
Abstract
Rigid polyurethane foam is a widely used insulation material in various industrial fields. Enhancing its insulating performance often involves controlling pore size to reduce radiative conductivity and, in some cases, decrease gas thermal conductivity through the Knudsen effect. While numerous studies have addressed reducing the pore size of thermoplastic polymer foam to the nano-scale, limited research has focused on rigid polyurethane foam. Although some studies have shown that pore size changes with the addition of nucleating agents, a comprehensive understanding of the mechanisms governing pore size remains elusive. Therefore, this study investigates the factors determining the pore size of rigid polyurethane foam. Our findings confirm that the size of air bubbles formed during the blending of prepolymers has the most significant impact on pore size. Furthermore, we demonstrate that regulating the size of air bubbles before the urethane reaction allows for control over the final pore size. Experimental details substantiating these findings are presented.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polymeric foams, recognized as quintessential insulation materials [1,2,3,4], exhibit superior mechanical properties despite their exceptionally low density. Their cost-effectiveness and ease of mass production have led to widespread use in various industrial applications [5,6,7,8,9]. The design of polymeric foam emphasizes achieving high insulation performance by increasing the gas fraction within the polymer matrix, which boasts lower thermal conductivity compared to metals or ceramics. However, a higher gas ratio leads to thinner polymer walls, compromising the foam’s mechanical integrity [10, 11]. Consequently, adjusting insulation performance through foam density entails an inevitable trade-off between insulation effectiveness and mechanical strength.
To optimize thermal insulation without significant degradation in mechanical properties, it is essential to fix the final density and adjust the thermal conductivity of the polymer matrix or the gas phase. The thermal conductivity of polymeric foam encompasses solid conduction, gas conduction, and radiative conduction [12]. With the typically high gas fraction, gas-phase conductivity has the greatest influence, increasing with decreasing foam density. Efforts to find gases with low thermal conductivity face limitations due to environmental regulations, making traditional choices like chlorofluorocarbon (CFC) or hydrogenated chlorofluorocarbon (HCFC) gases unsuitable for industrial use.
In response to these challenges, researchers have explored altering foam structure rather than gas composition. One strategy involves reducing pore size, which has two major thermal conductivity reduction effects. Firstly, it diminishes radiative conductivity [13, 14], dependent on cell wall thickness. Smaller cell sizes result in thicker walls, reducing radiative conduction [15]. Secondly, it invokes the Knudsen effect, where gas molecules collide with cell walls more frequently, decreasing gas-phase thermal conductivity [16]. While attempts to leverage the Knudsen effect have succeeded in thermoplastic polymeric foams, research on cross-linked thermoset foams is notably lacking.
For many thermoplastic polymeric foams, pore size reduction has been achieved, enabling the Knudsen effect [16, 17]. Typically, a high concentration of CO2 is used as a physical blowing agent by dissolving it [13,14,15,16,17,18,19,20,21,22,23,24]. This process results in polymeric foam with pores of several microns or nanometer scales, exhibiting the Knudsen effect. However, studies on reducing thermal conductivity through the Knudsen effect are limited to thermoplastic foams [17, 25,26,27,28,29,30,31], and to our knowledge, no research has been conducted on cross-linked thermoset foams. Rigid polyurethane foam, synthesized by combining polyol and polymeric isocyanate molecules, stands out as a representative thermoset polymer foam [32, 33]. While many studies have investigated the thermal conductivity of rigid polyurethane foam [34, 35], there is a dearth of research on controlling pore size. Most studies focus on pore sizes above 100 µm [8, 15, 36,37,38,39], and the mechanism determining pore size in rigid polyurethane foam remains unclear.
This study addresses the gap by describing a strategy to control pore size in rigid polyurethane foams. We first identify the mechanism determining pore size and then propose methods to reduce it. Due to the stiff mechanical properties of cross-linked polyurethane [40,41,42,43], volume expansion is almost impossible after the urethane reaction. The pore size is determined during the mixing of liquid reactants, polyol and polymer isocyanate, before solidification. Unlike traditional thermoplastic polymeric foams, rigid polyurethane foams experience non-classical nucleation due to air bubbles in the polyol. [36, 37] Additionally, it has been found that pore size can be controlled by changing the size of the air bubbles, although there is a lower limit that makes broad control challenging.
Materials and Methods
Rigid Polyurethane Foam Synthesis
The process for synthesizing rigid polyurethane foam is illustrated in Fig. 1. Polyurethane was synthesized using polyol and polymeric MDI (Methyl diphenyl diisocyanate), both supplied by KPX Chemical. Mixtures of polyols with different structures and varying numbers of hydroxyl functional groups were employed. The mixture system consists of 50% ester polyol and 50% ether polyol, with an average functionality of about 2.7. A polymeric MDI has the same average functionality as the polyol. The catalyst, Dimethyl cyclohexylamine (Polycat 8, Evonik), was used for the urethane reaction.

Scheme for the fabrication of a rigid polyurethane foam. a Chemical components used to synthesize rigid polyurethane foam. Both polyol and polymeric MDI have identical functionality. b By mixing polyol and MDI with a small amount of blowing agent, a rigid polyurethane foam can be synthesized
Rigid polyurethane foam was produced within a paper cup container. First, polyol, catalyst, and blowing agent (water) were mixed; then, a secondary mix was created by adding p-MDI. Finally, pour the mixture into a paper cup and wait for the urethane reaction to complete. The mixing procedures were carried out at 15,000 rpm with a homogenizer (PT 1300 E, KINEMATICA) following the established protocol. With a catalyst amount of 0.5%, the volume expansion ceased approximately 10 min after the second mix. Morphological analysis of the foam produced was conducted after aging for two hours.
Analysis of Foam Pore Size and Density
Pore size analysis of the rigid polyurethane foam was conducted using SEM imaging (SU 8230, HITACHI). In the foam manufacturing process, temperature differences between the core and the edges can lead to the formation of cells of different sizes, with the hotter core typically producing slightly larger cells. Therefore, only the central part was selected for SEM imaging. For the analysis of foam density, the mass of a 20 \(\times \) 20 \(\times \) 20 mm cube shape was measured. A sample was taken from the center, as the morphology at the edges may differ from that at the center.
Rheological Modification of the Polyol
Hydrophobic fumed silica particles (HDK H18, Wacker) were incorporated to regulate the rheological properties of the polyol. A suspension was formed by adding colloidal particles to the polyol at a concentration of 1 ~ 5% (g/100 g polyol) and stirring it at 70 °C for 1 h. The rheological properties of the silica suspension were measured using a rheometer (MCR302, Anton Paar) with a 25 mm parallel plate geometry. Viscosity was measured across a range of shear rates from 0.1 to 100 s⁻1. Modulus measurements were also taken using both a frequency sweep and a strain amplitude sweep. In the frequency sweep, the strain was fixed at 0.1%, and the frequency was varied from 100 to 0.1 rad/s. In the strain amplitude sweep, the frequency was fixed at 10 rad/s, and the strain was varied from 0.1 to 100%.
Interfacial Tension Measurement
The surface tension of the polyol and Novec oils (3 M) at the air interface, as well as the interfacial tension between the polyol and Novec oils, were measured using a custom-built pendant drop apparatus [44]. Polyol or Novec oils were suspended at the needle tip for surface tension measurements. For interfacial tension measurements, Novec oils were suspended as droplets at the needle tip in the polyol bath. All measurements were conducted at room temperature.
Eliminating Air Bubbles in Polyol
A significant number of air bubbles are trapped during the process of mixing high-viscosity polyol with a blowing agent and MDI. The addition of a co-solvent capable of dissolving both polyol and MDI can significantly reduce their viscosity and eliminate any trapped air bubbles. Moreover, polyol can be uniformly mixed with MDI without requiring additional mixing processes. Co-solvents such as acetone (ACS reagent, Sigma-Aldrich), diethyl ether (ACS reagent, Sigma-Aldrich), benzene (ACS reagent, Sigma-Aldrich), and dichloromethane (suitable for HPLC, Sigma-Aldrich) can be used to dissolve both polyol and MDI, resulting in a single phase. This allows for the simultaneous evaporation of the co-solvent and the urethane reaction over time.
To expedite solvent removal, we employed a method that selectively extracts only the co-solvent by introducing an additional solvent that is miscible with the co-solvent but immiscible with polyol and MDI. n-Hexane (Reagent Plus, Sigma-Aldrich) was used for extraction, given its low solubility for polyol and MDI but high miscibility with most co-solvents. Upon the addition of hexane to the co-solvent in which the polyol and MDI are dissolved, they separate into two phases. The relatively dense bottom phase contains polyol and MDI along with a small amount of co-solvent. The supernatant, containing a mixture of hexane and a significant amount of co-solvent, can be easily removed. The polyurethane foam with pores is formed by the evaporation of the remaining co-solvent in the bottom phase.
Result and Discussion
Influence of the Reaction Kinetics
The production of rigid polyurethane foam is a system in which the urethane reaction and blowing occur simultaneously, thus rendering kinetic influences on the reaction impossible to exclude. The rate of urethane reaction can be controlled by several variables, including temperature and catalyst. The higher the temperature, the faster the reaction, and the higher the amount of catalyst, the faster the reaction. However, since the urethane reaction is exothermic, the faster the reaction, the higher the temperature tends to be. Consequently, given the difficulty of maintaining a constant temperature during the reaction, it is preferable to control the amount of catalyst in order to quantitatively compare the effects of reaction speed in this study.
The foam was produced by varying the amount of dimethyl cyclohexylamine catalyst between 0.013 and 1%. The average cell density of the foam was obtained by dividing the average volume of the measured pores by the density of the foam. Figure 2 below illustrates the relative number of cells at 0.013% catalyst. It can be observed that the greater the catalyst concentration, the greater the number of cells within the foam, given the same mass of foam. This phenomenon is likely due to the fact that as the reaction rate increases, the walls harden rapidly, preventing the pores from escaping or merging with the surrounding pores.

Relative number of cells of rigid polyurethane foams with different amount of DMCHA catalyst. The cell density of the foam with a catalyst fraction of 0.013% was employed as reference
The reaction rate also affects the pore size, but the pore size remains above 100 µm even when the amount of catalyst is increased sufficiently. A simple calculation demonstrates that for the same amount of foaming agent to reduce the cell size by a factor of 10, the number of cells would need to be 1000 times larger, which is unlikely to be achieved by increasing the catalyst alone. Ultimately, it is our contention that other variables, in addition to reaction kinetics, must be considered, particularly a more precise comprehension of the cell formation process.
Change in Pore Size with Varying Amounts of Blowing Agent
The investigation focused on assessing the impact of varying amounts of blowing agent on the morphology of PU foam. The quantity of blowing agent employed emerges as the primary factor influencing the foam’s morphology, given that the blowing agent occupies the pores of the PU foam. To elucidate the blowing agent's impact on the foam’s structure, the pore size of rigid PU foam was measured while systematically reducing the blowing agent amount from 4 to 0.1%. Water served as the chemical blowing agent, with a 0.5% catalyst concentration. As depicted in Fig. 3(a), the pore size of the PU foam exhibits a proportional decrease with the decreasing amount of blowing agent. However, when the blowing agent amount falls below 0.5%, further reduction ceases, and the pore size reaches saturation. These findings indicate that the pore size of PU foam is not exclusively determined by the blowing agent quantity. Notably, certain pore sizes persist even in the absence of a blowing agent. It can be inferred that additional factors contribute to determining the pore size in rigid PU foam. However, it can be hypothesized that the saturation of the pore size is due to the residual moisture present within the polyol. Given that polyols are hygroscopic and invariably retain a certain amount of moisture, it is plausible that the quantity of blowing agent introduced was negligible in comparison to the residual moisture and had no impact on the pore size. To eliminate this possibility, further experiments were conducted.

The pore size or rigid PUs with different amount of blowing agents (water). a shows the pores size of foams with low blowing agent content. It is larger than 100 µm even in the absence of the blowing agent. b shows the pore size of foams prepared with different mixing procedures. As high energy is used, the smaller pores were observed
Another significant finding is that the pore size can be markedly altered during the mixing process of polyol, MDI, and water. The results presented in Fig. 3(b) illustrate the impact of varying the mixing power through hand mixing, blade-type stirrer, and homogenizer, respectively, on the pore size for the aforementioned mixing methods. The results demonstrate that hand mixing, which employs the lowest power, and homogenization, which employs the highest power, yield distinct foam characteristics. These findings indicate that the mixing process induces significant alterations in the system. In fact, the greater energy input during the mixing process of the polyol and water resulted in a more creamy polyol, as anticipated due to the substantial number of air bubbles captured within the polyol.
Air Bubble Entrapment in the Polyol
It has been verified that a substantial number of bubbles form when polyol is mixed with a blowing agent or isocyanate. These bubbles represent entrapped air bubbles generated during the mixing process of polyol, characterized by high viscosity (~ 2 Pa∙s). Consequently, when the polyol is heated to 70 °C to significantly reduce viscosity, the formation of bubbles is nearly eliminated. Based on these findings, it can be inferred that the pore sizes illustrated in Fig. 3(a) may originate from air bubbles, even in the absence of a blowing agent. To validate this blowing pattern, the number and size of bubbles generated after mixing polyol with a blowing agent and MDI were tracked over time. For this purpose, we positioned the small amount of the reacting mixture under an optical microscope. Then monitored the change of the air bubbles over time, as shown in Fig. 4(a), and the results are summarized in Fig. 4(b) and (c). Given that only a small proportion of the mixture is being observed in isolation, it is challenging for the overall system temperature to increase, which consequently slows down the reaction. Consequently, the expansion of the pores occurred at a slower rate than in a real foam system. The key insight here is that although the size of bubbles continued to grow over time, the number of bubbles did not change significantly. In other words, despite the addition of the blowing agent, no new nuclei were created.

a Optical microscopic images of air bubbles trapped in polyol. b Change in the number of air bubbles over time. c Change in bubble size over time
The absence of new nuclei formation can be explained by considering energy. According to classical nucleation theory, a nucleus requires free energy to overcome the high interfacial energy to form. Once the nucleus surpasses a critical size, it can continue to grow without additional energy. However, if it cannot exceed the critical size due to a lack of energy, it will be extinguished. The current system, however, already contains a significant number of air bubbles within the polyol, with a size exceeding 10 µm. Therefore, there is no incentive for new pores to form, as it is more energetically stable for the blowing agent to diffuse into the existing bubbles. This type of nucleation is referred to as non-classical nucleation. When non-classical nucleation occurs, the number of pores cannot be controlled by blowing agents because there are no newly formed small-sized nuclei. Therefore, adding blowing agents does not increase the number of pores, only their size. To effectively control the pore size of rigid PU foams, it is necessary to manage both the number and size of bubbles in the polyol, which serve as the nucleation sites for the pores.
Stability of Polyol Liquid Foam
Based on the preceding observations, the determination of pore size in rigid urethane foam initiates with the liquid polyol foam. To regulate bubble size in liquid foams, one must consider the stability of these bubbles. In liquid foams, the significant density disparity between the liquid continuous phase and the gas dispersed phase leads to creaming, wherein bubbles coalesce as they ascend. During this process, bubbles may continue to grow through coalescence or Ostwald ripening, jeopardizing the foam's stability by enabling easy movement of bubbles within the liquid continuous phase. Hence, minimizing bubble movement becomes crucial for prolonged liquid foam stability. Efforts have been made to enhance the viscosity of the continuous phase, specifically the polyol. Transforming the continuous phase into a yield stress fluid significantly enhances the stability of the liquid foam, as the yield stress fluid only flows under a certain stress, preventing foam collapse.
Colloidal particles were introduced to manipulate the rheological properties of the polyol. When dispersed in a liquid phase, these particles can increase viscosity with concentration, forming a gel with yield stress above a certain concentration. Variations in rheological properties may occur at the same concentration, depending on the affinity between particles and the solvent. Generally, lower affinity results in less uniform particle dispersion and increased connectivity, leading to higher viscosity at the same concentration [45]. Since polyols consist of ester and ether polymers with small molecular weights and multiple hydroxyl groups capable of hydrogen bonding, hydrophobic silica particles with surface hydroxyl groups replaced by methyl groups were added to the polyols.
Figure 5(a) illustrates the viscosity, G′, and G″ of silica polyol suspensions as a function of concentration. A steady increase in G′, G″, and viscosity is evident with rising concentration, especially above 5% silica addition, where G′ surpasses G″, indicating the fluid behaves like a solid under low shearing stress. Figure 5(b) depicts the pore size of rigid PU foams produced from polyols with modified rheological properties. Despite an increase in polyol viscosity, the foam’s pore size remains around 200 µm. The alteration in polyol rheological properties does not impact the pore size of the final foam. This is because phenomena such as coalescence and Ostwald ripening, which impede liquid foam stability, are not prevalent in polyol cases. Significant bubble destabilization is not anticipated during the PU foam production process due to the high viscosity of the polyol (~ 2 Pa·s) and the completion of the urethane reaction in approximately 10 min. Consequently, obtaining PU foam with small pores is expected by merely reducing the initial size of the bubbles in the polyol, without concerns about bubble stability.

a Rheological properties of polyol with addition of hydrophobic silica particles. b The pore size of polyurethane foams synthesized with rheology-modified polyols
Air Bubble Size Control in Polyol
In multiphase systems, such as liquid foams or emulsions, ensuring interfacial stability is crucial to reduce the size of the dispersed phase. Typically, this is achieved through the adsorption of surfactants or colloidal particles onto the interface. It is well-established that the size of the dispersed phase decreases with an increasing amount of stabilizer used. To stabilize highly hydrophobic bubbles in liquid foams, a hydrophobic stabilizer is necessary. However, if the stabilizer is excessively hydrophobic, it may pose challenges when used with polar solvents. To address this issue, a combination of hydrophobic particles and hydrophilic surfactants at the interface can enhance adsorption [46]. The hydrophilic surfactant adsorbs on the surface of the hydrophobic colloid, improving its ability to wet well in polar solvents. In this study, hydrophobic silica particles and polyacrylamide (Mn = 40,000) polymer were employed as the colloid and surfactant, respectively, in the current polyol system. Increasing the surfactant amount led to a decrease in bubble and pore size, reducing the pore size of the PU foam from 200 to 150 µm as shown in Fig. 6.

Change in pore size of the rigid polyurethane foam by controlling the size of air bubbles in polyol with colloidal particles and surfactants
Another strategy for stabilizing the bubble surface involves using a liquid instead of a solid colloid. Covering the bubble surface with a liquid that has lower surface tension than the air-polyol reduces the energy needed to form bubbles, enabling the creation of smaller bubbles with the same mixing energy. In this case, a total wetting liquid capable of completely covering the bubble's surface should be used. Novec oil, a fluorocarbon oil, has been confirmed to totally wet the air-polyol interface. The surface tension results in Fig. 7(a) show that Novec oils have lower surface tensions (12–14 mN/m) than the air-polyol interface (15 mN/m). Additionally, Novec oil has very low interfacial tension with polyols (1–3 mN/m). Based on these values, the spreading coefficient (S) can be calculated as follows.

a Interfacial tension of polyol and Novec oils. Black bars are surface tension with air interface, and red bars are interfacial tension with polyol interface. b Spreading parameter of Novec oils between air and polyol. c The pore size of polyurethane foams with addition of Novec oils in polyol. d Pore size change with Novec oil amount
\({\gamma }_{\text{a}-\text{p}}\), \({\gamma }_{\text{a}-\text{N}}\), and \({\gamma }_{\text{p}-\text{N}}\) is air-polyol surface tension, air-Novec oil surface tension, and polyol-Novec oil interfacial tension, respectively. If the spreading coefficient is positive, Novec oil can totally wet on the air-polyol interface. The spreading coefficient calculation in Fig. 7(b) indicates that Novec 7100 and 7200 are total wetting liquids, while Novec 7300 and 7500 are partial wetting liquids. The change in PU foam pore size with Novec oil addition is shown in Fig. 7(d), revealing that increasing the total wetting liquid reduces the pore size of rigid PU foam. However, with partial wetting liquids, there is minimal change in pore size. For instance, the foam made with 1% and 10% addition of Novec 7100 had pore sizes of approximately 200 µm and 150 µm, respectively. These findings suggest that total wetting liquids have the ability to stabilize air bubbles and generate smaller bubbles by increasing the liquid volume.
Previous experiments have demonstrated that reducing bubble size in liquid foam results in a smaller pore size in the final PU foam. However, even when the bubble size is reduced to the maximum, it remains around 10 µm. Achieving liquid foam sizes of 1 µm or nano-sized is an extremely challenging task. Consequently, reducing the pore size of rigid PU foam to below 100 µm presents a formidable challenge, not achievable solely by controlling the pore size. This may explain why most studies on rigid PU foam have focused on pore sizes above 100 µm.
Elimination of Air Bubbles in Polyol
The discovery of a limitation in significantly reducing the pore size of rigid PU foam when non-classical nucleation occurs due to air bubbles prompted an investigation into the potential for pore size reduction through induced classical nucleation. Classical nucleation requires the complete removal of all air bubbles trapped inside the polyol, necessitating a substantial lowering of polyol viscosity. To achieve this viscosity reduction, a low-viscosity solvent was introduced. In instances where the solvent served as a co-solvent capable of dissolving both polyol and MDI, it facilitated the uniform mixing of polyol and MDI without necessitating additional mixing steps.
For instance, dissolving both polyol and MDI in benzene, a co-solvent, effectively eliminated all air bubbles. However, a wet gel containing benzene persisted after the urethane reaction was complete, likely due to the high boiling point of benzene (80 °C), hindering its complete evaporation during the reaction. Conversely, the use of diethyl ether, with a lower boiling point of 34 °C, resulted in the majority of it evaporating during the urethane reaction, leaving behind very small 2 µm pores. (Fig. 8) This was attributed to the insufficient ability of the small amount of residual solvent to dissolve PU with a higher molecular weight, leading to phase separation and pore formation.

Morphology of polyurethane synthesized with co-solvent. Polyurethane wet-gel is made with benzene solvent. Microporous polyurethane foam is made with diethyl ether solvent
While classical nucleation proved capable of forming pores of several microns by rapidly removing the co-solvent after eliminating air bubbles, controlling the process through co-solvent evaporation posed challenges. Thus, a novel method involving the extraction of only the co-solvent was proposed. After the initial use of the co-solvent to dissolve polyol and MDI and remove air bubbles, an extraction solvent, miscible with the co-solvent but not with polyol and MDI, was introduced as shown in Fig. 9(a). This resulted in a separation into an upper phase containing the extraction solvent and extracted co-solvent, and a bottom phase containing polyol, MDI, and a small amount of residual co-solvent. The bottom phase transformed into polyurethane foam with micro-pores.

a Scheme of co-solvent extraction. Bottom phase which is composed of polyol, MDI, and small amount of co-solvent becomes polyurethane foam. b Pore size and c density of polyurethane foam. Acetone was used as co-solvent, and hexane was used as extraction solvent. d, e SEM images of polyurethane foam
The experimental setup used acetone as a co-solvent and hexane as an extraction solvent. Varying the amount of acetone allowed control over the residual co-solvent content, affecting the amount of volume expansion in the foam. The results which can be seen in Fig. 9(b–e) indicated that a greater amount of residual co-solvent led to more significant volume expansion and smaller pore sizes, approximately 5 to 10 µm. However, a notable challenge arose in terms of foam density, attributed to the hardness of the PU matrix. The rigid PU matrix’s high mechanical properties impeded substantial volume expansion during the urethane reaction, limiting the extent to which the final foam density could be reduced. This limitation presents challenges in utilizing the resulting material as an effective insulator.
Conclusion
This study investigates the foaming process in rigid polyurethane foam, identifying trapped air bubbles within the polyol as the key factor determining pore size. The polyol’s high viscosity ensures that once formed, these bubbles remain stable throughout the urethane reaction, as previously discussed. It has been verified that reducing the size of air bubbles leads to a corresponding reduction in the pore size of PU foam. Stabilizing air bubbles with colloids or total wetting liquids can achieve a reduction in their size to about 10 µm, allowing for the reduction of the pore size of PU foam to approximately 100 µm. While theoretically possible to reduce pore size further by creating nano-sized bubbles, practical implementation proves challenging due to the high interfacial energy of air bubbles.
Conversely, classical nucleation can be induced to form small pores if all air bubbles are effectively removed. This is accomplished by dissolving polyol and MDI in a low-viscosity co-solvent, facilitating mixing without any trapped air bubbles. The resulting PU foam synthesized using this method exhibits a very small pore size, measuring less than 10 µm. However, the material's density is remarkably high due to the rigid PU matrix, limiting volume expansion. In summary, the presence of air bubbles establishes a lower limit for pore size in rigid polyurethane foams. Paradoxically, the potential for non-classical bubble nucleation offers an avenue for producing low-density rigid polyurethane foams.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
R. Hasanzadeh, T. Azdast, P.C. Lee, C.B. Park, A review of the state-of-the-art on thermal insulation performance of polymeric foams. Therm. Sci. Eng. Prog. 41, 101808 (2023). https://doi.org/10.1016/j.tsep.2023.101808
J. Zhao, G. Wang, Z. Xu, A. Zhang, G. Dong, G. Zhao, C.B. Park, Ultra-elastic and super-insulating biomass PEBA nanoporous foams achieved by combining in-situ fibrillation with microcellular foaming. J. CO2 Util. 57, 101891 (2022). https://doi.org/10.1016/j.jcou.2022.101891
P. Buahom, P. Gong, C. Wang, H. Yu, J. Liu, C.B. Park, Carbon as a solution for nanocellular foam superinsulation. Carbon N. Y. 189, 319–338 (2022). https://doi.org/10.1016/j.carbon.2021.11.041
B.S. Kim, J. Choi, Y.S. Park, Y. Qian, S.E. Shim, Semi-rigid polyurethane foam and polymethylsilsesquioxane aerogel composite for thermal insulation and sound absorption. Macromol. Res. 30, 245–253 (2022). https://doi.org/10.1007/s13233-022-0026-8
B. Porkar, P.A. Atmianlu, M. Mahdavi, M. Baghdadi, H. Farimaniraad, M.A. Abdoli, Chemical modification of polystyrene foam using functionalized chitosan with dithiocarbamate as an adsorbent for mercury removal from aqueous solutions. Korean J. Chem. Eng. 40, 892–902 (2023). https://doi.org/10.1007/s11814-023-1387-1
D. Yun, J.H. Kim, Acoustic performance of flexible polyurethane composite foams filled with melamine particles. Korean J. Chem. Eng. 40, 3052–3058 (2023). https://doi.org/10.1007/s11814-023-1539-3
S.S. Yang, I. Jung, Y.J. Kang, Multiscale acoustical study on graphene oxide impregnated polyurethane foam. Macromol. Res. (2024). https://doi.org/10.1007/s13233-024-00281-7
D.H. Park, G.P. Park, S.H. Kim, W.N. Kim, Effects of isocyanate index and environmentally-friendly blowing agents on the morphological, mechanical, and thermal insulating properties of polyisocyanurate-polyurethane foams. Macromol. Res. 21, 852–859 (2013). https://doi.org/10.1007/s13233-013-1106-6
Y. Meng, X. Zhou, Z. Huang, Z. Wang, Pumping with modified polyurethane sponges: a rapid oil spill treatment technology. Korean J. Chem. Eng. 41, 2133–2142 (2024). https://doi.org/10.1007/s11814-024-00140-2
B. Nait-Ali, K. Haberko, H. Vesteghem, J. Absi, D.S. Smith, Thermal conductivity of highly porous zirconia. J. Eur. Ceram. Soc. 26, 3567–3574 (2006). https://doi.org/10.1016/J.JEURCERAMSOC.2005.11.011
G. Sung, H. Choe, Y. Choi, J.H. Kim, Morphological, acoustical, and physical properties of free-rising polyurethane foams depending on the flow directions. Korean J. Chem. Eng. 35, 1045–1052 (2018). https://doi.org/10.1007/s11814-017-0328-2
R. Hasanzadeh, T. Azdast, A. Doniavi, R.E. Lee, Multi-objective optimization of heat transfer mechanisms of microcellular polymeric foams from thermal-insulation point of view. Therm. Sci. Eng. Prog. 9, 21–29 (2019). https://doi.org/10.1016/j.tsep.2018.11.002
C.Y. Zhao, S.A. Tassou, T.J. Lu, Analytical considerations of thermal radiation in cellular metal foams with open cells. Int. J. Heat Mass Transf. 51, 929–940 (2008). https://doi.org/10.1016/j.ijheatmasstransfer.2007.10.010
B. Merillas, F. Villafañe, M.Á. Rodríguez-Pérez, Improving the insulating capacity of polyurethane foams through polyurethane aerogel inclusion: from insulation to superinsulation. Nanomaterials 12, 2232 (2022). https://doi.org/10.3390/nano12132232
H. Choe, Y. Choi, J.H. Kim, Threshold cell diameter for high thermal insulation of water-blown rigid polyurethane foams. J. Ind. Eng. Chem. 73, 344–350 (2019). https://doi.org/10.1016/j.jiec.2019.02.003
D. Schmidt, V.I. Raman, C. Egger, C. du Fresne, V. Schädler, Templated cross-linking reactions for designing nanoporous materials. Mater. Sci. Eng. C 27, 1487–1490 (2007). https://doi.org/10.1016/j.msec.2006.06.028
B. Notario, J. Pinto, E. Solorzano, J.A. De Saja, M. Dumon, M.A. Rodríguez-Pérez, Experimental validation of the Knudsen effect in nanocellular polymeric foams. Polymer (Guildf). 56, 57–67 (2015). https://doi.org/10.1016/j.polymer.2014.10.006
I. Tsivintzelis, A.G. Angelopoulou, C. Panayiotou, Foaming of polymers with supercritical CO2: an experimental and theoretical study. Polymer (Guildf). 48, 5928–5939 (2007). https://doi.org/10.1016/J.POLYMER.2007.08.004
C. Forest, P. Chaumont, P. Cassagnau, B. Swoboda, P. Sonntag, CO2 nano-foaming of nanostructured PMMA. Polymer (Guildf). 58, 76–87 (2015). https://doi.org/10.1016/J.POLYMER.2014.12.048
E. Di Maio, E. Kiran, Foaming of polymers with supercritical fluids and perspectives on the current knowledge gaps and challenges. J. Supercrit. Fluids 134, 157–166 (2018). https://doi.org/10.1016/j.supflu.2017.11.013
S. Liu, J. Duvigneau, G.J. Vancso, Nanocellular polymer foams as promising high performance thermal insulation materials. Eur. Polym. J. 65, 33–45 (2015). https://doi.org/10.1016/j.eurpolymj.2015.01.039
C. Yang, Z. Xing, Q. Zhao, M. Wang, G. Wu, A strategy for the preparation of closed-cell and crosslinked polypropylene foam. J. Appl. Polymer Sci. 135(6), 45809 (2017)
S.P. Nalawade, F. Picchioni, L.P.B.M. Janssen, Supercritical carbon dioxide as a green solvent for processing polymer melts: processing aspects and applications. Prog. Polym. Sci. 31, 19–43 (2006). https://doi.org/10.1016/J.PROGPOLYMSCI.2005.08.002
M. Sauceau, J. Fages, A. Common, C. Nikitine, E. Rodier, New challenges in polymer foaming: a review of extrusion processes assisted by supercritical carbon dioxide. Prog. Polym. Sci. 36, 749–766 (2011). https://doi.org/10.1016/J.PROGPOLYMSCI.2010.12.004
A.K. Bledzki, H. Kirschling, M. Rohleder, A. Chate, Correlation between injection moulding processing parameters and mechanical properties of microcellular polycarbonate. J. Cell. Plast. 48, 301–340 (2012). https://doi.org/10.1177/0021955X12441193
S. Pilla, A. Kramschuster, L. Yang, J. Lee, S. Gong, L.S. Turng, Microcellular injection-molding of polylactide with chain-extender. Mater. Sci. Eng. C 29, 1258–1265 (2009). https://doi.org/10.1016/J.MSEC.2008.10.027
H. Azimi, D. Jahani, S. Aghamohammadi, M. Nofar, Experimental and numerical investigation of bubble nucleation and growth in supercritical CO2-blown poly(vinyl alcohol). Korean J. Chem. Eng. 39, 2252–2262 (2022). https://doi.org/10.1007/s11814-022-1078-3
I.K. Hong, S. Lee, Microcellular foaming of silicone rubber with supercritical carbon dioxide. Korean J. Chem. Eng. 31, 166–171 (2014). https://doi.org/10.1007/s11814-013-0188-3
Y.K. Kwon, H.K. Bae, Production of microcellular foam plastics by supercritical carbon dioxide. Korean J. Chem. Eng. 24, 127–132 (2007). https://doi.org/10.1007/s11814-007-5022-3
I. Sánchez-Calderón, V. Bernardo, J. Martín-de-León, M.Á. Rodríguez-Pérez, Thermal conductivity of low-density micro-and nanocellular poly(methyl-methacrylate) (PMMA): experimental and modeling. Mater. Des. (2022). https://doi.org/10.1016/j.matdes.2022.110938
G. Wang, J. Zhao, L.H. Mark, G. Wang, K. Yu, C. Wang, C.B. Park, G. Zhao, Ultra-tough and super thermal-insulation nanocellular PMMA/TPU. Chem. Eng. J. 325, 632–646 (2017). https://doi.org/10.1016/j.cej.2017.05.116
G. Sung, J.H. Kim, Effect of high molecular weight isocyanate contents on manufacturing polyurethane foams for improved sound absorption coefficient. Korean J. Chem. Eng. 34, 1222–1228 (2017). https://doi.org/10.1007/s11814-016-0361-6
S. Huo, G. Wu, J. Chen, G. Liu, Z. Kong, Constructing polyurethane foams of strong mechanical property and thermostability by two novel environment friendly bio-based polyols. Korean J. Chem. Eng. 33, 1088–1094 (2016). https://doi.org/10.1007/s11814-015-0223-7
M.S. Han, S.J. Choi, J.M. Kim, Y.H. Kim, W.N. Kim, H.S. Lee, J.Y. Sung, Effects of silicone surfactant on the cell size and thermal conductivity of rigid polyurethane foams by environmentally friendly blowing agents. Macromol. Res. 17, 44–50 (2009). https://doi.org/10.1007/BF03218600
J.W. Kang, J.M. Kim, M.S. Kim, Y.H. Kim, W.N. Kim, W. Jang, D.S. Shin, Effects of nucleating agents on the morphological, mechanical and thermal insulating properties of rigid polyurethane foams. Macromol. Res. 17, 856–862 (2009). https://doi.org/10.1007/BF03218626
C. Brondi, E. Di Maio, L. Bertucelli, V. Parenti, T. Mosciatti, Competing bubble formation mechanisms in rigid polyurethane foaming. Polymer (Guildf). 228, 123877 (2021). https://doi.org/10.1016/j.polymer.2021.123877
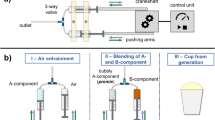
M. Hamann, S. Andrieux, M. Schütte, D. Telkemeyer, M. Ranft, W. Drenckhan, Directing the pore size of rigid polyurethane foam via controlled air entrainment. J. Cell. Plast. 59, 201–214 (2023). https://doi.org/10.1177/0021955X231152680
R. Jang, Y. Lee, K.H. Song, W.N. Kim, Effects of nucleating agent on the thermal conductivity and creep strain behavior of rigid polyurethane foams blown by an environment-friendly foaming agent. Macromol. Res. 29, 15–23 (2021). https://doi.org/10.1007/s13233-021-9003-x
H.J. Choi, J.H. Kim, Sound absorption improvement of polyurethane foam through sequential arrangement of its cellular morphology. Korean J. Chem. Eng. 39, 1072–1077 (2022). https://doi.org/10.1007/s11814-021-0974-2
M. Thirumal, D. Khastgir, N.K. Singha, B.S. Manjunath, Y.P. Naik, Mechanical, morphological and thermal properties of rigid polyurethane Foam: effect of chain extender, polyol and blowing agent. Cell. Polym. 28, 145–158 (2009). https://doi.org/10.1177/026248930902800202
D. Yan, L. Xu, C. Chen, J. Tang, X. Ji, Z. Li, Enhanced mechanical and thermal properties of rigid polyurethane foam composites containing graphene nanosheets and carbon nanotubes. Polym. Int. 61, 1107–1114 (2012). https://doi.org/10.1002/pi.4188
M.C. Hawkins, B. O’Toole, D. Jackovich, Cell morphology and mechanical properties of rigid polyurethane foam. J. Cell. Plast. 41, 267–285 (2005). https://doi.org/10.1177/0021955X05053525
F. Saint-Michel, L. Chazeau, J.Y. Cavaillé, E. Chabert, Mechanical properties of high density polyurethane foams: I. Effect of the density. Compos. Sci. Technol. 66, 2700–2708 (2006). https://doi.org/10.1016/J.COMPSCITECH.2006.03.009
J. Chae, S.Q. Choi, K. Kim, The role of excess attractive particles in the elasticity of high internal phase pickering emulsions. Soft Matter 18, 53–61 (2022). https://doi.org/10.1039/D1SM01338F
D.B. Genovese, Shear rheology of hard-sphere, dispersed, and aggregated suspensions, and filler-matrix composites. Adv. Colloid Interface Sci. 171–172, 1–16 (2012). https://doi.org/10.1016/J.CIS.2011.12.005
Y. Sheng, K. Lin, B.P. Binks, T. Ngai, Ultra-stable aqueous foams induced by interfacial co-assembly of highly hydrophobic particles and hydrophilic polymer. J. Colloid Interface Sci. 579, 628–636 (2020). https://doi.org/10.1016/J.JCIS.2020.06.098
Acknowledgements
This work was supported by Korea Evaluation Institute of Industrial Technology (20014762).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
There are no conflicts to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chae, J., Lee, Y. & Choi, S.Q. Pore Size Control Mechanism of a Rigid Polyurethane Foam. Korean J. Chem. Eng. (2024). https://doi.org/10.1007/s11814-024-00275-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11814-024-00275-2