
Opinion statement
Chimeric antigen receptor (CAR) T-cell therapy has become the standard of care for children and young adults with relapsed and refractory B-cell acute lymphoblastic leukemia (B-ALL), and it is a highly promising therapy under investigation for adults with relapsed disease. Despite having potentially life-threatening toxicities, such as cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome, the benefits of CAR T-cell therapy far outweigh these risks, particularly as increased experience and improved supportive care measures are mitigating these toxicities. CAR T cells can result in complete remission for significant proportion of patients with relapsed and refractory B-ALL and permit them to proceed to potentially curative allogeneic hematopoietic stem cell transplantation (allo-HSCT). CAR T cells may also be curative by themselves. Herein lie the greatest challenges and questions for clinical investigators, specifically, how are CAR T cells best employed and how do we overcome mechanisms of resistance to them? The primary clinical question is the timing and even the necessity of allo-HSCT. Relative to resistance, we know that target antigen loss, specifically CD19, is a major contributor to resistance. However, current investigations of alternative targets, such CD22, and CAR T cells expressing dual targeting antigen receptors have demonstrated encouraging initial results and provide a high degree of optimism that the efficacy and the broader application of CAR T-cell therapy will gradually increase in B-ALL. That optimism is not as high and the challenges are increased for the application of CAR T cells in T-cell leukemias and acute myeloid leukemia due to the relative lack of suitable leukemia surface targets that are not also expressed on normal hematopoietic progenitors. Despite these significant challenges, considerable research is being conducted into the development of CAR T cells for these diseases utilizing unique technologies, which may be applicable to other diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Leukemia is the most common oncologic diagnosis in pediatric patients and remains a significant cause of mortality in adult patients, particularly within the young adult population [1]. Although overall survival rates for acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) have surpassed 90% and 70%, respectively, even with the use of risk-stratified chemotherapy regimens, children with relapsed or refractory disease continue to have poor outcomes [2, 3]. Similarly, the survival rates of adults with leukemia are suboptimal, ranging from 40 to 50%, with increasingly dismal outcomes for those patients with relapsed disease [4, 5]. Over the past several years, the urgent need for novel treatments in these high-risk populations has led to the development of immunotherapies including chimeric antigen receptor (CAR) T-cell therapy. Anti-CD19 CAR T cells have revolutionized the treatment of B-cell malignancies, and ongoing studies seek to expand the application of CAR T-cell therapy by identifying new targets, improving efficacy, and reducing toxicity. We aimed to review the current status of therapeutic CAR T-cell options for both pediatric and adult patients with leukemia and identify treatment challenges associated with manufacturing and toxicities.
CAR T-cell therapy for acute lymphoblastic leukemia
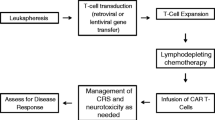
Early clinical trials of anti-CD19 CAR T-cell therapy in B-cell ALL (B-ALL) demonstrated outstanding response rates, with complete remissions noted in up to 90% of patients and 6-month event-free survival up to 70% [6,7,8,9]. These studies were among the first to comprehensively report the efficacy of second-generation CAR T cells manufactured with co-stimulatory domains from either the 4-1BB [6••, 8] or CD28 [7, 9••]. These studies were also the first to define diagnostic criteria for post-CAR T-cell infusion toxicities, including cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). The efforts of these studies, and in particular the ELIANA and JULIET trials which evaluated the efficacy and safety profile of anti-CD19 CAR T-cell therapy in children and young adults with relapsed or refractory (r/r) B-ALL and in adults with r/r diffuse large B-cell lymphoma (DLBCL), respectively, led to the first FDA-approved CAR T-cell product, tisagenlecleucel [10].
Since US Food and Drug Administration (FDA) approval of tisagenlecleucel in August 2017 for patients up to 25 years of age with B-ALL that is refractory or in second or later relapse, the Center for International Blood and Marrow Transplant Research and others have evaluated its “real-world” safety and efficacy in patients compared to those treated on clinical trials. Over 500 patients have been treated with the FDA-approved product for both B-ALL and r/r DLBCL, and data show that response rates and CRS and ICANS incidences are equivalent to patients treated on study. Notably, real-world use has expanded tisagenlecleucel access to pediatric patients less than 3 years of age, raised the median adult age by nearly 10 years, and led to less restrictive use of CAR T-cell therapy with regard to those with decreased organ function or lack of prior hematopoietic stem cell transplantation (HSCT) [11,12,13, 14•]. Additionally, more patients have been treated with tisagenlecleucel considered to be out of specification (OOS) per clinical trial standards due to lower viability, total nucleated cell count, or time from collection to infusion, and studies indicate that both safety and efficacy remain largely unaffected by OOS use [14•, 15]. Such conclusions have recently been extended by the Pediatric Real World Consortium to include evaluation of 38 patients who received tisagenlecleucel via the Managed Access Program, which provides tisagenlecleucel to patients with an initial product that is OOS and for whom leukapheresis is not feasible, as well as patients who received it as a single-patient Investigational New Drug application [15]. Neither the efficacy nor incidences of CRS or ICANS in these patients were different than those who received an in-specification product.
While the FDA approval of tisagenlecleucel has made CAR T-cell therapy more widely available to patients with B-ALL, ongoing clinical trials continue to investigate different variables affecting outcomes, including CAR T cell construct, T cell origins, other target antigens, and presence or site of disease burden (Table 1). Studies have demonstrated that CAR constructs with 4-1BB co-stimulatory domains lead to improved persistence of CAR T cells, while constructs with CD28 co-stimulation result in rapid proliferation and early anti-leukemia effects post-infusion [16, 17]. Additionally, a recent meta-analysis of clinical trials utilizing anti-CD19 CAR T cells suggests that products with 4-1BB co-stimulatory constructs may lead to improved minimal residual disease (MRD) negativity when compared to products with CD28 co-stimulation, but that progression-free survival rates at 1 year do not differ between constructs [18]. This analysis also noted superior CR rates in patients treated with autologous CAR T cells compared to those who received CAR T-cell products made from allogeneic T cells. However, the relatively low number of studies utilizing allogeneic CAR T cells makes it difficult to draw definitive conclusions with regard to efficacy. While commercialization has allowed for the delivery of tisagenlecleucel without disease-specific clinical study limitations, it remains unclear if the amount of disease or the site of disease affects overall efficacy. A retrospective analysis of 200 patients who received tisagenlecleucel off-study suggests that patients with documented high disease burden (> 5% bone marrow lymphoblasts, peripheral blood lymphoblasts, CNS3 status, or non-CNS extramedullary disease) have lower CR rates early post-CAR T infusion and lower overall survival at 6 and 12 months compared to those patients with low disease burden [19]. Additional studies are currently underway to determine how the anatomical site, molecular characteristics of disease, and timing of CAR T-cell infusion during upfront treatment impact survival following anti-CD19 CAR T-cell treatment (Table 1).
Despite the outstanding response rates in B-ALL patients treated with anti-CD19 CAR T-cell therapy, relapses can still occur in over 60% of patients within the first 12 months post-infusion [9, 20]. Up to 40% of these relapses are CD19-negative with associated immune evasion due to antigen loss or decreased antigen expression post-infusion [7,8,9, 21,22,23]. Whether this is from escape variants of CD19-negative cells with other oncogenic mutations (e.g., TP53), outgrowth of preexisting CD19-negative clones, or from induced CD19 acquired mutations is an area of active investigation. This clinical scenario has led to the pursuit of alternate targets, such as CD22, as well as bivalent CAR T cells. As with CD19, the majority of B-cell lymphoblasts express CD22, making it a promising target [24]. A recent phase 1 study of children and young adults treated with a CD22-targeted CAR T-cell product resulted in potent anti-leukemic effects in patients with CD19dim or CD19− relapse, even though the majority had already been treated with previous anti-CD19 CAR T-cell therapy [25]. Complete remissions were seen in 70% of patients, and the median relapse-free survival was 6 months [25, 26]. However, relapses post-CD22-CAR T cells were associated with decreased CD22 expression, again raising concerns for immune evasion and long-term efficacy of monovalent CAR therapy. Recent studies have used dual expressing CAR T cells targeting both CD19 and CD22, hypothesizing that a bivalent product could reduce relapses in which antigen downregulation or loss occurs. Anti-leukemic activity and CR rates with this construct have been comparable to monovalent CAR T-cell therapy, and patients experiencing relapse have blasts that retain antigen positivity, indicating that poor CAR T cell persistence, rather than immune evasion, may be the cause of disease recurrence in these patients [27, 28]. Similar studies using bispecific CAR T-cell therapy against B-ALL are ongoing and will require long-term follow-up to determine the efficacy of these products as compared to monovalent CAR T cells.
T-cell leukemias, while not as common as B-cell leukemias, are seen in both pediatric and adult populations and have limited treatment options for relapsed disease [29, 30]. The urgent need for novel therapeutics for these diseases has raised the question of translating CAR T-cell therapy to a treatment modality for r/r T-cell malignancies. However, many challenges arose during pre-clinical studies. Selecting an antigen with minimal off-target expression remains a limiting factor in the development of immunotherapies for this disease entity, as many T-lymphoblast antigens are also found on healthy T cells, making prevention of long-term T-cell lymphopenia challenging [31]. Sparing normal T cells is essential, as a resulting T-cell aplasia would be detrimental to full immune reconstitution and prevention of infections, leading to life-threatening immune deficiency. Additionally, CAR T cells and normal T cells share common antigens, which can lead to product contamination with leukemic cells, as well as CAR T fratricide [32, 33]. Early phase trials extended from patients with T-cell lymphomas are targeting antigens expressed predominantly on malignant cells, including CD30, TRBC1, and CCR4 [34,35,36,37]. Other open trials are evaluating outcomes in patients treated with CAR T cells directed at pan-T-cell antigens, such as CD5 and CD7. Pre-clinical studies have shown that anti-CD5 CAR T cells can downregulate their own CD5 expression, thus limiting fratricide, with off-target toxicity that is restricted to a minor population of immune cells including thymocytes and peripheral T cells [38]. Anti-CD7 CAR T cells can be engineered to prevent self-destruction by genomic disruption of the CD7 gene, thus allowing for robust expansion. Although CD7 is expressed on healthy T cells, the CAR T cells themselves appear to retain antiviral activity [39]. Other such examples are increasingly being tested such as CAR T cells directed to CD1a, which is restricted to developing cortical thymocytes along with T-ALL blasts, have shown promising pre-clinical results against patient-derived primary T-ALL blasts and because CD1a is not expressed on mature T cells, appear to be fratricide resistant [33]. Lastly, donor-derived T cells would represent an ideal T cell source for CAR T-cell therapy, such as those from patients following allogeneic HSCT (allo-HSCT) or through the use of “off the shelf” allogeneic-tolerant cells. Preliminary results have shown potential using genetically edited fratricide-resistant allogeneic CAR T cells directed against human T-ALL [40, 41]. Results of these phase 1 clinical trials and long-term follow-up of patients will be critical for determining outcomes and the effects on T-cell-mediated immunity.
CAR T-cell therapy for acute myeloid leukemia
Relapsed and refractory AML remains a difficult clinical scenario. Allo-HSCT, which preferentially requires that patients are in remission, is the standard of care and only potentially curative option for this group of patients, highlighting the need for additional therapeutics. The development of CAR T-cell therapy for use in AML faces many of the same challenges as T-ALL, if not more, particularly due to the lack of an AML-specific targetable antigen not expressed on normal myeloid progenitors. Currently, there are several clinical trials studying the safety and efficacy of CAR T cells targeting CD33, CLL-1, or CD123 antigens expressed on most myeloblasts. CD33 has proved an effective target for immunotherapy against AML, perhaps highlighted best with the FDA approval gemtuzumab ozogamicin, an anti-CD33 antibody-drug conjugate [42]. Propelled by these results, pre-clinical studies have showed potent anti-leukemic effects of 3 different anti-CD33 CAR T cells using differing constructs [43]. Early reports have shown responses in 2 patients, one of whom had a CR with the use of a dual anti-CD33/CLL-1 CAR T-cell product prior to proceeding to transplant [44, 45]. Another potential AML-targeted antigen, CD123, while expressed by normal hematopoietic cells and endothelial cells, is also overexpressed on leukemic myeloblasts in comparison to healthy myeloid cells. Pre-clinical data supporting in vivo anti-leukemic activity of anti-CD123 CAR T cells led to a first-in-human clinical trial using second-generation CAR T-cells with a CD28 co-stimulatory domain [46]. Early results showed upfront responses in 5 out of 6 patients with refractory AML following allo-HSCT, 3 of which experienced CR [47]. In an effort to mitigate long-term severe myelosuppression, another trial utilized “biodegradable” anti-CD123 CAR T-cells that were manufactured using T cells that were electroporated with anti-CD123 CAR mRNA and administered as multiple infusions in patients with r/r AML. While the off-target myelosuppressive effects were minimal, the lack of product expansion provided a poor anti-leukemic effect, and the trial was halted due to lack of efficacy [48]. However, the positive safety profile of this product supported the development of a more traditional lentiviral transduction approaches using anti-CD123 CAR T cells with either 4-1BB or CD28 co-stimulatory domains [49]. These and additional clinical studies using CD33 and CD123-specific CAR T cells are ongoing (Table 2).
In the race to manufacture an effective and minimally toxic CAR T-cell therapy for AML, the primary limiting factor remains lack of a leukemia-specific antigen. Unlike anti-CD19 CAR-T therapy, which results in a relatively benign and manageable toxicity of B-cell aplasia, the destruction of healthy myeloid precursors could lead to profound neutropenia, anemia, and thrombocytopenia. One strategy to mitigate these potential toxicities, while still allowing for expansion and an anti-leukemic effect, is to incorporate a “safety switch.” An example of this is a current phase 1 study is using anti-CD19 CAR T cells that co-express EGFRt, which can be targeted with the EGFR-specific monoclonal antibody, cetuximab [50]. Other approaches that have been used in CAR T cells targeting B-cell antigens include the use of suicide genes such as iCasp9, HSV-TK, and TMPK, but this technology has yet to be translated over to anti-AML CAR T cells [51, 52]. Despite these mechanisms for reducing off-target toxicity and other barriers that limit the use of CAR T cells against AML such as its inherent heterogeneity and immunosuppressive microenvironment, a continued search for tumor-specific antigens remains of utmost importance.
Treatment challenges
An expanding myriad of challenges have emerged regarding the use of CAR T-cell therapies against leukemias as the use of CAR T cells against B-ALL has expanded through clinical studies and real-world experiences. We highlight some of these challenges and current exploration of solutions to address them below.
Challenges in manufacturing CAR T cells
Collection of adequate T cells from heavily pre-treated patients continues to be a barrier to the production of CAR T cells. The exact number of peripheral lymphocytes (absolute lymphocyte count; ALC) and CD3+ T cells needed for in-specification expansion remains unclear, as most patients have an ALC that is skewed towards the normal range [53]. Experience of Shah and colleagues at the National Cancer Institute suggests that collection of patients with CD3+ cell counts as low as 150 cells/μL is feasible for generating CAR T-cell products [26]. Expanding CAR T cells has also proven difficult from certain patients. Chemotherapies commonly used to treat leukemias, such as clofarabine, doxorubicin, cyclophosphamide, and cytarabine, appear to lead to poor-quality CAR T cells and selective reduction of early lineage T cells in the product [54, 55]. In fact, the optimal ratio of CD4+ and CD8+ T cells and their developmental lineage remain questions that are currently under investigation. Expansion protocols, timing, and cytokines used for production also appear to alter the potency of anti-leukemic CAR T cells. For instance, early work found that anti-CD19 CAR T cells made from patients with CD19+ malignancies were preferentially expanded using a combination of IL-7 and IL-15 rather than IL-2 [56]. This and other work has also shown that anti-CD19-CAR T cells derived from long-lived, self-renewing, multipotent T memory stem cells (TSCM) mediate superior anti-leukemic responses compared to those generated from more terminally differentiated T cells [57, 58•]. Continual optimization of tisagenlecleucel production from clinical trials to real-world experiences highlights the importance of these issues and how such production challenges are likely to be different and must be tailored for CAR T cells directed at dissimilar leukemic targets [59].
Mechanisms of resistance to CAR T-cell therapy
Most data regarding CAR T-cell resistance come from patients treated with anti-CD19 CAR T cells against B-ALL and r/r DLBCL. There are two patterns of post-CAR T-cell relapse, antigen positive (i.e., CD19+) or antigen negative (i.e., CD19−). The key mechanisms leading to antigen positive leukemic relapse lies in poor in vivo persistence and limited potency of CAR T cells. There are many putative mechanisms for such impotent CAR T-cell products, some of which have been discussed above. They range from the co-stimulatory domain used, with that from 4-1BB showing greater persistence than those from CD28, and starting T cell phenotype and exhaustion characteristics [16, 17, 60•]. Long-term follow-up analysis of patients treated with anti-CD19 CAR T cells for B-ALL also suggests that children and young adults have longer event-free survival than adult patients, implying that T cell age may play a role [20]. There are now a number of pre-clinical and early phase clinical trials testing the ex vivo addition of small molecules targeting T cell signal transduction pathways, such as BTK and PI3K, to alter differentiation, exhaustion, and metabolic status of CAR T cells against ALL and AML [61,62,63].
Approximately 10–20% of patients with B-ALL will experience CD19-negative relapse following anti-CD19 CAR T treatment [64]. There are various known mechanisms responsible for B-ALL antigen loss following CAR T-cell therapy including, most commonly, CD19 gene mutation and RNA splice variants [23, 65]. Interestingly, in contrast to CD19, downregulation of CD22 appears to be a major mechanism of escape following anti-CD22 CAR T-cell therapy [25]. As detailed above, in an effort to overcome such antigen loss, dual expressing anti-CD19/CD22 CAR T cells have been developed against B-ALL. However, recent data suggest that anti-CD22 CAR T cells induce both CD19 and CD22 downregulation and that a method to combat such generalized immune evasion may be through concomitant treatment with epigenetic modifying agents [66]. Heterogeneity of CD19 and CD22 expression on leukemic cell surface exists pre-CAR therapy as well and likely impacts responses to such targeted therapies and is responsible for outgrowth of antigen dim/loss minor leukemic populations [67]. Lastly, genetic instability from leukemic driver mutations also leads to antigen downregulation following mono- or dual-specific CAR T-cell treatment, as was recently detected in TP53-mutated B-ALL in children treated with anti-CD19 CAR T cells followed by either allo-HSCT or anti-CD22 CAR T cells [68]. It is all but certain that we will discover additional mechanisms of resistance in the future following CAR T-cell therapy against different leukemic targets given what we have learned regarding those responsible for anti-CD19 and anti-CD22 CAR T-cell failure.
Toxicities associated with CAR T-cell therapy
A number of significant toxicities have been associated with CAR T-cell therapy for leukemias. Early toxicities include CRS, ICANS, and hemophagocytic lymphohistiocytosis and toxicities directly associated with lymphodepleting chemotherapy [69]. Later toxicities include prolonged cytopenias and hypogammaglobulinemia. The incidence, onset, severity, and duration of these toxicities are dependent on the target antigen, the CAR construct, the specific disease being treated, and disease burden. The incidence and severity of both CRS and ICANS have been higher in anti-CD19 CAR T-cell therapy for B-ALL, as compared with other diseases.
CRS is the most common toxicity reported with CAR T-cell trials for leukemia. Pathophysiologically, CRS results from high levels of inflammatory cytokines (e.g., IL-6 and IFN-γ) following T-cell activation and proliferation. Clinically, it is manifested by development of fever, headaches, myalgias, hypotension, and hypoxia. The reported overall and ≥ Grade 3 incidences of CRS in the ELIANA trial were 77% and 47%, respectively [9••]. In adult B-ALL trials, the incidences of overall and ≥ Grade 3 CRS have been 85–93% and 26–93%, respectively [20, 70]. A high leukemic burden, higher doses of CAR T cells, and CAR constructs containing a CD28 co-stimulatory domain have been associated with an increased risk and severity of CRS. In early CAR T-cell trials, CRS was primarily managed with tocilizumab, as corticosteroids were generally avoided over concerns that they may impede CAR T-cell efficacy. However, subsequent studies demonstrated that these concerns were relatively unwarranted, and corticosteroids are utilized earlier in the course of CRS, particularly if there is lack of early response to tocilizumab. There is generally complete resolution of CRS symptoms, and it is rare for CRS symptoms to persist beyond the first month after CAR T-cell infusion.
The second most common complication of CAR T-cell therapy for leukemia is ICANS, which presents clinically as confusion, delirium, expressive aphasia, weakness, tremor, headache, seizures, altered level of consciousness, and the most serious manifestation, cerebral edema. Cerebral edema can occur relatively suddenly and has been lethal in most cases. ICANS generally does not occur without CRS preceding it, and increased severity of CRS correlates with the risk of developing ICANS. Although generally occurring shortly after CRS and resolving in most cases within 2–4 weeks, ICANS can occur beyond a month after infusion and can persist for several weeks to months. Corticosteroids have been successfully used for the management of ICANS.
Early and late infections have been observed in a significant proportion of leukemia patients treated with CAR T-cell therapy. In the ELIANA trial alone, 43% of patients experienced an infection including 24% experiencing ≥ Grade 3 infections. Early infections can be partially attributed to the immuno- and myelosuppressive effects of lymphodepleting chemotherapy given prior to CAR T-cell infusion, resulting primarily in bacterial infections. Late infections, primarily viral and including CMV, have been attributed to delayed T- and B-cell recovery. The latter is a result of ongoing effects against cells expressing CD19, resulting in clinically significant hypogammaglobulinemia. Bacterial, fungal, and viral prophylaxis has become standard, and intravenous immunoglobulins are commonly employed in patients with hypogammaglobulinemia and recurrent infections. With growing experience, the toxicities associated with CAR T-cell therapy have increasingly become manageable; however, they can be life-threatening and require careful monitoring, adequate supportive care measures, and clinical expertise.
A bridge to transplant or stand-alone therapy?
Despite relatively high remission rates in patients with r/r B-ALL following anti-CD19 CAR T-cell therapy, relapses remain common. In fact, up to 50% of patients will eventually relapse, most within the first 12 months following infusion [9••, 20]. This begs the question if long-term survival of patients with leukemia would be improved with post-CAR consolidation with allo-HSCT or followed by treatment with alternatively specific CAR T cells. While allo-HSCT is the only established curative treatment for aggressive leukemias to date, there is no consensus on its role following CAR T-cell therapy, and its use as a consolidative treatment remains institutionally specific and may differ between pediatric and adult patients and dependent upon the CAR T-cell product. Ten percent of patients with negative MRD in the initial phase 1 clinical trial of the anti-CD19/4-1BB CAR T product underwent allo-HSCT, and in the extended ELIANA trial, this rose to 14% [9••, 71]. However, in another phase 1 study using a different anti-CD19/4-1BB product, median duration of B-cell aplasia was shorter, and 28% of patients underwent allo-HSCT [53]. Consistent with CD28-based anti-CD19 CAR T cells, two phase 1 studies using such products found relatively shorter periods of B-cell aplasia and thereby a larger percentage of patients underwent allo-HSCT, 75% and 83%, respectively [8, 72, 73]. Published phase 1 data in adults is somewhat similar with 39% of patients infused with an anti-CD19/CD28 product and 45% of those infused with an anti-CD19/4-1BB product having undergone allo-HSCT following CAR T-cell infusion [20, 74]. Not surprisingly, one observation from these trials is that patients with shorter periods of B-cell aplasia (~3–6 months) generally do better following allo-HSCT than those who do not undergo transplant. Because shorter periods of B-cell aplasia are found following CD28-based anti-CD19 CAR T-cells, some investigators have proposed that such products should be used to place patients in deep remission prior to allo-HSCT. Conversely, there exists some controversy on what to do following infusion of 4-1BB-based anti-CD19 CAR T-cells and when to “pull the trigger” on allo-HSCT based on surveillance bone marrow detection of hematogones or peripheral B-cell recovery. There currently are no risk stratifications or timeline to B-cell recovery guidelines for pediatric or adult patients who may benefit from allo-HSCT. Until these are determined, the decision to use CAR T cells as a bridge to transplant will remain patient specific and institutional. Regardless of CAR T-cell product, risk stratification should be based upon if patients have a prior history of allo-HSCT, the biology of their disease, their age, and if future CAR T-cell products that target different leukemic antigens show efficacy post-initial CAR T cell failure. Firm establishment of disease risk factors and outcomes of ongoing clinical trials and evidence provided by real-world data will be key to answering these questions moving forward.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. https://doi.org/10.3322/caac.21551.
Alexander TB, Wang L, Inaba H, Triplett BM, Pounds S, Ribeiro RC, et al. Decreased relapsed rate and treatment-related mortality contribute to improved outcomes for pediatric acute myeloid leukemia in successive clinical trials. Cancer. 2017;123(19):3791–8. https://doi.org/10.1002/cncr.30791.
Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373(16):1541–52. https://doi.org/10.1056/NEJMra1400972.
Pulte D, Jansen L, Castro FA, Brenner H. Changes in the survival of older patients with hematologic malignancies in the early 21st century. Cancer. 2016;122(13):2031–40. https://doi.org/10.1002/cncr.30003.
Tamamyan G, Kadia T, Ravandi F, Borthakur G, Cortes J, Jabbour E, et al. Frontline treatment of acute myeloid leukemia in adults. Crit Rev Oncol Hematol. 2017;110:20–34. https://doi.org/10.1016/j.critrevonc.2016.12.004.
•• Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. https://doi.org/10.1126/scitranslmed.3008226 This study was the first report of efficacy in 16 patients with r/r B-ALL treated with a CAR T cell product using the CD28 co-stimulatory domain and led the way for a larger multicenter phase II trial using the same product. This study also was one for the fist to define dignostic criteria for severe CRS.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–18. https://doi.org/10.1056/NEJMoa1215134.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28. https://doi.org/10.1016/S0140-6736(14)61403-3.
•• Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48. https://doi.org/10.1056/NEJMoa1709866 This study reported the results from the ELIANA trial, a phase 2, single-cohort, multicenter global study on the efficacy of tisagenlecleucel in 75 patients with r/r B-ALL.
O’Leary MC, Lu X, Huang Y, Lin X, Mahmood I, Przepiorka D, et al. FDA approval summary: tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin Cancer Res. 2019;25(4):1142–6. https://doi.org/10.1158/1078-0432.CCR-18-2035.
Cahill KE, Leukam MJ, Riedell PA. Refining patient selection for CAR T-cell therapy in aggressive large B-cell lymphoma. Leuk Lymphoma. 2020;61(4):799–807. https://doi.org/10.1080/10428194.2019.1691201.
Grupp S, Hu Z-H, Zhang Y, Keating A, Pulsipher MA, Philips C, et al. Tisagenlecleucel chimeric antigen receptor (CAR) T-cell therapy for relapsed/refractory children and young adults with acute lymphoblastic leukemia (ALL): real world experience from the Center for International Blood and Marrow Transplant Research (CIBMTR) and cellular therapy (CT) registry. Blood. 2019;134(Supplement_1):2619. https://doi.org/10.1182/blood-2019-129279.
Jaglowski S, Hu Z-H, Zhang Y, Kamdar M, Ghosh M, Lulla P, et al. Tisagenlecleucel chimeric antigen receptor (CAR) T-cell therapy for adults with diffuse large B-cell lymphoma (DLBCL): real world experience from the Center for International Blood and Marrow Transplant Research (CIBMTR) cellular therapy (CT) registry. Blood. 2019;134(Supplement_1):766. https://doi.org/10.1182/blood-2019-130983.
• Pasquini MC, Hu ZH, Curran K, Laetsch T, Locke F, Rouce R, et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 2020;4(21):5414–24. https://doi.org/10.1182/bloodadvances.2020003092 This study reports initial experience with tisagenlecleucel in a real-world setting and describes remission rates, survival outcomes, toxicities, and differences between in- and out-of-specification products.
Rossoff J, Baggott C, Prabhu S, Pacenta H, Phillips CL, Stefanski H, et al. Real-world treatment of pediatric patients with relapsed/refractory B-cell acute lymphoblastic leukemia using tisagenlecleucel that is out of specification for commercial release. Blood. 2020;136(Supplement 1):42–4. https://doi.org/10.1182/blood-2020-136674.
Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–90. https://doi.org/10.1038/nm.3838.
Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415–28. https://doi.org/10.1016/j.ccell.2015.09.004.
Anagnostou T, Riaz IB, Hashmi SK, Murad MH, Kenderian SS. Anti-CD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: a systematic review and meta-analysis. Lancet Haematol. 2020;7(11):e816–e26. https://doi.org/10.1016/S2352-3026(20)30277-5.
Schultz LM, Baggott C, Prabhu S, Pacenta H, Phillips CL, Rossoff J, et al. Disease burden impacts outcomes in pediatric and young adult B-cell acute lymphoblastic leukemia after commercial tisagenlecleucel: results from the pediatric real world CAR consortium (PRWCC). Blood. 2020;136(Supplement 1):14–5. https://doi.org/10.1182/blood-2020-134472.
Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–59. https://doi.org/10.1056/NEJMoa1709919.
Fousek K, Watanabe J, Joseph SK, George A, An X, Byrd TT, et al. CAR T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia. 2021;35(1):75–89. https://doi.org/10.1038/s41375-020-0792-2.
Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127(20):2406–10. https://doi.org/10.1182/blood-2015-08-665547.
Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282–95. https://doi.org/10.1158/2159-8290.CD-15-1020.
Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121(7):1165–74. https://doi.org/10.1182/blood-2012-06-438002.
Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–8. https://doi.org/10.1038/nm.4441.
Shah NN, Highfill SL, Shalabi H, Yates B, Jin J, Wolters PL, et al. CD4/CD8 T-cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: updated results from a phase I anti-CD22 CAR T-cell trial. J Clin Oncol. 2020;38(17):1938–50. https://doi.org/10.1200/JCO.19.03279.
Schultz LM, Muffly LS, Spiegel JY, Ramakrishna S, Hossain N, Baggott C, et al. Phase I trial using CD19/CD22 bispecific CAR T cells in pediatric and adult acute lymphoblastic leukemia (ALL). Blood. 2019;134(Supplement_1):744. https://doi.org/10.1182/blood-2019-129411.
Shalabi H, Yates B, Shahani S, Qin H, HIghfill SL, Panch S, et al. Abstract CT051: safety and efficacy of CD19/CD22 CAR T cells in children and young adults with relapsed/refractory ALL. Cancer Res. 2020;80(16 Supplement):CT051-CT. https://doi.org/10.1158/1538-7445.Am2020-ct051.
Litzow MR, Ferrando AA. How I treat T-cell acute lymphoblastic leukemia in adults. Blood. 2015;126(7):833–41. https://doi.org/10.1182/blood-2014-10-551895.
McMahon CM, Luger SM. Relapsed T cell ALL: current approaches and new directions. Curr Hematol Malig Rep. 2019;14(2):83–93. https://doi.org/10.1007/s11899-019-00501-3.
Fleischer LC, Spencer HT, Raikar SS. Targeting T cell malignancies using CAR-based immunotherapy: challenges and potential solutions. J Hematol Oncol. 2019;12(1):141. https://doi.org/10.1186/s13045-019-0801-y.
Cooper ML, DiPersio JF. Chimeric antigen receptor T cells (CAR-T) for the treatment of T-cell malignancies. Best Pract Res Clin Haematol. 2019;32(4):101097. https://doi.org/10.1016/j.beha.2019.101097.
Sanchez-Martinez D, Baroni ML, Gutierrez-Aguera F, Roca-Ho H, Blanch-Lombarte O, Gonzalez-Garcia S, et al. Fratricide-resistant CD1a-specific CAR T cells for the treatment of cortical T-cell acute lymphoblastic leukemia. Blood. 2019;133(21):2291–304. https://doi.org/10.1182/blood-2018-10-882944.
Maciocia PM, Wawrzyniecka PA, Philip B, Ricciardelli I, Akarca AU, Onuoha SC, et al. Targeting the T cell receptor beta-chain constant region for immunotherapy of T cell malignancies. Nat Med. 2017;23(12):1416–23. https://doi.org/10.1038/nm.4444.
Perera LP, Zhang M, Nakagawa M, Petrus MN, Maeda M, Kadin ME, et al. Chimeric antigen receptor modified T cells that target chemokine receptor CCR4 as a therapeutic modality for T-cell malignancies. Am J Hematol. 2017;92(9):892–901. https://doi.org/10.1002/ajh.24794.
Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest. 2017;127(9):3462–71. https://doi.org/10.1172/JCI94306.
Scherer LD, Brenner MK, Mamonkin M. Chimeric antigen receptors for T-cell malignancies. Front Oncol. 2019;9:126. https://doi.org/10.3389/fonc.2019.00126.
Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126(8):983–92. https://doi.org/10.1182/blood-2015-02-629527.
Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130(3):285–96. https://doi.org/10.1182/blood-2017-01-761320.
Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32(9):1970–83. https://doi.org/10.1038/s41375-018-0065-5.
Rasaiyaah J, Georgiadis C, Preece R, Mock U, Qasim W. TCRalphabeta/CD3 disruption enables CD3-specific antileukemic T cell immunotherapy. JCI Insight. 2018;3(13). https://doi.org/10.1172/jci.insight.99442.
Appelbaum FR, Bernstein ID. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood. 2017;130(22):2373–6. https://doi.org/10.1182/blood-2017-09-797712.
Li S, Tao Z, Xu Y, Liu J, An N, Wang Y, et al. CD33-specific chimeric antigen receptor T cells with different co-stimulators showed potent anti-leukemia efficacy and different phenotype. Hum Gene Ther. 2018;29(5):626–39. https://doi.org/10.1089/hum.2017.241.
Liu F, Cao Y, Pinz K, Ma Y, Wada M, Chen K, et al. First-in-human CLL1-CD33 compound CAR T cell therapy induces complete remission in patients with refractory acute myeloid leukemia: update on phase 1 clinical trial. Blood. 2018;132(Supplement 1):901. https://doi.org/10.1182/blood-2018-99-110579.
Wang L, Zhang Y, Anderson E, Kumar R, Lamble AJ, Orentas RJ. CD22 CAR-T induces both CD19 and CD22 surface down-modulation: defining a mechanism of generalized immune evasion and the effects of epigenetic modifiers. Blood. 2020;136(Supplement 1):22–3. https://doi.org/10.1182/blood-2020-134930.
Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang X, Budde LE, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122(18):3138–48. https://doi.org/10.1182/blood-2012-12-474056.
Budde L, Song JY, Kim Y, Blanchard S, Wagner J, Stein AS, et al. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: a first-in-human clinical trial. Blood. 2017;130(Supplement 1):811. https://doi.org/10.1182/blood.V130.Suppl_1.811.811.
Cummins KD, Frey N, Nelson AM, Schmidt A, Luger S, Isaacs RE, et al. Treating relapsed/refractory (RR) AML with biodegradable anti-CD123 CAR modified T cells. Blood. 2017;130(Supplement 1):1359. https://doi.org/10.1182/blood.V130.Suppl_1.1359.1359.
Mardiana S, Gill S. CAR T cells for acute myeloid leukemia: state of the art and future directions. Front Oncol. 2020;10:697. https://doi.org/10.3389/fonc.2020.00697.
Paszkiewicz PJ, Frassle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest. 2016;126(11):4262–72. https://doi.org/10.1172/JCI84813.
Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, et al. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol Ther. 2017;25(3):580–92. https://doi.org/10.1016/j.ymthe.2017.01.011.
Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105(11):4247–54. https://doi.org/10.1182/blood-2004-11-4564.
Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322–31. https://doi.org/10.1182/blood-2017-02-769208.
Das RK, Storm J, Barrett DM. Abstract 1631: T cell dysfunction in pediatric cancer patients at diagnosis and after chemotherapy can limit chimeric antigen receptor potential. Cancer Res. 2018;78(13 Supplement):1631. https://doi.org/10.1158/1538-7445.Am2018-1631.
Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med. 2016;8(320):320ra3. https://doi.org/10.1126/scitranslmed.aad5222.
Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR. CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123(24):3750–9. https://doi.org/10.1182/blood-2014-01-552174.
Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. 2017;23(1):18–27. https://doi.org/10.1038/nm.4241.
• Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128(4):519–28. https://doi.org/10.1182/blood-2015-11-683847 This pre-clinical study demonstrated that CD19-CAR T cells generated from modified multipotent T memory stem cells (TSCM) had superior anti-tumor responses compared to conventionally generated, and clinically used, CD19-CAR T cells.
Tyagarajan S, Spencer T, Smith J. Optimizing CAR-T cell manufacturing processes during pivotal clinical trials. Mol Ther Methods Clin Dev. 2020;16:136–44. https://doi.org/10.1016/j.omtm.2019.11.018.
• Gardner R, Finney O, Brakke H, Rhea S, Hicks R, Doolittle D, et al. Starting T cell and cell product phenotype are associated with durable remission of leukemia following CD19 CAR-T cell immunotherapy. Blood. 2018;132(Supplement 1):4022. https://doi.org/10.1182/blood-2018-99-117493 Analysis of products from the initial phase 1 anti-CD19 CAR T cell trail found that long-term CAR T cell efficacy could be predicted by the starting phenotype and functional characteristics of the initial T cell product.
Fan F, Yoo HJ, Stock S, Wang L, Liu Y, Schubert ML, et al. Ibrutinib for improved chimeric antigen receptor T-cell production for chronic lymphocytic leukemia patients. Int J Cancer. 2021;148(2):419–28. https://doi.org/10.1002/ijc.33212.
Ruella M, Kenderian SS, Shestova O, Fraietta JA, Qayyum S, Zhang Q, et al. The addition of the BTK inhibitor ibrutinib to anti-CD19 chimeric antigen receptor T cells (CART19) improves responses against mantle cell lymphoma. Clin Cancer Res. 2016;22(11):2684–96. https://doi.org/10.1158/1078-0432.CCR-15-1527.
Zheng W, O’Hear CE, Alli R, Basham JH, Abdelsamed HA, Palmer LE, et al. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia. 2018;32(5):1157–67. https://doi.org/10.1038/s41375-017-0008-6.
Xu X, Sun Q, Liang X, Chen Z, Zhang X, Zhou X, et al. Mechanisms of relapse after CD19 CAR T-cell therapy for acute lymphoblastic leukemia and its prevention and treatment strategies. Front Immunol. 2019;10:2664. https://doi.org/10.3389/fimmu.2019.02664.
Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24(10):1504–6. https://doi.org/10.1038/s41591-018-0146-z.
Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015;23(1):184–91. https://doi.org/10.1038/mt.2014.164.
Rosenthal J, Naqvi AS, Luo M, Wertheim G, Paessler M, Thomas-Tikhonenko A, et al. Heterogeneity of surface CD19 and CD22 expression in B lymphoblastic leukemia. Am J Hematol. 2018;93(11):E352–E5. https://doi.org/10.1002/ajh.25235.
Pan J, Tan Y, Deng B, Tong C, Hua L, Ling Z, et al. Frequent occurrence of CD19-negative relapse after CD19 CAR T and consolidation therapy in 14 TP53-mutated r/r B-ALL children. Leukemia. 2020;34(12):3382–7. https://doi.org/10.1038/s41375-020-0831-z.
Maus MV, Alexander S, Bishop MR, Brudno JN, Callahan C, Davila ML, et al. Society for immunotherapy of cancer (SITC) clinical practice guideline on immune effector cell-related adverse events. J Immunother Cancer. 2020;8(2). https://doi.org/10.1136/jitc-2020-001511.
Shah BD, Bishop MR, Oluwole OO, Logan A, Baer MR, Donnellan WB, et al. End of phase I results of ZUMA-3, a phase 1/2 study of KTE-X19, anti-CD19 chimeric antigen receptor (CAR) T cell therapy, in adult patients (pts) with relapsed/refractory (R/R) acute lymphoblastic leukemia (ALL). J Clin Oncol. 2019;37(15_suppl):7006. https://doi.org/10.1200/JCO.2019.37.15_suppl.7006.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17. https://doi.org/10.1056/NEJMoa1407222.
Curran KJ, Margossian SP, Kernan NA, Silverman LB, Williams DA, Shukla N, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood. 2019;134(26):2361–8. https://doi.org/10.1182/blood.2019001641.
Lee DW III, Stetler-Stevenson M, Yuan CM, Shah NN, Delbrook C, Yates B, et al. Long-term outcomes following CD19 CAR T cell therapy for B-ALL are superior in patients receiving a fludarabine/cyclophosphamide preparative regimen and post-CAR hematopoietic stem cell transplantation. Blood. 2016;128(22):218. https://doi.org/10.1182/blood.V128.22.218.218.
Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–38. https://doi.org/10.1172/JCI85309.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Katherine Harris declares that she has no conflict of interest. James L. LaBelle declares that he has no conflict of interest. Michael R. Bishop has received research funding from Novartis, Kite Pharma/Gilead, and CRISPR Therapeutics and has received compensation for service as a consultant from Novartis, Kite Pharma/Gilead, Bristol-Myers Squibb/Juno Therapeutics, and Autolus Therapeutics.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Leukemia
Rights and permissions
About this article

Cite this article
Harris, K., LaBelle, J.L. & Bishop, M.R. Current Status of CAR T Cell Therapy for Leukemias. Curr. Treat. Options in Oncol. 22, 62 (2021). https://doi.org/10.1007/s11864-021-00859-8
Accepted:
Published:
DOI: https://doi.org/10.1007/s11864-021-00859-8