
Abstract
Statins are the first-line therapy in LDL-Cholesterol (LDL-C) reduction and its clinical use has contributed to significant prevention and treatment of atherosclerotic vascular disease. Yet, a significant proportion of patients remain at high risk. Recently, a number of new therapies have been developed to further lower LDL-C. These agents may provide clinical benefit on top of statin therapy in patients with high residual risk, severe hypercholesterolemia or as an alternative for patients who are intolerant to statins. We review four novel approaches based on the inhibition of proprotein convertase subtilisin/kexin type 9 (PCSK9), apolipoprotein-B100 (apoB), Cholesteryl ester transport protein (CETP) and microsomal triglyceride transfer protein (MTP). ApoB and MTP inhibitors (Mipomersen and Lomitapide) are indicated only for homozygous familial hypercholesterolemia patients. The results of ongoing trials with CETP and PCSK9 inhibitors may warrant a wider employment in different categories of patients at high risk for cardiovascular disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Coronary artery disease (CAD) is the leading cause of death in most countries and is associated with a significant economic and health care burden [1]. Reduction in low-density lipoprotein cholesterol (LDL-C) levels has been shown to reduce the risk of CAD and death. Currently, statins are the agents primarily used to lower LDL-C.
Statin efficacy has been proven both in patients with cardiovascular and cerebrovascular disease in secondary prevention, and in high cardiovascular (CV) risk patients in primary prevention [2, 3]. Despite the widespread use of statin therapy, alone or in combination with other agents, many individuals do not achieve LDL-C goals, and the rates of CV events and mortality remain high [4]. Additional therapies, such as nicotinic acid, fenofibrate and ezetimibe, which also lower LDL-C, often serve as adjunctive therapies. The role of nicotinic acid and fenofibrates for the primary and secondary prevention of cardiovascular diseases is limited although these agents can improve a patient’s lipid profile, their effects on clinical outcomes are often neutral or restricted to subgroups of subjects [5, 6].
The IMPROVE-IT trial is scheduled for completion and data collection in late 2014 and will compare simvastatin monotherapy with simvastatin plus ezetimibe in high-risk patients after an acute coronary syndrome [7, 8]. This trial will provide the still lacking evidence regarding the efficacy and safety of this drug on top of statin therapy for reducing cardiovascular events in high-risk patients.
Among high-risk patients, particular care is required for subjects with Familial Hypercholesterolemia (FH). In fact, patients affected by this common genetic disorder (one in 500 individuals in the heterozygous form) characterized by elevation of serum LDL-C, develop CAD that usually becomes evident in the fourth or fifth decade of life [9].
Therefore, alternative therapies have been explored to lower LDL-C and to further reduce cardiovascular events in patients with established coronary disease or at risk for cardiovascular events.
Emerging Therapies
Recently, a number of new therapies have been developed that rely on novel mechanisms to further lower LDL-cholesterol in patients with high risk for CAD, including patients with inherited disorder of lipid metabolism (FH). These therapies include mipomersen, inhibitors of PCSK9 and cholesteryl ester transfer protein (CETP), as well as lomitapide, a microsomal triglyceride transfer protein (MTP) inhibitor.
Mipomersen
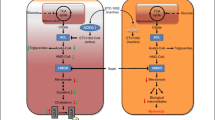
Apolipoprotein B-100 (apoB-100) is one of most important apolipoprotein and an essential component of all atherogenic lipoproteins (VLDL, LDL, intermediate-density lipoprotein (IDL), and Lp (a) [10, 11]. For this reason, the inhibition of apoB-100 synthesis is a promising way to decrease circulating levels of atherogenic lipoproteins.
The recent advances in the understanding of RNA biology have opened new opportunities in developing strategies directed to RNA targeting for therapeutic intervention.
Antisense oligonucleotides (ASOs) represent one of these promising therapeutic approaches. ASOs are short (usually 20 nucleotides in length), single stranded analogues of nucleic acids that can complementary bind directly to target mRNA promoting its degradation and in turn prevent translation [12].
Recent advances in the antisense technology have enabled design and synthesis of highly specific ASOs that are chemically modified to be stable against digestion by hydrolyzing enzymes [13].
Mipomersen (mipomersen sodium, Kynamro™, Genzyme, Cambridge, MA), developed under the name ISIS 301012, is a second-generation ASO complementary and specific to human apoB-100 mRNA.
After its administration by subcutaneous injection in a formulation with 0.9 % sodium chloride, mipomersen acts mainly in the liver, where it binds to APOB mRNA causing its cleavage by the action of RNase H and prevents the synthesis of apoB-100 protein [14].
Mipomersen in Clinical Trials
In Phase I trials efficacy and tolerability of mipomersen was tested in individuals with mild dyslipidemia [15].
In these trials mipomersen was administered at a weekly dose of 50 to 400 mg for four weeks and rapid and dose-dependent reductions in plasma apoB, LDL-C, total cholesterol, and triglycerides (47 %, 40 %, 41 %, and 63 %, respectively, with the highest dose of 400 mg/wk) was observed. The median dose (200 mg/wk) was also associated with substantial reductions in the above-mentioned lipid parameters (42 %, 27 %, 34 %, and 27 %, respectively). Notably, reduced plasma levels of apoB and LDL-C were sustained up to three months after administration of the last dose. Subsequently mipomersen has been evaluated in phase II and phase III trials in different settings of patients.
Akdim et al. [16] evaluated in a phase II trial the efficacy of subcutaneous administration (dose range, 50–400 mg/wk) over a 13-week treatment period in subjects with mild to moderate hyperlipidemia. The results showed dose-dependent and prolonged reductions of apoB (46 % to 61 %) and LDL-C (45 % to 61 %). In addition, a median reduction of 53 % of plasma trygliceride was also observed. In patients with Heterozygous Familial Hypercholesterolemia (HeFH) on statin therapy, mipomersen (50–300 mg/wk) resulted in a dose dependent 23 % to 33 % reduction in apoB and a concomitant 21 % to 34 % reduction in LDL-C. The effects were greater at the 200 and 300 mg/wk doses [16].
Consistently, Visser et al. [17] reported that mipomersen therapy (200 mg/wk) for 13 weeks in HeFH patients reduced apoB, LDL-C, and Lp(a) by 20 %, 22 %, and 20 %, respectively.
In two recently published Phase III trials in HeFH patients with coronary artery disease [18] and severe hyperlipidemia [19], treatment with mipomersen (200 mg/wk) was associated with significant decrements in plasma apoB (26 %–36 %), LDL-C (28 %–36 %), total cholesterol (19 %-28 %) and Lp(a) (21 % - 33 %). HDL-C concentrations were not affected in both trials.
Moreover, in statin-intolerant patients, including those with HeFH, mono-therapy with mipomersen demonstrated a significant reduction of LDL-C and apoB (47 % and 46 %, respectively) [20].
In the Phase III setting, Raal et al., assessed the efficacy and safety of 200 mg/week of mipomersen in patients with Homozygous Familial Hypercholesterolemia (HoFH) in a randomized, double blind placebo controlled trial [21••]. In the mipomersen group on maximum tolerated dose of a lipid-lowering drug, except apheresis, a 25 % mean reduction of LDL-C of was observed. However, there was considerable broad range of variability in observed LDL-C changes (- 2 % to - 82 %) that appeared to be independent of baseline LDL-C levels, age, race, or gender.
Finally, the first phase III study of mipomersen in non-FH patients with high cardiovascular risk due to prior CAD events and/or concurrent Type 2 Diabetes Mellitus (T2DM) demonstrates a significant reduction of LDL-C, apoB and Lp(a) in patients with hypercholesterolemia with, or at high risk for, coronary heart disease not controlled by existing therapies [22•].
An interesting finding from the studies with mipomersen is the consistent observation that overall there is a significant reduction of Lp(a) while triglycerides and HDL-cholesterol levels were not significantly affected.
Tolerability
Mipomersen neither is metabolized by CYP450 nor inhibits any of the major enzymes of this drug-metabolizing system (CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) [23]. In addition, mipomersen does not exhibit relevant pharmacokinetic interactions when coadministered with simvastatin or ezetimibe [23].
Phase II and Phase III studies have shown a satisfactory safety profile of mipomersen. The most common adverse event was the injection site reactions, seen in a substantial number of patients. The erythematous lesions occur within 24 hours of drug injection [15–20, 21••, 22•]. Although injection site reactions are not considered to be serious adverse events they may affect patient compliance. Flu-like symptoms have been observed in 30 % to 50 % of treated patients with mipomersen [16, 18, 19, 21••].
Finally one of the concerns is that in some individuals mipomersen increases hepatic fat [17, 18], and there are no data about the progression of hepatic steatosis to a more advanced chronic liver disease.
Fatty liver observed in patients treated with mipomersen could parallel the hepatic fat accumulation naturally occurring in familial hypobetalipoproteinemia (FHBL) due to mutations of the APOB gene in which the development of hepatic complications is rarely observed [24].
However the recent description of a family carrying a nonsense mutation of APOB gene causing FHBL with a massive history of severe steatosis associated with development of hepatocellular carcinoma in carriers of this mutation [25] may raise some concerns.
PCSK9 Inhibitors
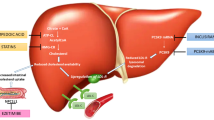
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a serine protease which is expressed in hepatocytes, kidney mesenchymal cells, intestine as well as in embryonic brain telencephalon neurons [26]. PCSK9 has a central role in regulation of cholesterol homeostasis by enhancing the endosomal and lysosomal degradation of hepatic low-density lipoprotein receptor (LDLR) [27].
Dominant gain of function mutations in the PCSK9 gene cause a phenotype similar to familial hypercholesterolemia (FH) [28], while loss of function variants are associated with hypocholesterolemia and protection against coronary artery disease [29–31].
After the observation by Dubuc et al., that PCSK9 expression is up-regulated in vitro in isolated hepatocytes treated with different statins [32], a similar significant increase in plasma PCSK9 has been described in both in mice [33] and in humans [34•] treated with statins. Statins, and also ezetimibe [27], increase simultaneously the cell membrane LDL- receptor numbers and also their PCSK9-mediated degradation.
It has been shown that these effects are mediated via the SREBP-2- related transcriptional activation that regulates both LDLR and PCSK9 [21••, 33].
These data support the rationale that a combination therapy with statins and a PCSK9 inhibitor could potentiate the statin cholesterol lowering efficacy.
Although several approaches to inhibit PCSK9 have been pursued for the treatment of hypercholesterolemia, to date the use of fully humanized human monoclonal antibodies (mAbs) targeting PCSK9 seems to be the more successful [34•].
The effective and sustained reduction of LDL-C mediated by the intravenous injection of fully humanized PCSK9 mAbs was first reported by two proof-of-concept studies in nonhuman primates [35, 36] and these results kicked off testing the efficacy in Phase I and Phase II trials in humans.
Clinical Trials with PCSK9 mAbs
The fully humanized monoclonal antibody SAR236553/REGN727 (Sanofi/Regeneron, Alirocumab) has been shown to reduce effectively LDL-C in healthy individuals (33-46 %) [37, 39] as well as in patients with HeFH and individuals with polygenic hypercholesterolemia in conjunction with statins (40-72 % and 38-66 %, respectively) [38, 40].
Moreover, treatment with SAR236553/REGN727 has been able to show a substantial reduction of apoB [38], Lp(a) [40] and triglycerides [38] of 56 %, 34 %, and 19 %, respectively, and a small increase of HDL-C concentrations (by a maximum of 8.5 %) [38].
Similar reductions in LDL-C have also been observed with AMG145 (Amgen), another fully humanized antibody (up to 64 % relative to placebo in healthy individuals) [41]. The administration of 140 mg of AMG145 antibody every two weeks reduced LDL-C by 51 % in patients with FH [42], while in patients treated with statins with or without ezetimibe, AMG145 decreased LDL-C levels in a dose dependent fashion by up to 66 % [43, 44].
Besides LDL-C reduction, AMG145 decreased Lp(a) concentrations by 9 % to 27 % and modestly elevated HDL-C by 5 % to 12 % [42–44].
Thus, both the SAR236553/REGN727 and AMG145 mAbs injected every two weeks appear to display similar efficacies in terms of LDL-C reduction both as monotherapy or in conjunction with statins. Moreover, anti-PSCK9 mAbs treatment resulted in the achievement of optimal target levels of LDL-C (<70 mg/dl) in the majority of the heterozygous familial hypercholesterolemia patients enrolled in these studies [37, 42].
Very recently, a pooled analysis of data from 1,359 patients in four phase II trials assessed the effects of AMG145 (evolocumab) on Lp(a) levels, the relationships between Lp(a) and lowering of LDL-C and apoB, and finally the influence of background statin therapy [45].
AMG145 treatment for 12 weeks resulted in significant mean dose-related reductions in Lp(a) of 29.5 % and 24.5 % with 140 mg and 420 mg every two and four weeks, respectively. Lipoprotein (a) reductions were significantly correlated with percentage reductions in LDL-C and apoB. Moreover, a trend toward a major reduction was observed in patients treated in combination with statins.
Finally, the TESLA proof-of-concept study (Trial Evaluating PCSK9 Antibody in Subjects With LDL Receptor Abnormalities) was designed to evaluate the safety, tolerability and efficacy of AMG 145 in a particular setting of subjects with HoFH in which it was unknown if PCSK9 inhibition could work [46].
Eight HoFH patients with null or defective mutations if the LDL-receptor were recruited and treated with AMG145 (420 mg every four weeks for ≥12 weeks, followed by every two weeks for an additional 12 weeks) [46].
After 12 weeks treatment, a 16.5 % of LDL-C mean reduction from baseline was observed in the treated patients. However the patients who experienced significant reduction of LDL-C (up to 43.6 % from baseline) were carriers of defective mutations in the LDLR gene while no significant changes from baseline were observed in HoFH patients carriers of null mutations (receptor negative patients) [46].
These results suggest for the first time that PCSK9 mAbs may be useful in the lowering LDL-C in HoFH carriers of defective mutations in LDLR gene.
Tolerability of PCSK9 mAbs
Phase I/II trials have shown that both REGN727 and AMG145 were well tolerated and not associated with serious adverse events.
The most frequent complaints were mild injection-site reactions. There have been only sporadic reports of increase of serum levels of creatininekinase [37], generalized pruritus due to hypersensitivity [39], and one case of leukocytoclastic vasculitis [38].
Although the efficacy and safety results of these studies are promising, the ongoing phase III PCSK9 mAbs clinical trials will provide more information on efficacy and safety including the risk of immune complex-mediated disorders related to the long-term administration of monoclonal antibodies.
Currently, a number of Phase III trials of PCSK9 mAbs are underway or were recently completed including two clinical outcomes trials, ODYSSEY Outcomes (NCT01663402) with REGN727 and FOURIER (NCT01764633) with AMG145.
Other Approaches to PCSK9 Inhibition
Along with mAbs, other approaches for PCSK9 inhibition are under consideration.
These include the use of mimetic peptides and ASOs. Mimetic peptides are structurally similar to the EGF-A domain of LDLR and compete with endogenous PCSK9 for receptor binding [47–50]. Other types of mimetic peptides are also under development, including truncated forms of PCSK9 that contain the receptor binding C-terminal domain but that lack the functional segment [51]. Data regarding the clinical efficacy and tolerability of these agents are not available. With ASOs, experimental findings have shown up-regulation of LDLR expression by 2 to 3 fold and a 40 % to 50 % reduction in LDL-C concentrations following treatment [52–54].
CETP Inhibitors
Low HDL cholesterol is a risk factor for CV disease [55•]. Nevertheless, the lack of specific HDL-cholesterol-raising drugs has prevented the possibility of proving that the CV risk can be reduced by increasing HDL cholesterol. HDLs are the main effectors of the reverse cholesterol transport (RCT), a physiological mechanism responsible for cholesterol removal from periphery (including atheroma) and its transport to the liver. Liver cholesterol is either excreted into bile or it is packed into nascent lipoproteins [56]. Cholesteryl ester transport protein (CETP), one of the key enzymes of RCT, was identified about fifteen years ago [57]. CETP transfers cholesteryl esters (CE) from HDLs to LDLs in exchange with triglycerides. LDL-CE is then removed by the blood stream by the LDL receptor mediated pathway. CETP deficiency increase HDL-C by inhibiting CETP function [58]. The first cases of CETP gene deficiency have been discovered in a cluster of Japanese subjects with very high HDL-C levels [57] but several CETP deficiency cases have been detected worldwide [59]. Survival CV curves according to plasma HDL-C levels showed that subjects with CETP deficiency are at CV risk [60], suggesting that CETP function is athero-protective. Based on the assumption that partial CETP inhibition could be beneficial in low HDL-C patients, drug companies have identified CETP as a suitable drug target. Up to date, four CETP inhibitors have been synthesized and tested in humans: torcetrapib, dalcetrapib, and more recently anacetrapib and evacetrapib.
Torcetrabib was the first compound to be subjected to preclinical studies. Torcetrapib is a potent CETP inhibitor able to increase HDL cholesterol in humans [61]. The largest trial with torcetrapib was the “Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events” (ILLUMINATE) trial. This trial included 15,067 subjects followed for 1.5 years [62]. Torcetrapib was administred on top of atorvastatin treatment and it was able to raise HDL-C by 72 % while LDL-C was reduced by 25 %. In spite of the improvement of the lipid profile, torcetrapib significantly increased the composite primary CV endpoint occurrence, i.e., CV-related death and hospitalization for CV events and these results caused the premature termination of the trial. The causes of the CV events excess were attributed mainly to the rise of blood pressure observed in the trial. The post-hoc analyses showed that torcetrapib caused a sustained increase in aldosterone levels [63], with negative effects on water and salt body balance and a consequential increase in blood pressure. The ILLUSTRATE trial [64] showed that atheroma extension was not affected by torcetrapib by intravascular ultrasonography (IVUS), confirming that torcetrapib is not beneficial in high risk CV patients.
Dalcetrapib was the second CETP inhibitor tested in humans. It is an irreversible low-potency CETP antagonist [65]. Tough dalcetrapib was not able to increase blood pressure or aldosterone levels [65], it failed to show improvement of the CV endpoint in the phase III trials. The Dal-OUTCOMES trial [66] included 15,981 patients with recent history of acute coronary syndrome. Dalcetrapib was administered against placebo resulting in a 40 % increase of HDL-C. However, no difference in the composite CV endpoint was observed over the 2.6 years mean observation period. Dal-PLAQUE [67] and Dal-VESSEL [68] failed to demonstrate respective improvements of atheroma volume investigated by MRI and surrogate markers of endothelial dysfunction as CRP, plasma selectins and adhesion molecules. In conclusion, the use of dalcetrabip does not seem beneficial in high CV risk patients.
Anacetrapib is a powerful CETP inhibitor, similar to torcetrapib. The effects of anacetrapib were tested in the “Determining the Efficacy and Tolerability of CETP Inhibition with Anacetrapib” (DEFINE) trial [69]. A total of 1,623 high CV risk patients on statin therapy were randomized to receive anacetrapib vs. placebo. The CV endpoint was a composite endpoint that included CV death and events, plus hospitalization for angina. Patients LDL-C levels at baseline were below 100 mg/dL on statin therapy. Anacetrabip determined a further 40 % LDL-C levels reduction and a 138 % increase of HDL-C levels over the 18 months observation period. Composite CV endpoint was not improved, but anacetrapib was shown not to be harmful to the patients. The trial was followed by a study evaluating the safety profile of the patients that discontinued anacetrapib after the DEFINE trial [70•]. Anacetrapib plasma concentration reached 40 % of the trial plasma levels after 12 weeks of drug discontinuation, while traces of anacetrapib were detected in the plasma after two years of discontinuation in a small cohort. Neither liver enzymes elevations nor significant adverse events were observed in the discontinuation period. The ambitious “ Randomized Evaluation of the Effects of Anacetrapib Through Lipid-modification” (REVEAL) trial will evaluate the effect of anacetrapib on hard CV endopoints, including CV death and events [71]. The investigators plan to enroll over 30,000 high CV risk subjects and to follow them for about five years.
Evacetrapid is the last CETP inhibitor to be developed. In a small trial including 156 subjects evacetrapib significantly decreased LDL-C and increased HDL-C without any increase of plasma aldosterone or blood pressure [72]. The “A Study of Evacetrapib in High-Risk Vascular Disease” (ACCELERATE) trial is to randomize more than 11,000 high CV-risk patients to receive evacetrapib vs. placebo [73]. The trial will show the effect of evecetrapib on hard CV endpoints (CV death and event, CAD hospitalization) after the study completion, expected in 2015.
MTP Inhibitors
Microsomal triglyceride transfer protein (MTP) is an intracellular lipid-transfer protein essential for the assembly and secretion of very-low-density lipoprotein (VLDL) particles in hepatocytes and chylomicron particles in enterocytes. MTP initiates the incorporation of lipids into apoB and acts as a chaperone to assist in the proper folding of the apoB protein [74]. The role of MTP in lipid transport and metabolism was revealed by studies demonstrating that a genetic defect in MTP gene causes abetalipoproteinemia, an autosomal recessive disorder characterized by lack of production of ApoB containing lipoproteins (chylomicrons and VLDL).
Studies demonstrating that defects in MTP cause abetalipoproteinemia suggested that inhibition of MTP could be a novel new mechanism to lower plasma lipid levels. One would predict that MTP inhibitors would be capable of inhibiting the production of both VLDL and chylomicrons.
Pharmacological inhibition of MTP represents a novel therapeutic strategy to treat severe forms of hyperlipidemia [75].
Research efforts led to the development of a compound named BMS-201038/AEGR-733, a synthetic small molecule benzimidazole-based analogue, that was recently approved by FDA and EMA (Juxtapid® in the US and Lojuxta® in the EU) as adjunctive treatment of Homozygous Familial Hypercholesterolemia.
Clinical Trials with Lomitapide
Lomitapide was administered alone or in combination with ezetimibe 10 mg daily in a phase II double-blind placebo-controlled trial in 85 hypercholesterolemic patients at moderate or high CV risk [76]. In patients who were treated with lomitapide alone, a significant reduction of LDL-C and apoB (30 % and 24 %, respectively) was observed at a dose of 10 mg/day. Combination therapy produced similar but larger dose-dependent decreases of LDL-C levels (46 % at 10 mg/day of both lomitapide and ezetimibe).
Further, the efficacy of lomitapide was tested in the severe forms of familial hypercholesterolemia (homozygous FH - HoFH).
HoFH is a rare inherited disease, which has a prevalence worldwide of 1: 1,000,000, though recent studies suggest that the prevalence of HoFH may be higher than previously thought [77, 78].
The biochemical phenotype of HoFH is expressed at birth and clinical manifestations of coronary heart disease due to chronic exposure (since birth) to very high cholesterol levels, occur in the 1st-2nd decade of life.
After a proof-of-concept phase II trial [79], lomitapide has been tested in a multinational single-arm, open-label, 78-week, phase 3 trial [80••].
Lomitapide effectively reduced mean plasma LDL-C levels by 50 % from baseline in 23 adults with HoFH over a 26 week treatment period and this reduction was sustained for an additional 52 weeks of lomitapide treatment.
The phase III trial also demonstrated that 46 % patients (six of 13) patients interrupted or reduced the frequency of apheresis treatments because of an important and stable reduction of LDL-C.
Tolerability of Lomitapide
Lomitapide Phase II studies [76, 79] have shown that a high dose of the drug was associated with severe and frequent hepatic and gastrointestinal side effects, both due to the drug mechanism of action. The Phase III trial results at the safety end point (week 78) indicate that overall lomitapide is safe and well tolerated. The adverse events (AE) were mainly gastrointestinal (GI) including nausea, heartburn, flatulence, diarrhea and fecal urgency. The majority of the patients in the efficacy phase experienced GI AEs of mild or moderate intensity. Interestingly, a decrease in the frequency and intensity of GI AEs was observed over time, maybe due to an intestinal adaptation to the drug or to a better self-management of the dietary fat intake. The transaminases (ALT and/or AST) levels in the majority of patients remained <2× ULN; only three patients had ALT and/or AST levels >5× ULN - ≤10 × ULN and one patient >10× ULN - ≤20 × ULN. No patients showed changes in bilirubin and alkaline phosphatase levels. Lomitapide, by inhibiting MTP, reduces the hepatic triglycerides export leading to fatty liver. In the Phase III trial, hepatic fat accumulation was monitored by nuclear magnetic resonance spectroscopy (NMRS). In the trial period, the hepatic fat content increased from 1 % at baseline to 8.6 % at end of the efficacy end point (week 26), remaining stable at the end of safety endpoint (week 78). During the entire trial only one patient showed a hepatic fat content >20 % to ≤25 % and two patients >25 %.
At this time, lomitapide is indicated only in adult patients with HoFH in addition to a low-fat diet, other lipid-lowering medications and with or without LDL-apheresis treatment. Treatment should be initiated and monitored by a physician experienced in the treatment of lipid disorders.
In order to minimize gastrointestinal adverse reactions associated with the use lomitapide, patients should follow a low fat diet (less than 20 % of energy from fat) and dietary counseling should be provided.
It is reasonable to expect that in the future an increasing number of HoFH patients will be treated with lomitapide and that more evidence on long-term safety will be available. Evidence is needed on the possible benefits of an early start of the lomitapide treatment in HoFH pediatric patients. The ultimate long-term challenge for lomitapide will be to show a reduction of cardiovascular events and mortality in HoFH [81].
Conclusions
We have reviewed the clinical and safety data regarding new therapies for lowering plasma LDL-C. levels. In Table 1 the main characteristics of the reviewed compounds are summarized. All of the described agents produce potent reductions in plasma LDL-C and apoB on top of statin and/or other lipid lowering therapies in high-risk individuals with hypercholesterolemia, and in FH patients.
At this time Mipomersen and Lomitapide are indicated only for HoFH and in the future could beneficially affect the natural history of this severe and rare disease.
Anti-PCSK9 mAbs show a good efficacy associated with an excellent safety profile; and this could warrant a wider employment in the future in different categories of patients at high risk for CVD as patients with: a- LDL-C levels not at target on maximal doses of statins; b-high residual risk on statins; c- intolerance to statins; and d- familial hypercholesterolemia.
The results of ongoing trials with CETP inhibitors may warrant and support their use especially when statins are not tolerated or statin therapy fails to achieve adequate reductions in LDL-cholesterol level, or for patients with low HDL-cholesterol and high triglyceride levels.
Overall, longer term efficacy, tolerability and impact of all these compounds on reducing cardiovascular events are currently being investigated in large-scale end point trials and all end-points need to be fulfilled.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Go AS, Mozaffariam D, Roger VL, on behalf of the American Heart Association Statistics Committee and Stroke Statistics subcommittee, et al. Heart disease and stroke statistics: 2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–245.
Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002;106:3143–421.
Smith Jr SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation. Circulation. 2011;124:2458–73.
Waters DD, Brotons C, Chiang CW, et al. Lipid treatment assessment project 2: a multinational survey to evaluate the proportion of patients achieving low-density lipoprotein cholesterol goals. Circulation. 2009;120:28–34.
The AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67.
The ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–74.
Cannon CP, Giugliano RP, Blazing MA, et al. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimbe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J. 2008;156:826–32.
Califf RM, Lokhnygina Y, Cannon CP, et al. An update on the IMProved reduction of outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) design. Am Heart J. 2010;159:705–9.
Bertolini S, Pisciotta L, Rabacchi C, et al. Spectrum of mutations and phenotypic expression in patients with autosomal dominant hypercholesterolemia identified in Italy. Atherosclerosis. 2013;227(2):342–8.
Packard CJ, Demant T, Stewart JP, et al. Apolipoprotein B metabolism and the distribution of VLDL and LDL subfractions. J Lipid Res. 2000;41:305–18.
Elovson J, Chatterton JE, Bell GT, et al. Plasma very low density lipoproteins contain a single molecule of apolipoprotein B. J Lipid Res. 1998;29:1461–73.
Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–93.
Kurreck J. Antisense technologies. Improvement through novel chemical modifications. Eur J Biochem. 2003;270:1628–44.
Thomas T, Ginsberg H. Targeting ApoB as a therapeutic approach for the treatment of dyslipidemia: the potential role of mipomersen. Clin Lipidol. 2010;5:457–64.
Kastelein JJ, Wedel MK, Baker BF, et al. Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation. 2006;114(16):1729–35.
Akdim F, Stroes ES, Sijbrands EJ, et al. Efficacy and safety of mipomersen, an antisense inhibitor of apolipoprotein B, in hypercholesterolemic subjects receiving stable statin therapy. J Am Coll Cardiol. 2010;55:1611–8.
Visser ME, Akdim F, Tribble DL, et al. Effect of apolipoprotein-B synthesis inhibition on liver triglyceride content in patients with familial hypercholesterolemia. J Lipid Res. 2010;51:1057–62.
Stein E, Dufour R, Gagne C, et al. A randomized, double-blind, placebo-controlled study to assess efficacy and safety of mipomersen as add-on therapy in heterozygous familial hypercholesterolemia patients with coronary artery disease. Eur Heart J. 2010;31:S898.
Tardif JC, Mcgowan M, Ceska R, et al. Apolipoprotein B synthesis inhibition by mipomersen reduces LDL-C when added to maximally tolerated lipid-lowering medication in patients with severe heterozygous hypercholesterolemia. J Am Coll Cardiol. 2011;57:E492.
Visser ME, Wagener G, Baker BF, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, lowers low-density lipoprotein cholesterol in high-risk statin-intolerant patients: a randomized, double-blind, placebo-controlled trial. Eur Heart J. 2012;33(9):1142–9.
Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375(9719):998–1006. This paper demonstrated that mipomersen is effective in reducing LDL-C levels in homozygous familial Hypercholesterolemia.
Thomas GS, Cromwell WC, Ali S, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2013;62(23):2178–84. First phase III study testing mipomersen in patients at high cardiovascular risk.
Yu RZ, Geary RS, Flaim JD, et al. Lack of pharmacokinetic interaction of mipomersen sodium (ISIS 301012), a2’-O-methoxyethyl modified antisense oligonucleotide targeting apolipoproteinB-100 messenger RNA, with simvastatin and ezetimibe. Clin Pharmacokinet. 2009;48:39–50.
Tarugi P, Averna M, Di Leo E, et al. Molecular diagnosis of hypobetalipoproteinemia: an ENID review. Atherosclerosis. 2007;195:e19–27.
Cefalù AB, Pirruccello JP, Noto D, et al. A novel APOB mutation identified by exome sequencing cosegregates with steatosis, liver cancer, and hypocholesterolemia. Arterioscler Thromb Vasc Biol. 2013;33(8):2021–5.
Seidah NG, Benjannet S, Wickham L, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A. 2003;100:928–33.
Lambert G, Sjouke B, Choque B, et al. The PCSK9 decade. J Lipid Res. 2012;53(12):2515–24.
Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154–6.
Fasano T, Cefalù AB, Di Leo E, et al. A novel loss of function mutation of PCSK9 gene in white subjects with low-plasma low-density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol. 2007;27(3):677–81.
Cohen JC, Boerwinkle E, Mosley Jr TH, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–72.
Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79(3):514–23.
Dubuc G, Chamberland A, Wassef H, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–9.
Rashid S, Curtis DE, Garuti R, et al. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking PCSK9. Proc Natl Acad Sci U S A. 2005;102:5374–9.
Catapano AL, Papadopoulos N. The safety of therapeutic monoclonal antibodies: Implications for cardiovascular disease and targeting thePCSK9 pathway. Atherosclerosis. 2013;228:18–28. This is a comprehensive review on PCSK9 function and therapeutic opportunities.
Chan JC, Piper DE, Cao Q, et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and non human primates. Proc Natl Acad Sci U S A. 2009;106:9820–5.
Ni YG, Di MS, Condra JH, et al. A PCSK9-binding antibody that structurally mimics the EGF(A) domain of LDL-receptor reduces LDL cholesterol in vivo. J Lipid Res. 2011;52:78–86.
Stein EA, Mellis S, Yancopoulos GD, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366(12):1108–18.
McKenney JM, Koren MJ, Kereiakes DJ, et al. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59(25):2344–53.
Stein EA, Gipe D, Bergeron J, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380(9836):29–36.
Roth EM, McKenney JM, Hanotin C, et al. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367(20):1891–900.
Dias CS, Shaywitz AJ, Wasserman SM, et al. Effects of AMG 145 on low density lipoprotein cholesterol levels: results from 2 randomized, double-blind, placebo-controlled, ascending-dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. J Am Coll Cardiol. 2012;60(19):1888–98.
Koren MJ, Scott R, Kim JB, et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): a randomised, double-blind, placebo-controlled, phase 2 study. Lancet. 2012;380(9858):1995–2006.
Raal F, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol lowering effects of AMG145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation. 2012;126(20):2408–17.
Giugliano RP, Desai NR, Kohli P, et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet. 2012;380(9858):2007–17.
Raal FJ, Giugliano RP, Sabatine MS, Koren MJ, Langslet G, Bays H, et al. Reduction in lipoprotein(a) with the PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of over 1300 patients in 4 phase 2 trials. J Am Coll Cardiol. 2014. doi:10.1016/j.jacc.2014.01.006.
Stein EA, Honarpour N, Wasserman SM, et al. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128(19):2113–20.
Shan L, Pang L, Zhang R, et al. PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem Biophys Res Commun. 2008;375:69–73.
Mc Nutt MC, Kwon HJ, Chen C, et al. Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J Biol Chem. 2009;284:10561–70.
Schroeder CI, Swedberg JE, Withka JM, et al. Design and synthesis of truncated EGF-A peptides that restore LDL-R recycling in the presence of PCSK9 in vitro. Chem Biol. 2014. doi:10.1016/j.chembiol.2013.11.014.
Zhang Y, Eigenbrot C, Zhou L, et al. Identification of a small peptide that inhibits PCSK9 protein binding to the low density lipoprotein receptor. J Biol Chem. 2014;289(2):942–55.
Du F, Hui Y, Zhang M, et al. Novel domain interaction regulates secretion of proprotein convertase subtilisin/kexin type 9 (PCSK9) protein. J Biol Chem. 2011;286:43054–61.
Graham MJ, Lemonidis KM, Whipple CP, et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res. 2007;48:763–7.
Gupta N, Fisker N, Asselin MC, et al. A locked nucleic acid antisense oligonucleotide (LNA) silences PCSK9 and enhances LDLR expression in vitro and in vivo. PLoS One. 2010;5:10682. doi:10.1371/journal.pone.0010682.
Lindholm MW, Elmen J, Fisker N, et al. PCSK9 LNA antisense oligonucleotides induce sustained reduction of LDL cholesterol in nonhuman primates. Mol Ther. 2012;20:376–81.
Barter P. HDL-C: role as a risk modifier. Atheroscler Suppl. 2011;12(3):267–70. This is a review of evidence of the role of HDL-C on cardiovascular risk.
Klerkx AH, El Harchaoui K, van der Steeg WA, et al. Cholesteryl ester transfer protein (CETP) inhibition beyond raising high-density lipoprotein cholesterol levels: pathways by which modulation of CETP activity may alter atherogenesis. Arterioscler Thromb Vasc Biol. 2006;26(4):706–15.
Brown ML, Inazu A, Hesler CB, et al. Molecular basis of lipid transfer protein deficiency in a family with increased high-density lipoproteins. Nature. 1989;342(6248):448–51.
Kurasawa T, Yokoyama S, Miyake, et al. Rate of cholesteryl ester transfer between high and low density lipoproteins in human serum and a case with decreased transfer rate in association with hyperalphalipoproteinemia. J Biochem. 1985;98:1499–508.
Cefalù AB, Noto D, Magnolo L, et al. Novel mutations of CETP gene in Italian subjects with hyperalphalipoproteinemia. Atherosclerosis. 2009;204(1):202–7.
Zhong S, Sharp DS, Grove JS, et al. Increased coronary heart disease in Japanese-American men with mutation in the cholesteryl ester transfer protein gene despite increased HDL levels. J Clin Invest. 1996;97:2917–23.
Brousseau ME, Schaefer EJ, Wolfe ML, et al. Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. N Engl J Med. 2004;350(15):1505–15.
Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–22.
Clerc RG, Stauffer A, Weibel F, et al. Mechanisms underlying off-target effects of the cholesteryl ester transfer protein inhibitor torcetrapib involve L-type calcium channels. J Hypertens. 2010;28(8):1676–86.
Nissen SE, Tardif JC, Nicholls SJ, et al. Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. 2007;356(13):1304–16.
Stein EA, Roth EM, Rhyne JM, et al. Safety and tolerability of dalcetrapib (RO4607381/JTT-705): results from a 48-week trial. Eur Heart J. 2010;31(4):480–8.
Schwartz GG, Olsson AG, Abt M, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367(22):2089–99.
Fayad ZA, Mani V, Woodward M, et al. Safety and efficacy of dalcetrapib on atherosclerotic disease using novel non-invasive multimodality imaging (dal-PLAQUE): a randomised clinical trial. Lancet. 2011;378(9802):1547–59.
Lüscher TF, Taddei S, Kaski JC, et al. Vascular effects and safety of dalcetrapib in patients with or at risk of coronary heart disease: the dal-VESSEL randomized clinical trial. Eur Heart J. 2012;33(7):857–65.
Cannon CP, Shah S, Dansky HM, et al. Determining the Efficacy and Tolerability Investigators. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363(25):2406–15.
Gotto Jr AM, Cannon CP, Li XS, et al. Evaluation of lipids, drug concentration, and safety parameters following cessation of treatment with the cholesteryl ester transfer protein inhibitor anacetrapib in patients with or at high risk for coronary heart disease. Am J Cardiol. 2014;113(1):76–83. This paper confirms that anacetrapib is effective in increasing HDL-C and safe for patients at high-risk.
ClinicalTrials.gov. 2013. REVEAL: Randomized Evaluation of the Effects of Anacetrapib through Lipid- Modification. http://clinicaltrials.gov/show/NCT01252953.
Nicholls SJ, Brewer HB, Kastelein JJ, et al. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: a randomized controlled trial. JAMA. 2011;306(19):2099–109.
ClinicalTrials.gov. 2013. ACCELERATE: A Study of Evacetrapib in High-Risk Vascular Disease. http://www.clinicaltrials.gov/ct2/show/NCT01687998
Raabe M, Véniant MM, Sullivan MA, et al. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J Clin Invest. 1999;103(9):1287–98.
Gordon DA, Jamil H. Progress towards understanding the role of microsomal triglyceride transfer protein in apolipoprotein-B lipoprotein assembly. Biochim Biophys Acta. 2000;1486(1):72–83.
Samaha FF, McKenney J, Bloedon LT, et al. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2008;5(8):497–505.
Benn M, Watts GF, Tybjaerg-Hansen A, et al. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab. 2012;97(11):3956–64.
Nordestgaard BG, Chapman MJ, Humphries SE, for the European Atherosclerosis Society Consensus Panel, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478–90a.
Cuchel M, Bloedon LT, Szapary PO, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356(2):148–56.
Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381(9860):40–6. This phase III trial demonstrates safety and efficacy of lomitapide in homozygous FH patients.
Aegerion Pharmaceuticals Inc. US prescribing information for JuxtapidTM (lomitapide). 2013. http://www.juxtapid.com/_pdf/Prescribing_Information.pdf.
Compliance with Ethics Guidelines
Conflict of Interest
Maurizio Averna, Angelo B. Cefalù, and Davide Noto have served as clinical investigators in several hypolipidemic drug trials for Aegerion Merck, Pfizer, Sanofi, and Astra Zeneca. MR Averna is a member of Aegerion Europe and Italy Advisory Boards. The authors have no other relevant affiliations and financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or material discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Cardiovascular Disease and Stroke
Rights and permissions
About this article

Cite this article
Noto, D., Cefalù, A.B. & Averna, M.R. Beyond Statins: New Lipid Lowering Strategies to Reduce Cardiovascular Risk. Curr Atheroscler Rep 16, 414 (2014). https://doi.org/10.1007/s11883-014-0414-4
Published:
DOI: https://doi.org/10.1007/s11883-014-0414-4