
Abstract
Sulfosuccination of castor oil-derived methyl ricinoleate and methyl 12-hydroxy stearate have been carried out in the present work. Synthesis involves malenization of secondary alcohol of methyl ricinoleate/methyl 12-hydroxy stearate followed by sulfonation of maleic monoester to generate double-headed dianionic surfactant with carboxylate and sulfosuccinate functionalities in the head group region. Various reaction conditions were optimized for maximum production of these two sulfosuccinates. Both compounds were evaluated for surface and detergency properties. The surface tension study indicated that the critical micelle concentration of sulfosuccinated methyl ricinoleate and methyl 12-hydroxy stearate is 0.26 and 0.11 mM, respectively. The detergency property of these two surfactants indicated that they were excellent in wetting time emulsification and Ca-tolerance. However, these two surfactants exhibited very poor foam height and foam stability.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sulfosuccinate is a mild anionic surfactant having wide application in the personal care industry. Mostly they are used in conditioning and anti-dandruff shampoos and other cleansing formulations as additives to reduce eye and skin irritation [1, 2]. They are also used as wetting agents in textile, paint, leather and printing industries. Versatility, biodegradability, and good wetting properties make this class of surfactant attractive industrially [2]. Salts of the monoester of sulfosuccinic acid are in general obtained by reacting maleic acid, fumaric acid, or maleic anhydride with fatty alcohol, alkoxylated fatty alcohol, or fatty acid alkanolamides. The resulting butanedioic acid half ester is sulfonated with sulphite, pyrosulfite or bisulfite of alkaline earth metal to get the desired salts of monoester of sulfosuccinic acid [2–4].
Sulfosuccinates could be of the diester type, but require harsher reaction conditions for the second esterification. Diester sulfosuccinates are rarely used in the personal care industry due to their several disadvantages like poor solubility, poor foaming, and skin irritating nature. However, they are used in the industry as wetting agents and dispersants and are commercialized under the name aerosol surfactants, the most familiar among them is 2-ethylhexyl sulfosuccinate. Because of their solubility in water, organic solvents and even hydrocarbons, diester sulfosuccinate are also used in dry-cleaning formulations. Different types of sulfosuccinates and their synthesis, properties, and industrial applications are reviewed in the literature [2, 3]. Most of the succinate monoesters are synthesized by reacting maleic anhydride with primary alcohol, and there are very few reports wherein secondary alcohol was reacted with maleic anhydride to synthesize branched succinate monoester [2–6]. The present work is directed towards the synthesis of branched succinate monoester, which may provide an ideal building block for newer surfactants.
Synthesis of structurally novel surfactants has attracted organic and physical chemists to study their surface properties. On the other hand, there is a demand for newer surfactants from renewable sources, presumably due to environmental concerns about petrochemical-based surfactants. Castor oil is an industrially important renewable resource for diverse kinds of oleochemicals and may play a vital role in developing newer surfactants. This is due to its easy availability, inexpensiveness, ecofriendly nature, and, most importantly, the presence of an unusual major fatty acid, called ricinoleic acid (RA) or 12-hydroxyoctadec-9-ene-1-oic acid. Castor oil contains nearly 85–88 % of RA, and the rest are other non-hydroxy fatty acids. The presence of a double bond as well as a hydroxyl moiety makes RA an attractive molecule to carry out chemical modifications to generate high value oleochemicals. The overviews on utility of the castor oil-based feedstock are thoroughly discussed in several review articles [7, 8]. 12-Hydroxy stearic acid (12-HSA) is the waxy saturated counterpart of RA, obtained through catalytic hydrogenation of RA [9]. 12-HSA is used mostly as lubricating grease in the industry and also as thixotropic gellants in the paint and coating industries [10]. Thus, the secondary alcohol of RA and 12-HSA has given us the opportunity to design a new class of branched succinate monoesters, which, upon sulfonation, generate dianionic surfactants with carboxylate and sulfosuccinate as head groups.
In the present work, malenization of methyl ricinoleate and methyl 12-hydroxy stearate was carried out initially to get a monoester of maleate, which was subsequently sulfonated to get two new dianionic sulfosuccinates, disodium-4-[(18-methoxy-18-oxooctadec-9-en-7-yl)oxy]-4-oxo-2-sulfonatobutanoate (RSS) and disodium-4-[(18-methoxy-18-oxooctadecan-7-yl)oxy]-4-oxo-2-sulfonatobutanoate (HSS). Both these surfactants were evaluated for surface and thermodynamic properties such as critical micelle concentration (CMC), surface tension at cmc (γ cmc), efficiency of surface adsorption (pC 20), surface excess (Γmax), minimum area per molecule at the air–water interface (A min), free energy of adsorption (ΔG°ads) and micellization (ΔG°mic), wetting time, foaming, emulsion stability, and calcium tolerance.
Experimental Methods
Materials
Castor oil and hydrogenated castor oil were procured from a local industry. Maleic anhydride and sodium bisulphite were purchased from Finar Chemicals Ltd. Sodium hydroxide, sodium sulphate, sulphuric acid, paraffin liquid (light), hyamine, methylene blue, and other organic solvents were procured from SD Fine Chem (Mumbai, India).
Synthesis
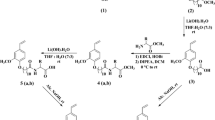
Synthesis was carried out as per Scheme 1. The hydroxyl group present at 12th position in both RA and 12-HSA was esterified initially with maleic anhydride at 90 °C under solvent-free and catalyst-free conditions. Various reaction conditions such as the molar ratio of substrates, reaction time, and mode of addition of maleic anhydride were varied to optimize the conditions for maximum conversion. Initially esterification was conducted by reacting methyl ricinoleate and maleic anhydride at 1.0:1.5 M ratios to make up loss due to sublimation of maleic anhydride at 90 °C for 3 h. The isolated yield of monomaleate ester was found to be 43.95 %. However, on increasing the reaction time to 5 h, increases the yield of isolated product to 61.1 %. Beyond 5 h, no further improvement in yield of the product was observed. Regarding substrate ratio, any attempt to reduce the molar equivalence of maleic anhydride (up to equimolar equivalence to alcohol) affects the overall yield of the product. On the other hand, increasing the molar equivalence of maleic anhydride to 1.6 and 1.8 Eq to alcohol showed a marginal increase of isolated yields to 77.39 and 78.91 %, respectively.

Synthesis of disodium-4-[(18-methoxy-18-oxooctadec-9-en-7-yl)oxy]-4-oxo-2-sulfonatobutanoate (RSS) and disodium-4-[(18-methoxy-18-oxooctadecan-7-yl)oxy]-4-oxo-2-sulfonatobutanoate (HSS)
After confirming the reaction time and substrate ratio, the mode of addition of maleic anhydride was changed from one time addition to instalment addition (three equal instalments over a period of 0.5 h). There is a significant increase in isolated yield of the product, from 61.1 to 76.0 %. Finally, the resultant maleate monoester was sulfonated using an equimolar amount of NaHSO3 at 70 °C and finally neutralized with aqueous NaOH solution
Sulfosuccination of Methyl Ricinoleate
Synthesis of 4-(18-Methoxy-18-Oxooctadec-9-En-7-Yl)Oxy-4-Oxobut-2-Enoic Acid (RME)
Methyl ricinoleate (10.36 g, 0.033 M) was weighed in a 100 mL round bottom flask and stirred at 90 °C for 15 min. This was followed by the addition of maleic anhydride (4.88 g, 0.0498 M) in five equal instalments at 0.5 h time intervals. The progress of reaction was monitored by TLC (using hexane:ethyl acetate, 90:10 v/v as eluent). After 5 h, the crude reaction mixture was dissolved in hexane and filtered to remove maleic acid, produced from unreacted maleic anhydride during the progress of the reaction. The filtrate was concentrated by rotary evaporator and subjected to silica gel column chromatography using hexane and ethyl acetate as eluting solvent to get maleic monoester as viscous liquid (10.26 g, 75.44 %). The compound was characterized by 1H NMR and mass spectroscopy. 1H NMR (500 MHz, CDCl3, ppm): δ 6.35–6.45 (dd, 2H; –OOC–CH=CH–COOH), 5.5 (m, 1H; –CH=CH–CH2), 5.3 (m, 1H; –CH=CH–CH2–), 5.0 (m, 1H; HOOC–CH2=CH–CO–O–CH–), 3.65 (s, 3H; –CH2–COO–CH 3 ), 2.3–2.5 (m, 4H; –CH 2–COOCH3 and –CH=CH–CH 2–CH–O–), 2 (m, 2H; –CH 2–CH=CH–CH2–O–), 1.65 (m, 4H; –CH 2–CH2–COOCH3 and –CH=CH–CH2–CHO–CH 2–), 1.3 (broad s, 16H; chain –CH 2), 0.9 (t, 3H; –(CH)2–CH 3); ESI MS: m/z 411 (M+1), 433 (M+Na), 449 (M+K), 295 (M–OOCCH=CH–COOH).
Synthesis of Disodium-4-[(18-Methoxy-18-Oxooctadec-9-En-7-Yl)Oxy]-4-Oxo-2-Sulfonatobutanoate
In a 100 mL round bottom flask attached to a water condenser, about 10.0 g of RME (0.024 M) was taken and stirred at 70 °C. About 2.0 mL of 50 % aqueous sodium hydroxide solution (0.024 M) was added into the stirred solution in order to maintain the pH of the reaction mixture to be in the range of 5.5–6 followed by the addition of isopropanol (7.0 mL). After 15 min, aqueous solution of sodium bisulphite (2.53 g, 0.024 mol) was added in three instalments (0.25, 0.5, and 0.5 % aqueous solution) at 0.5 h intervals. Progress of the reaction was monitored by TLC (using hexane:ethyl acetate, 90:10 v/v as eluent) as well as estimation of %SO3 by the hyamine method. After 2 h of total reaction time, the TLC profile indicated the complete disappearance of starting material. The hyamine method showed 14.2 % SO3 fixation indicating 95.4 % conversion (theoretical value for %SO3 is 14.92 for complete conversion).
After completion of reaction, water and IPA were evaporated using a rotary evaporator and kept under vacuum for 0.5 h at 70 °C for complete removal of solvent. The crude reaction mixture was dissolved in 100 mL of chloroform and filtered to remove traces of sediments. Then chloroform was evaporated using a rotary evaporator to obtain a white solid, which was washed with hexane (50 mL) for three times to remove any unreacted starting material. Finally the solid was dried in desiccators under a vacuum for 5 h (12.14 g, yield 92.9 %). The product was a white amorphous solid having a melting point in the range of 239–241 °C. The compound was characterized by IR, NMR (1H, and 13C) and mass spectroscopy. IR (cm−1): 3,415 (O–H stretching), 3,020, 2,928, 2,856 (aliphatic C–H stretching), 1,724 (methyl ester carbonyl stretching), 1,612 (succinic carbonyl stretching), 1,401 (C–H scissoring and S=O stretching), 1,216, 1,048 (C–O stretching), 766 (C–H rocking); 1H NMR (500 MHz, CDCl3, ppm): δ 5.25–5.5 (broad, 2H; –CH=CH–CH2), 4.8 (broad, 1H; HOOC–CH2=CH–CO–O–CH–), 3.65 (s, 3H; –CH2–COO–CH 3 ), 3.60–2.82 (broad, 3H; NaOOC–CH(SO3Na)–CH 2–CO–O–CH–), 2.29 (broad, 4H; –CH 2–COOCH3 and –CH=CH–CH 2–CH–O–), 1.98 (broad, 2H; –CH 2–CH=CH–CH2–O–), 1.62 (broad, 2H; –CH 2–CH2–COOCH3) 1.48 (broad, 2H; –CH=CH–CH2–CHO–CH 2–), 1.25 (broad s, 16H; chain –CH 2), 0.86 (t, 3 –(CH)2–CH 3);13C NMR (75 MHz, CDCl3): 174 (3 carbonyl carbons C=O), 132 (–CH=CH–CH2–CH–O), 124 (–CH=CH–CH2–CH–O), 75 (OOC–CH2–CH–SO3), 51 (–CH2–COOCH3), 34 (2 CH2–C=O), 29–22 (chain –CH2–), 14 (–(CH)2–CH3); ESI MS: m/z 409 [M–SO3], 491 [M+1], 513 [M+Na]; HRMS (m/z) calculated for C23H38O9S is 491.22508, found 491.22474.
Sulfosuccination of Methyl-12-Hydroxy Stearate
Synthesis of 4-(18-Methoxy-18-Oxooctadecan-7-Yl)Oxy-4-Oxobut-2-Enoic Acid (HSME)
Methyl-12-hydroxy stearate (6.6 g, 0.021 M) was reacted with maleic anhydride (3.09 g, 0.0315 M) according to the procedure mentioned above. After 5 h, the crude reaction mixture was dissolved in hexane, filtered to remove maleic acid, concentrated, and purified by column chromatography to get maleic monoester as a viscous liquid (6.44 g, 74.3 %). 1H NMR (500 MHz, CDCl3): δ (ppm) 6.43–6.31 (dd, 2H; –OOC–CH=CH–COOH), 5.01 (m, 1H; HOOC–CH2=CH–CO–O–CH–), 3.66 (s, 3H; –CH2–COO–CH 3 ), 2.30 (t, 2H; –CH 2–COOCH3), 1.61 (broad, 6H; –CH 2–CH2–COOCH3 and –CH2–CH2–CH 2–CHO–CH 2–), 1.26 (broad s, 22H; chain –CH 2), 0.88 (t, 3H; –(CH)2–CH 3); ESI MS: m/z 413 (M+1), 435 (M+Na), 451 (M+K), 297 (M–OOCH=CH–COOH).
Synthesis of Disodium-4-[(18-Methoxy-18-Oxooctadecan-7-Yl)Oxy]-4-Oxo-2-Sulfonatobutanoate
In a 100 mL round bottom flask attached to a water condenser, about 6.4 g of HSME (0.0155 M) was obtained and stirred at 70 °C. About 1.25 mL of 50 % aqueous sodium hydroxide solution (0.0155 M) was added into the stirred solution followed by the addition of isopropanol (6.0 mL). After 15 min, an aqueous solution of sodium bisulphite (1.61 g, 0.0155 M) was added in three equal instalments (0.25, 0.5, and 0.5 % aqueous solution) at 0.5 h intervals. Progress of the reaction was monitored by TLC (using hexane:ethyl acetate, 90:10 v/v as eluent) as well as estimation of %SO3 by the hyamine method. After 2 h of total reaction time, the TLC profile indicated the complete disappearance of starting material. The hyamine method showed 14.1 % SO3 fixation indicating 94.6 % conversion (theoretical value for %SO3 is 14.86 for complete conversion).
After completion of reaction and usual workup as mentioned above, the solid was dried in desiccators under vacuum for 5 h and weighed to obtain 7.78 g (isolated yield, 93.2 %) of white amorphous solid having a melting point in the range of 255–257 °C. The compound was characterized by IR, NMR (1H and 13C), and mass spectroscopy. IR (cm−1): 3,415 (O–H stretching), 3,020, 2,928, 2,856 (aliphatic C–H stretching), 1,724 (methyl ester carbonyl stretching), 1,612 (succinic carbonyl stretching) 1,401 (C–H scissoring and S=O stretching), 1,216, 1,048 (C–O stretching), 766 (C–H rocking); 1H NMR (500 MHz, CDCl3): δ (ppm) 4.77 (broad, 1H; NaOOC–CH(SO3Na)–CH2–CO–O–CH–), 3.66 (s, 1H; –CH2–COO–CH 3 ), 3.00 (broad, 3H; NaOOC–CH(SO3Na)–CH 2–CO–O–CH–), 2.29 (t, 2H; –CH 2–COOCH3), 1.6 (broad, 2H; –CH 2–CH2–COOCH3) 1.5 (broad, 4H; –CH2–CH2–CH 2–CHO–CH 2–), 1.25 (broad s, 22H; chain –CH 2), 0.88 (t, 3H; –(CH)2–CH 3); ESI MS: m/z 411 [M–SO3]+, 493 [M+1]+, 515 [M+Na]+; 13C NMR (75 MHz, CDCl3): 174 (3 carbonyl carbons C=O), 75 (OOC–CH2–CH–SO3), 51 (–CH2–COOCH3), 34 (2 CH2–C=O), 31–22 (chain –CH2–), 14 (–(CH)2–CH3); HRMS (m/z) calculated for C23H40O9S is 493.23859, found 493.23884.
Analytical Methods
Purity of methyl esters of RA and 12-HSA was determined by a GC (Agilent 6890N series gas chromatograph) equipped with a flame ionization detector (FID) on a split injector with a split ratio of 50:1. A non-polar capillary column (HP-1, 30 m × 0.25 mm ID × 0.25 µm) was used for separation. The oven temperature was programmed at 150 °C for 2 min, increased to 300 °C at 10 °C/min and held for 20 min at 300 °C. The injector and detector temperatures were maintained at 280 and 300 °C, respectively. The nitrogen gas was used as carrier gas, and the flow rate was maintained at 1 mL/min. 1H and 13C NMR spectra were recorded on 500 and 75 MHz (Varian, Palo Alto, USA) spectrometers, respectively. HRMS data were recorded on a Thermo Scientific Exactive Orbitrap Mass spectrometer (Germany) and are given in mass units (m/z). A Waters LC–MS mass spectrometer (Palo Alto, USA) was used to record ESI MS spectra, and the data was recorded in the ESI mode, represented in mass units (m/z). Melting point determination was carried out using a Branstead electrothermal melting point apparatus.
Synthesized compounds were crystallized from ethanol before evaluating their surface properties. The surface tension was measured using a Kruss K100 Tensiometer (Krüss GmbH, Hamburg, Germany) equipped with a platinum ring having a mean circumference of 6 cm. The surface tension (γ) was measured at different concentrations by adding a subsequent volume of stock surfactant solution with a 765 Dosimat (Metrohm), connected with the system. All surface tension measurements were made at 27 °C.
The hyamine method [11] was used for the estimation of %SO3 in the sulfosuccinate, and a brief description of the method is given below:
-
(a)
Preparation of indicator solution dissolve 30 g of Na2SO4 with 6.6 mL of conc H2SO4 in water followed by the addition of 0.03 g of methylene blue powder. The entire mixture was made up to 1 L with water.
-
(b)
Hyamine solution 1.788 g of hyamine was solubilized in 1 L of water.
-
(c)
Standardization of hyamine solution 0.5 % Na-lauryl sulfate solution (prepared by dissolving 0.5 g of SLS in 100 mL of water) was obtained in a conical flask followed by the addition of 25 mL of chloroform, 25 mL of methylene blue indicator solution and shaken well. The blue color will be in the bottom layer. The entire solution was then titrated against the standard hyamine solution till the intensity of blue colour in top and bottom layer is identical.
$$\begin{aligned} {\text{Strength}}\;{\text{of}}\;{\text{hyamine}}\;{\text{solution}}\;({\text{Hyamine}}\;{\text{factor}}) = 27.4/{\text{Titre}}\;{\text{value}} \hfill \\ (\% SO_3 \;in\;SLS\;is\;27.4,\;on\;the\;basis\;of\;99\,\% \;purity) \hfill \\ \end{aligned}$$ -
(d)
Procedure 0.5 % solution of the reaction mixture was prepared in water. About 10 mL of the above solution was obtained in a conical flask followed by the addition of 25 mL of chloroform, 25 mL of methylene blue indicator solution, and shaken well. The blue colour will be in the bottom layer. The entire solution was then titrated against the standard hyamine solution till the intensity of blue color in top and bottom layer is identical.
$$\% {\text{SO}}_3 \;{\text{content}}\;{\text{in}}\;{\text{the}}\;{\text{sample}} = \;{\text{Titre}}\;{\text{value}} \times {\text{Hyamine}}\;{\text{factor}} .$$
Foaming properties were evaluated at ambient temperature using Ross-Miles Pour Foam apparatus [12], having a jacketed cylindrical column of 90 cm height and 5 cm internal diameter. The studied surfactant solution (0.025 %) was taken in a 200 mL pipette with an orifice of 3 mm and fixed at the top of the column containing the same test solution (50 mL). The surfactant solution was allowed to drop from the top to the same solution obtained in the column. Foam height obtained initially and after a regular interval of time was recorded on the scale attached to the column. Emulsification properties of the studied surfactant solutions (0.025 %) were determined according to the method described in the literature [13]. Equal volumes (40 mL) of surfactant solution and paraffin liquid (light) were obtained in a 500-mL Erlenmeyer flask, and the mixture was given ten downward strokes and transferred immediately to a 100-mL measuring cylinder. Time taken for the separation of 10 and 20 mL of the aqueous phase solution was determined. For estimating the wetting time, the Draves–Clarkson method was employed [14]. Briefly, skeins of 34 cm circumference weighing 5.0 ± 0.05 g were prepared from unbleached grey carded Indian yarn of single 20’s. A hook weighing 4.5 g carrying a lead anchor weighing 27.1 g was attached to the skeins and sinking times were determined on a surfactant solution (0.025 %) obtained in a 500 mL measuring cylinder. Calcium tolerance of a surfactant solution is defined as the amount of Ca2+ ion required to make 1 mL of surfactant solution turbid and was determined by a modified Hart’s method [15]. Surfactant solution (0.025 %, 50 mL) was obtained in a conical flask and titrated against 1 % calcium acetate solution in water, obtained in a 50 mL burette. Titration was carried out till the turbidity of the solution just obscured a strip of printed paper fastened to one side of the beaker.
Statistical Analysis
Results reported in the present work are the mean of three measurements (presented as mean ± SD) and were analyzed by a paired Student’s t test to evaluate the level of statistical significance. Differences were assessed by one-way analysis of variance. A P value <0.05 was considered significant.
Results and Discussion
Mono- and di-alkyl sulfosuccinates are one of the most important industrial anionic surfactants [1, 2]. Synthesis involves malenization of alcohol, mostly linear followed by sulfonation of the maleate monoester/diester. RA or methyl ricinoleate is a specialty fatty acid present in castor oil. The malenization of methyl ricinoleate is not new and has been reported in the literature in synthesizing monoester. A Chinese patent [16] synthesized ricinoleate maleate monoester sodium salt and used it as a monomer for the preparation of lipophilic acrylic resin having application in the leather industry. There are some literature reports wherein malenization of castor oil as such or methyl ricinoleate/methyl isoricinoleate was carried out for their subsequent application in the detergent formulation [5, 17]. There is no report wherein the surface properties of sulfosuccinated methyl ricinoleate or its saturated analogue has been studied. In the present work, we report synthesis of methyl ricinoleate and methyl-12-hydroxy stearate-based disodium sulfosuccinates and evaluated their surface properties using a surface tensiometer.
Synthesis involves initially ring opening of maleic anhydride with fatty alcohol followed by sulfonation over the double bond of maleic ester. Methyl ricinoleate and methyl-12-hydroxystearate was prepared, respectively, from castor oil and hydrogenated castor oil following a base catalyzed transesterification [18, 19]. Both esters were purified by column chromatography and analyzed by GC. The purity of methyl ricinoleate and methyl-12-hydroxystearate was found to be 99.3 and 97.2 %, respectively. As both RA and 12-HSA possess secondary alcohol at the C-12 position, initial esterification reaction conditions were optimized for maximum formation of monomaleate. Final optimized conditions are: 1.0:1.5 M ratio of methyl ricinoleate and maleic anhydride; temperature, 90 °C; time 5 h, and slow addition of maleic anhydride (in three equal instalments) over 0.5 h.
Surface Activity
All test solutions were prepared in Milli-Q water and measured for surface tension and CMC using Krüss K100 tensiometer. Variation of surface tension as a function of the logarithm of surfactant concentration is shown in Fig. 1. The CMC is determined from the intersection of two linearly fitted lines. Each measurement is an average of three independent measurements. Surface properties of RSS and HSS obtained by the surface tension method are given in Table 1. It was found that the CMC of RSS is roughly twice the CMC of HSS (P < 0.001). Such an effect of unsaturation in the hydrophobic chain of an ionic surfactant on its CMC has been reported earlier in the literature [20]. The surface tensions at CMC of the two synthesized surfactants are found to be in the range of 36–42 mN/m. The pC 20 is an index of surface tension reduction efficiency, where C 20 is the molar concentration of surfactant require to reduce the surface tension of water by 20 mN/m. the effect of unsaturation is also apparent in pC 20 values, showing lower values for RSS than HSS. However, both surfactants possess higher efficiency of reduction of surface tension, which can be assessed by their higher pC 20 values. Higher values of pC 20 (>3) indicates a higher hydrophobic character of the surfactant, resulting in higher efficiency of reduction of surface tension [21].

Plot of surface tension (γ) versus ln C of the two studied surfactants (RSS and HSS)
The surface excess, Γmax (mol/cm2), i.e., the amounts of surfactant adsorbed per unit area at the air/water interface after complete monolayer formation and minimum surface area occupied by the each surfactant molecule (A min) were calculated from the slope of the linear part of the surface tension plot using Gibbs adsorption isotherm [22].
where R is the gas constant (8.314 J/mol K), T the absolute temperature, γ the surface tension, C the surfactant concentration, n the number of species constituting the surfactant adsorbed at the interface, and N is Avogadro’s number. The value of n is considered as 3 due to the dual charged nature of the studied surfactant with univalent counterions [23, 24].
Both Γmax and A min values of the two sulfosuccinates are given in Table 1. These two parameters predict the packing pattern and orientation of the surfactant molecule at the air–water interface and, hence, are dependent on alkyl chain length or hydrophobicity of the surfactant. As HSS is the saturated analog of RSS, there are no significant differences in the values of Γmax and A min. However, a marginal decrease in Γmax value and increase in A min value for HSS compared to RSS is observed. Higher A min value of saturated analogue indicates that the molecule is less tightly packed at the interface compared to its unsaturated counterpart. Lower A min value of RSS also indicates its effectiveness to be adsorbed at the air–water interface compared to HSS. Such minor compactness and effectiveness of RSS may be attributed to the double bond in its hydrophobic chain. HSS possess slight flexibility in the hydrocarbon chain compared to RSS at the air–water interface.
The adsorption of a surfactant at the air–water interface and its micellization in the bulk aqueous solution occurs simultaneously. The free energy of adsorption (ΔG°ads) and free energy of micellization (ΔG°mic) of the synthesized surfactants at the air–water interface was evaluated using the following equations [25, 26].
where R is gas constant 8.314 J/mol K, n = 3 and T is absolute temperature.
The presence of two ionogenic groups in the head group region increases repulsion and is responsible for the low free energy of micellization [27]. However, there is no significant difference in values of ΔG°ads and ΔG°mic (Table 1) between RSS and HSS. The negative values of ΔG°ads and ΔG°mic indicates that both adsorption and micellization has happened spontaneously at 27 °C. Generally, a surfactant with high ΔG°ads and low ΔG°mic will be more favourable for adsorption rather than micellization and, hence, will exhibit higher CMC and vice versa. In this case, low ∆G°ads value compared to ∆G°mic indicates that adsorption of both RSS and HSS at the interface is associated with a decrease in the free energy of the system and, hence, exhibited low CMC.
The 0.1 % (w/v) aqueous solutions of RSS and HSS were also evaluated for surface active properties such as wetting time, calcium tolerance, emulsion stability, and foamability and compared with sodium lauryl sulphate (SLS) (Table 2). The foamability is nothing but the ability to produce foam immediately after agitation, and the foam stability is estimated by studying the foam volume after some time. Both RSS and HSS exhibited moderate foam height and poor foam stability compared to SLS. This is expected as sulfosuccinates are, in general, mild surfactants, and mildness and foamability is inversely related [2]. Emulsification properties of both RSS and HSS are better than SLS. Sulfosuccinates are reported to possess moderate wetting power and are dependent on hydrophilicity of the sulfosuccinates [2]. The wetting time of RSS is more than HSS, but later matched well with SLS. Hard water resistance of this class of surfactants is generally very good. In the present study, both RSS and HSS exhibited excellent calcium tolerance, better than SLS.
Conclusions
Two branched dianionic sulfosuccinates were synthesized from renewable castor oil. A new strategy was developed for efficient synthesis of maleic esters of secondary alcohol. Synthesized surfactants differ by the presence of a double bond in the hydrophobic part, which is found to influence CMC. Both the surfactants exhibited excellent calcium tolerance, emulsion and wetting properties, but possess moderate foam height and poor foam stability. Low wetting times of these surfactants provide the possibility of substituting the potential wetting agents like aerosols, which have similar structural features. Preferential orientation of the hydrophobic tail might serve as an interesting and flexible platform for further studies on the self assembly of aggregates formed by these surfactants. These two surfactants may find applications in some industry, especially the textile industry, where it is desirous to have a surfactant with good wetting and low foam properties alongside mild surface activity.
References
Rosen MJ (2004) Surfactants and interfacial phenomenon: characteristic feature of surfactant, chap 1, 3rd edn. Willey, NJ, pp 1–32
Domsch A, Irrgang B (1995) Sulfosuccinates. In: Stache HW (ed) Anionic surfactant: organic chemistry. Surfactant science series. CRC Press, New York, pp 502–546
Deepika Tyagi VK (2006) Sulfosuccinates as mild surfactants. J Oleo Sci 55:429–439
Bloch M, Inglis RP, Koebner A (1977) Process for preparing sulfosuccinate. US Patent 4,039,562
Ahmad I, Mahajan V, Bhalla M (2003) Reaction of maleic anhydride with methyl ricinoleate and isoricinoleate. J Oil Technol Assoc India 35:99–102
Rogerio SM, Regina CC, Claudia L, Luciana F (2006) A non-rinse-off cosmetic composition for skin and lips comprising ricinoleyl monomaleate triglyceride and palmitic acid. PCT Int. Appl. WO 2006105628
Ounniyi DS (2006) Castor oil: a vital industrial raw material. Biores Technol 97:1086–1091
Mutlu H, Meier MAR (2010) Castor oil as a renewable source for the chemical industry. Eur J Lipid Sci Technol 112:10–30
Trivedi RK, Vasistha AK (1988) Low pressure hydrogenation of castor oil. J Am Oil Chem Soc 65:1467
Schwitzer MK (1990) Castor oil. In: Applewhite TH (ed) Proceedings of world conference on oleochemicals into the 21st century. AOCS Press, Champaign, pp 111–118
ASTM D1681-05 (2014) Standard test method for synthetic anionic active ingredient in detergents by cationic titration procedure. doi:10.1520/d1681-05
Zocchi G (1999) In: Broze G (ed) Handbook of detergents, part A—surfactant properties. Surfactant science series, vol 82. Marcel Dekker, NY, pp 424–427
Subrahmanyam VVR, Achaya KT (1961) Structure and surfactance—evaluation of ricinoleyl alcohol. J Chem Eng Data 6:38–42
Indian Standard No. 1185 (1957) Bureau of Indian Standards, New Delhi
Wilkes BG, Wickert JN (1937) Synthetic aliphatic penetrants. Ind Eng Chem 29:1234–1239
Ding X, Xue Q, Peng Y (2013) Preparation method of soft acrylic resin leather retanning agent. Chinese patent CN 103255243 B
Vibhute BP, Khotpal RR, Karadbhajane VY, Kulkarni AS (2013) Preparation of maleinized castor oil (MCO) by conventional method and its application in the formulation of liquid detergent. Int J ChemTech Res 5:1886–1896
Christie WW (1972) The preparation of the alkyl esters from fatty acids and lipids. In: Gunstone FD (ed) Topics in lipid chemistry. Elek Sci, London, pp 171–197
Karuna MSL, Reddy JRC, Rao BVSK, Prasad RBN (2005) Lipase-mediated synthesis of alkyl ricinoleates and 12-hydroxy stearates and evaluation of the surfactant properties of their sulfated sodium salts. J Surfactants Deterg 8:271–276
Shobha J, Balasubramanian D (1986) Hairpin looping of terminally functionalized carboxylate surfactants. J Phys Chem 90:2800–2802
Sreenu M, Nayak RR, Prasad RBN, Sreedhar B (2014) Synthesis, surface and micellar properties of N-oleoyl amino acid. Colloids Surf A Physicochem Eng Asp 449:74–81
Kanjilal S, Maiti S, Kaimal TNB (1999) Synthesis and physicochemical studies of methyl-12-[1′-β-d-lactosyl]-octadec-9-ene-1-oate: a biosurfactant analog. J Surfactants Deterg 2:531–538
Li X, Hu Z, Zhu H, Zhao S, Cao D (2010) Synthesis and properties of novel alkyl sulfonate gemini surfactants. J Surfactants Deterg 13:353–359
Javadian S, Aghdastinat H, Tehrani-Bagha A, Gharibi H (2013) Self-assembled nano structures of cationic ester-containing gemini surfactants: the surfactant structure and salt effects. J Chem Thermodyn 62:201–210
Mukherjee P (1967) The nature of the association equilibria and hydrophobic bonding in aqueous solutions of association colloids. Adv Colloid Interface Sci 1:242–275
Moulik SP, Haque ME, Jana PK, Das AR (1996) Micellar properties of cationic surfactants in pure and mixed states. J Phys Chem 100:701–708
Kolesnikova EN, Glukhareva NA (2008) Micellization in solutions of anionic surfactants with two ionogenic groups. Colloids J 7:184–188
Acknowledgments
PPK gratefully acknowledge the Senior Research Fellowship from the Council of Scientific and Industrial Research (CSIR), Government of India.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Kumar, P.P., Ramesh, P. & Kanjilal, S. Sulfosuccination of Methyl Ricinoleate and Methyl 12-Hydroxy Stearate Derived from Renewable Castor Oil and Evaluation of Their Surface Properties. J Surfact Deterg 19, 447–454 (2016). https://doi.org/10.1007/s11743-016-1804-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11743-016-1804-0