAS, and BRAF mutations showed that there was no overall difference between the methods and the three tested genes in assay performance; the average analysis accuracy was 97% [60]. Since testing for KRAS, BRAF, MSI/dMMR, MLH1, and possibly other genes constitutes a prerequisite of CRC diagnostics, panel-based assays are understandably more prominent in current pathological practice—in Germany, all major university and non-university institutions are now using focused NGS for this purpose. Frequently used platforms include Illumina (MiSeq™ or NextSeq™) and Thermo Fisher (Ion Gene Studio S5™) [61, 62, 63]: in a multicenter validation study across Germany, a high level of consistency between different NGS platforms and gene panels was shown; apart from CRC, samples of lung and breast cancers were also tested [61].
Immunohistochemical methods
Besides DNA analytical methods, protein-based analyses using the VE1 antibody may provide an alternative to molecular pathological testing for BRAFV600E; the latter is widely regarded as the gold standard in BRAF mutation testing [50, 64]. At the same time, protein-based immunohistological detection is the only reasonably practicable method for determining the expression level of mutant BRAF protein. This method is also applicable for MSI testing. It is characterized by a specificity of 98–100%, a sensitivity of 85–100% [49, 50], and an in-lab turnaround time of 1 day. Thus, the method is generally reliable, but some challenges remain, such as establishing a standardized scoring of protein expression, which is needed to avoid a substantial number of misclassifications [64]. Immunostaining is in principle a fast and cost-effective method for the determination of BRAF-mutant protein; however, as CRC meanwhile requires the determination of multiple alterations, DNA analytical methods should certainly be preferred nowadays.
Chu et al. investigated outcomes of immunohistochemical (IHC) and NGS testing in a cohort of almost 1900 CRC patients [65]. The rate of false-positive IHC tests was 17%; however, confirmatory re-testing by NGS was performed in only 43% of the IHC-tested patients. NGS-tested patients had a favorable median OS, at younger age, and a lower rate of synchronous metastases and a higher rate of therapy. The authors concluded that NGS should be considered as standard testing but that IHC might serve as an optional screening test if NGS testing is not available in a timely manner. This is underlined by the rapid availability of results via IHC and the finding that reflexive IHC testing made it possible to identify 57% more BRAF-mutated mCRC than standard NGS methods.
Conclusions for clinical practice
-
The detection of a BRAFV600E mutation, particularly in the presence of MSS status, is associated with a dismal prognosis, indicating an aggressive molecular subtype of CRC.
-
For Patients with BRAFV600E-mutated CRC and their “high unmet medical need,” encorafenib plus cetuximab now constitutes a chemotherapy-free, targeted therapy standard after systemic treatment; combined BRAF and EGFR blockade is expected to be included in the German and European treatment algorithms and guidelines in the coming months.
-
For adequate planning of the therapy sequence, BRAF testing is therefore an absolute necessity in all patients with mCRC before initiating first-line therapy.
-
For testing, a large variety of methods is available, although panel diagnostics with NGS should be preferred to integrate testing for various molecular alterations.
Notes
Regarding the guidelines of the European Society of Medical Oncology (ESMO), there are not only the Consensus Guidelines, latest edition published in 2016, for metastatic CRC (see Ref. [4]), but also the Pan-Asian Adapted ESMO Consensus Guidelines published in early 2018. Although this article refers to the somewhat older ESMO guideline, the reference always applies to both guidelines and the current study evidence shown therein.
With the “CE label,” the manufacturer, distributor, or EU authorized representative declares under EU Regulation 765/2008 “that the product meets the applicable requirements stipulated in the harmonization legislation of the Community on its affixing.”
References
Ferlay J, Colombet M, Soerjomataram I et al (2018) Cancer incidence and mortality patterns in Europe: estimates for 40 countries and 25 major cancers in 2018. Eur J Cancer 103:356–387
Dienstmann R, Salazar R, Tabernero J (2018) Molecular subtypes and the evolution of treatment decisions in metastatic colorectal cancer. Am Soc Clin Oncol Educ Book 38:231–238
Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF) (2019) S3-Leitlinie Kolorektales Karzinom, Langversion 2.1. http://www.leitlinienprogramm-onkologie.de/leitlinien/kolorektales-karzinom/. Accessed: 20. Mai 2020 (AWMF registration number: 021/007OL)
Van Cutsem E, Cervantes A, Adam R, Sobrero A et al (2016) ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 27:1386–1422
Dankner M, Rose AAN, Rajkumar S et al (2018) Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene 37:3183–3199
Sanz-Garcia E, Argiles G, Elez E et al (2017) BRAF mutant colorectal cancer: prognosis, treatment, and new perspectives. Ann Oncol 28:2648–2657
Cancer Genome Atlas Network (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487:330–337
Davies H, Bignell GR, Cox C et al (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954
Tannapfel A, Neid M, Aust D et al (2010) The origins of colorectal carcinoma: specific nomenclature for different pathways and precursor lesions. Dtsch Arztebl Int 107:760–766
Baretton GB, Tannapfel A, Schmitt W (2011) Standardized and structured histopathological evaluation of colorectal polyps: a practical checklist against the background of the new WHO classification. Pathologe 32:289–296
Bylsma LC, Gillezeau C, Garawin TA et al (2020) Prevalence of RAS and BRAF mutations in metastatic colorectal cancer patients by tumor sidedness: a systematic review and meta-analysis. Cancer Med 9:1044–1057
Tran B, Kopetz S, Tie J et al (2011) Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 117:4623–4632
Roth AD, Tejpar S, Delorenzi M et al (2010) Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC‑3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 28:466–474
Safaee Ardekani G, Jafarnejad SM, Tan L et al (2012) The prognostic value of BRAF mutation in colorectal cancer and melanoma: a systematic review and meta-analysis. PLoS One 7:e47054
Taieb J, Lapeyre-Prost A, Laurent Puig P et al (2019) Exploring the best treatment options for BRAF-mutant metastatic colon cancer. Br J Cancer 121:434–442
Venderbosch S, Nagtegaal ID, Maughan TS et al (2014) Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res 20:5322–5330
Lochhead P, Kuchiba A, Imamura Y et al (2013) Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 105:1151–1156
Taieb J, Shi Q, Pederson L et al (2019) Prognosis of microsatellite instability and/or mismatch repair deficiency stage III colon cancer patients after disease recurrence following adjuvant treatment: results of an ACCENT pooled analysis of seven studies. Ann Oncol 30:1466–1471
Lee S, Cho NY, Choi M et al (2008) Clinicopathological features of CpG island methylator phenotype-positive colorectal cancer and its adverse prognosis in relation to KRAS/BRAF mutation. Pathol Int 58:104–113
Samowitz WS, Sweeney C, Herrick J et al (2005) Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res 65:6063–6069
Sahin IH, Kazmi SM, Yorio JT et al (2013) Rare though not mutually exclusive: a report of three cases of concomitant KRAS and BRAF mutation and a review of the literature. J Cancer 4:320–322
Morris V, Overman MJ, Jiang ZQ et al (2014) Progression-free survival remains poor over sequential lines of systemic therapy in patients with BRAF-mutated colorectal cancer. Clin Colorectal Cancer 13:164–171
Ulivi P, Capelli L, Valgiusti M et al (2012) Predictive role of multiple gene alterations in response to cetuximab in metastatic colorectal cancer: a single center study. J Transl Med 10:87
Pietrantonio F, Petrelli F, Coinu A et al (2015) Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer 51:587–594
Rowland A, Dias MM, Wiese MD et al (2015) Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br J Cancer 112:1888–1894
Modest DP, Martens UM, Riera-Knorrenschild J et al (2019) Folfoxiri plus panitumumab as first-line treatment of RAS wild-type metastatic colorectal cancer: the randomized, open-label, phase II Volfi study (AIO KRK0109). J Clin Oncol 35:3401–3411
Loupakis F, Cremolini C, Salvatore L et al (2014) Folfoxiri plus bevacizumab as first-line treatment in BRAF mutant metastatic colorectal cancer. Eur J Cancer 50:57–63
Cremolini C, Loupakis F, Antoniotti C et al (2015) Folfoxiri plus bevacizumab versus folfiri plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol 16:1306–1315
Cremolini C, Antoniotti C, Lonardi S et al (2019) Updated results of TRIBE2, a phase III, randomized strategy study by GONO in the first- and second-line treatment of unresectable mCRC. J Clin Oncol 37(15):3508
Cremolini C, Antoniotti C, Stein A et al (2020) Folfoxiri/bevacizumab (bev) versus doublets/bev as initial therapy of unresectable metastatic colorectal cancer (mCRC): a meta-analysis of individual patient data (IPD) from five randomized trials. J Clin Oncol 38(15):4015
Karapetis CS, Liu H, Sorich M et al (2020) Impact of molecular markers status on treatment effects comparing EGFR and VEGF monoclonal antibodies (mAbs) in untreated metastatic colorectal cancer (mCRC): Pooled individual patient data (IPD) analysis of randomized trials from the ARCAD database. Ann Oncol 31(4):S426 (Abstr. 434P)
Kopetz S, Desai J, Chan E et al (2015) Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol 33:4032–4038
Prahallad A, Sun C, Huang S et al (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483:100–103
Corcoran RB, Ebi H, Turke AB et al (2012) EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2:227–235
Poulikakos PI, Zhang C, Bollag G et al (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464:427–430
Bokemeyer C, Van Cutsem E, Rougier P et al (2012) Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the crystal and opus randomised clinical trials. Eur J Cancer 48:1466–1475
Douillard JY, Oliner KS, Siena S et al (2013) Panitumumab-Folfox4 treatment and RAS mutations in colorectal cancer. N Engl J Med 369:1023–1034
Stintzing S, Miller-Phillips L, Modest DP et al (2017) Impact of BRAF and RAS mutations on first-line efficacy of folfiri plus cetuximab versus folfiri plus bevacizumab: analysis of the FIRE‑3 (AIO KRK-0306) study. Eur J Cancer 79:50–60
Peeters M, Smith Oliner K, Price TJ et al (2014) Updated analysis of KRAS/NRAS and BRAF mutations in study 20050181 of panitumumab (pmab) plus folfiri for second-line treatment (tx) of metastatic colorectal cancer (mCRC). J Clin Oncol 32(5):3568
Seymour MT, Brown SR, Middleton G et al (2013) Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (Piccolo): a prospectively stratified randomised trial. Lancet Oncol 14:749–759
Peeters M, Oliner KS, Parker A et al (2013) Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res 19:1902–1912
Karapetis CS, Jonker D, Daneshmand M et al (2014) PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer—results from NCIC CTG/AGITG CO.17. Clin Cancer Res 20:744–753
European Commission (2020) Zusammenfassung der Produkteigenschaften von Braftovi. https://www.ema.europa.eu/en/documents/product-information/braftovi-epar-product-information_de.pdf. Accessed: 20. Aug. 2020
Kopetz S, Grothey A, Yaeger R et al (2019) Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med 381:1632–1643
Kopetz S, Grothey A, Van Cutsem E et al (2020) Encorafenib plus cetuximab with or without binimetinib for BRAF V600E metastatic colorectal cancer: updated survival results from a randomized, three-arm, phase III study versus choice of either irinotecan or folfiri plus cetuximab (beacon CRC). J Clin Oncol 38(15):4001
Büttner R, Friedrichs N (2019) Hereditary colon cancer in Lynch syndrome/HNPCC syndrome in Germany. Pathologe 40:584–591
Rau TT, Dawson H, Hartmann A et al (2017) Hereditary colorectal cancer: an update on genetics and entities in terms of differential diagnosis. Pathologe 38:156–163
Bucksch K, Zachariae S, Aretz S et al (2020) Cancer risks in Lynch syndrome, Lynch-like syndrome, and familial colorectal cancer type X: a prospective cohort study. BMC Cancer 20:460
Ihle MA, Fassunke J, König K et al (2014) Comparison of high resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional Sanger sequencing for the detection of p.V600E and non‑p.V600E BRAF mutations. BMC Cancer 14:13
Cheng L, Lopez-Beltran A, Massari F et al (2018) Molecular testing for BRAF mutations to inform melanoma treatment decisions: a move toward precision medicine. Mod Pathol 31:24–38
Marchant J, Mange A, Larrieux M et al (2014) Comparative evaluation of the new FDA approved THxID™-BRAF test with high resolution melting and sanger sequencing. BMC Cancer 14:519
Qu K, Pan Q, Zhang X et al (2013) Detection of BRAF V600 mutations in metastatic melanoma: comparison of the Cobas 4800 and Sanger sequencing assays. J Mol Diagn 15:790–795
Schiefer AI, Parlow L, Gabler L et al (2016) Multicenter evaluation of a novel automated rapid detection system of BRAF status in formalin-fixed, paraffin-embedded tissues. J Mol Diagn 18:370–377
Jørgensen JT, Hersom M (2018) Clinical and regulatory aspects of companion diagnostic development in oncology. Clin Pharmacol Ther 103:999–1008
DAkkS (2016) Leitfaden des Sektorkomitees Pathologie/Neuropathologie für die Validierung von Untersuchungsverfahren in der Molekularpathologie. https://www.dakks.de/sites/default/files/dokumente/71_sd_4_037_leitfaden_validierung_molpath_20161004_v1.1.pdf. Accessed: 30. Mai 2020
Dietmaier W, Hummel M (2018) Quality assurance in molecular pathology. Pathologe 39:178–180
FDA (2017) Discussion paper on laboratory developed tests (LDTs). https://www.fda.gov/media/102367/download. Accessed: 30. Mai 2020
Genzen JR (2019) Regulation of laboratory-developed tests. Am J Clin Pathol 152:122–131
FDA (2017) List of cleared or approved companion diagnostic devices (in vitro and imaging tools). https://www.fda.gov/medical-devices/vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-vitro-and-imaging-tools. Accessed: 30. Mai 2020
Kim AS, Bartley AN, Bridge JA et al (2018) Comparison of laboratory-developed tests and FDA-approved assays for BRAF, EGFR, and KRAS testing. JAMA Oncol 4:838–841
Hirsch B, Endris V, Lassmann S et al (2018) Multicenter validation of cancer gene panel-based next-generation sequencing for translational research and molecular diagnostics. Virchows Arch 472:557–565
Udar N, Lofton-Day C, Dong J et al (2018) Clinical validation of the next-generation sequencing-based extended RAS panel assay using metastatic colorectal cancer patient samples from the phase 3 prime study. J Cancer Res Clin Oncol 144:2001–2010
Kwon D, Kim B, Shin HC et al (2019) Cancer panel assay for precision oncology clinic: results from a 1-year study. Transl Oncol 12:1488–1495
Dvorak K, Higgins A, Palting J et al (2019) Immunohistochemistry with anti-BRAF V600E (VE1) mouse monoclonal antibody is a sensitive method for detection of the BRAF V600E mutation in colon cancer: evaluation of 120 cases with and without KRAS mutation and literature review. Pathol Oncol Res 25:349–359
Chu JE, Johnson B, Kugathasan L et al (2010) Population-based screening for BRAF V600E in metastatic colorectal cancer reveals increased prevalence and poor prognosis. Clin Cancer Res 26:4599–4605
Funding
Editorial and medical writing assistance was provided by Dr Markus Hartmann from Ecc-Oncology (Trier, Germany), funded by Pierre Fabre Pharma GmbH (Freiburg im Breisgau, Germany).
Funding
Open Access funding enabled and organized by Projekt DEAL
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
A. Vogel declares having received honoraria in the last 3 years, although not in relation to the present review, from the following pharmaceutical companies: Bristol-Myers Squibb, Merck, MSD, Pierre Fabre, and Roche. M. Hummel received support from Novartis, BMS, MSD, and Pierre Fabre for various activities. S. Hegewisch-Becker and J.H.L. Neumann declare that they have no competing interests.
For this article no studies with human participants or animals were performed by any of the authors. All studies performed were in accordance with the ethical standards indicated in each case.
The supplement containing this article is not sponsored by industry.
Additional information
Editor
H. Baba, Essen
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hummel, M., Hegewisch-Becker, S., Neumann, J.H.L. et al. BRAF testing in metastatic colorectal carcinoma and novel, chemotherapy-free therapeutic options. Pathologe 42 (Suppl 1), 98–109 (2021). https://doi.org/10.1007/s00292-021-00946-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00292-021-00946-5