
Abstract
Nonionic–zwitterionic hybrid surfactants, namely N,N-dimethyl-N-dodecyl polyoxyethylene (3) amine oxide, and the corresponding betaine and/or sulfobetaine, have been synthesized. Their molecule structures were characterized by means of electrospray ionization mass spectrometry and 1H nuclear magnetic resonance. Compared to the structurally related conventional amine oxide, betaine and sulfobetaine, the critical micelle concentrations of the three hybrid surfactants are one order of magnitude smaller than those of traditional ones. The polyoxyethylene segment between the hydrophilic head group and the hydrophobic tail makes the surface activities of the hybrid surfactants superior to their structure-related counterparts.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ethoxylated surfactants, especially those having a chain segment of polyoxyethylene between hydrophilic head group and hydrophobic tail, are called hybrid surfactants [1, 2]. The traditional representatives are alcohol ether sulfates (AES) [3, 4], alcohol ether carboxylates (AEC) [5] and sodium alkyl-benzyl polyoxyethylenated propane sulfonates (ABEPS) [2]. These nonionic–ionic hybrid surfactants always present combined properties of nonionic and ionic surfactants [2] and have better electrolyte and water hardness tolerance. However, the literature reports on hybrid surfactants are mainly on nonionic–anionic surfactants, the synthesis and the surface activity of nonionic–zwitterionic hybrid surfactants have seldom been reported up till now.
One US patent [6] reveals the synthesis of N,N-dimethyl-N-alkyl polyoxyethylene amine and the corresponding amine oxides from commercial available AES. One Japanese patent [7] reveals the synthesis of N,N-dimethyl-N-alkyl polyoxyethylene amine-based betaines from commercial available fatty alcohols. However, the typical physicochemical properties of these hybrid surfactants, such as the critical micelle concentration (CMC), the surface tension at CMC (γ CMC), the adsorption efficiency (pC 20), the surface pressure at the CMC (Π CMC), the maximum surface excess (Γ m), the minimum cross-sectional area (A min) have not been reported. Due to the length distribution of the alkyl tail and the polyoxyethylene segment in commercially available AES and polyethoxylated fatty alcohols, the corresponding amine oxides and betaines are mixtures of different molecules. In order to get the typical surface activity parameters of nonionic–zwitterionic hybrid surfactants and to compare them with structure-related traditional ones, three hybrid surfactants, namely N,N-dimethyl-N-dodecyl polyoxyethylene (3) amine-based amine oxide (C12EO3AO), and the corresponding betaine (C12EO3Be) and sulfobetaine (C12EO3SBe) were synthesized (see Scheme 1). The molecular structures of the main intermediates and objective surfactants were confirmed by means of electrospray ionization mass spectrometry (ESI–MS), and 1H nuclear magnetic resonance (1H NMR), respectively. The typical physicochemical properties of CMC, γ CMC, pC 20, Γ m, and A min were determined and calculated from the results of γ-logc (concentration of surfactant) curves.

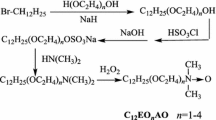
The synthesis routes of C12EO3AO, C12EO3Be and C12EO3SBe
Experimental Section
Materials
1-Bromododecane (Br-C12), triethylene glycol (EO3), sodium hydride (NaH), dimethylamine (DMA), sodium chloroacetate, 1,3-propanesultone and H2O2 (30 wt%) were used as received without further purification. Deionized water was obtained from a Millipore Milli-Q water purification system (Millipore, USA).
Synthesis of C12EO3
As shown in Scheme 1 (Step 1), NaH (4.80 g) and EO3 (72.08 g) were added to a 250-mL three-neck round-bottom flask. Br-C12 (26.54 g) was added drop wise to the flask with stirring at 100 °C. The reaction was protected by N2 and completed in 48 h. Then, 80 mL ethyl acetate was added to the flask. The excess EO3 and the by-product polyethylene glycol were washed off using saturated NaCl aqueous solution. The faint yellow crude product was obtained after the solvent was removed by rotary evaporator under reduced pressure. Finally, the crude C12EO3 was purified by silica gel column chromatography. A mixture of mineral ether–ethyl acetate (volume ratio 2:1) was used as the eluent. A colorless liquid of C12EO3 was obtained with a yield of 61 %.
Synthesis of C12EO3A
HSO3Cl (12.82 g) was added dropwise to C12EO3 (31.8 g) at 25–30 °C, after that, the system was aged for 0.5 h. After being neutralized by 15 % NaOH aqueous solution, the product of C12EO3SD (Scheme 1, Step 2) was obtained. According to the method in [6, 8], a light yellow oil liquid of C12EO3A (Scheme 1, Step 3) was synthesized with a yield of 94 %.
Synthesis of C12EO3AO
As shown in Scheme 1 (Step 4), C12EO3A (34.51 g) was oxidized by 30 % H2O2 (45.37 g, dropwise) at 55–65 °C with a trace amount of citric acid. The crude C12EO3AO was extracted by CHCl3 and was purified by alumina column chromatography. A mixture of CHCl3–CH3OH (volume ratio 60:1) was used as the eluent. C12EO3AO was obtained with a yield of 91 %.
Synthesis of C12EO3Be
In a 250-mL round-bottom flask, C12EO3A (17.31 g) and sodium chloroacetate (5.83 g) were dissolved in 100 mL methanol–H2O (volume ratio 1:1), the pH of the above system was adjusted to 7–8 by NaHCO3. The reaction was carried out at 70 °C for 2 h, then 80 °C for a further 2 h. The solvent was removed by rotary evaporation under reduced pressure. The residue was dried in a vacuum oven at 70 °C. The dried residue was dissolved in isopropanol, and inorganic salts were filtered off. The crude C12EO3Be was purified by silica gel column chromatography. A mixture of ethyl acetate–CH3OH (volume ratio 3:1) was used as the eluent. C12EO3Be was obtained with a yield of 89 %.
Synthesis of C12EO3SBe
In a 250-mL round-bottom flask, C12EO3A (17.31 g) was dissolved in 80 mL acetone in an ice-water bath, then 6.72 g 1,3-propanesultone in 80 mL acetone was added dropwise to the flask with stirring. After the addition of 1,3-propanesultone, the temperature of the system was elevated to room temperature naturally. The reaction was carried out for 5 days at room temperature with stirring. The white crude C12EO3SBe was purified by silica gel column chromatography. A mixture of CHCl3–CH3OH (volume ratio 4:1) was used as the eluent. C12EO3SBe was obtained with a yield of 76 %.
Analytical Methods
The ESI–MS measurements were carried out using a Maldi Synapt Q-TOF MS (Waters) in the positive ion mode (Fig. 1, ESM Figure 1S). The 1H NMR spectra were obtained with an Avance III 400 MHz digital NMR spectrometer (Bruker) using CDCl3 as solvent (Fig. 2). And the NMR peak of CDCl3 (δ = 7.26 ppm) was used as the reference in determining the chemical shifts (δ, ppm) of the protons.

The ESI–MS spectrum of C12EO3AO with positive ion mode

The 1H NMR spectrum of a C12EO3, b C12EO3A, c C12EO3AO, d C12EO3Be and e C12EO3SBe
The surface tension (γ, mN m−1) measurements were conducted with a drop volume tensiometer at 25 ± 0.1 °C. The outer radius of the glass capillary was 0.66 mm. During the procedure of γ measurements, a sufficient aging time was necessary for the pendant drop surface to reach an equilibrium state. The aging time of the drop surface was determined by the plot of γ versus the drop detachment time t (min) (Fig. 3). Finally, the drop volume was corrected by the Harkins–Brown method [9]. The surface activity parameters, such as the CMC, the γ at CMC (γ CMC), the adsorption efficiency (pC 20), the surface pressure at the CMC (Π CMC), the maximum surface excess (Γ m), and the minimum cross-sectional area (A min) were obtained or calculated [10] from the γ-logc curves (Fig. 4).

The plot of surface tension (γ in mN m−1) versus drop surface aging time (t in min) for C12EO3AO and C12EO3Be aqueous solution at 25 °C, the concentration of the surfactant was 3.0 × 10−5 mol L−1

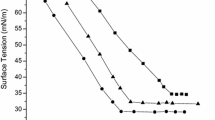
The plots of the surface tension (γ in mN m−1) versus logc (concentration of surfactant in mol L−1) at 25 °C
Results and Discussion
Structural Characterization of C12EO3, C12EO3A, C12EO3AO, C12EO3Be and C12EO3SBe
The ESI–MS technique was employed to measure the molecule weight of the products in this work. The ESI–MS spectrum of C12EO3AO is shown in Fig. 1. With the positive ion mode, various ions of [M+H]+, [M+Na]+ and [2M+H]+ were detected. The ESI–MS spectra of the rest samples (surfactants of C12EO3Be and C12EO3SBe, intermediates of C12EO3 and C12EO3A) can be seen in Figure 1S as ESM. The results obtained of the mass-to-charge ratio (m/z) for the three hybrid surfactants match well with the calculated results. The interpretation of the ESI–MS spectra can be seen in detail in the ESM.
In order to obtain the detailed molecule structural information, the 1H NMR spectra of the objective compound were obtained (Fig. 2). C 12 EO 3 1H NMR [400 MHz, (CDCl3)] δ = 0.86–0.90 (t, 3H, –CH 3), δ = 1.26–1.29 (m, 18H, –(CH 2)9–), δ = 1.54–1.61 (m, 2H, C10–CH 2–CH2–O), δ = 2.66 (s, 1H, –OH), δ = 3.43–3.47 (t, 2H, C10–CH2–CH 2–O), δ = 3.58–3.74 (m, 12H, C12–(OC2 H 4)3–). C 12 EO 3 A 1H NMR [400 MHz, (CDCl3)] δ = 0.86–0.90 (t, 3H, –CH 3), δ = 1.26–1.29 (m, 18H, –(CH 2)9–), δ = 1.56–1.59 (m, 2H, C10–CH 2–CH2–O), δ = 2.26 (s, 6H, N–(CH 3)2), δ = 2.49–2.52 (t, 2H, –CH 2–N), δ = 3.43–3.46 (t, 2H, C11–CH 2 –O), δ = 3.57–3.66 (m, 10H, C12–(OC2 H 4)2–OCH 2–). C 12 EO 3 AO 1H NMR [400 MHz, (CDCl3)] δ = 0.86–0.90 (t, 3H, –CH 3), δ = 1.26–1.29 (m, 18H, –(CH 2)9–), δ = 1.55–1.59 (m, 2H, C10–CH 2–CH2–O), δ = 3.25 (s, 6H, N–(CH 3)2), δ = 3.40–3.44 (t, 2H, C11–CH 2–O), δ = 3.48–3.52 (t, 2H, –CH 2–N), δ = 3.55–3.65 (m, 8H, C12–(OC2 H 4)2–O), δ = 4.00 (t, 2H, –O–CH 2CH2N). C 12 EO 3 Be 1H NMR [400 MHz, (CDCl3)] δ = 0.86–0.89 (t, 3H, –CH 3), δ = 1.26 (m, 18H, –(CH 2)9–), δ = 1.52–1.57 (m, 2H, C10H21–CH 2–CH2–O), δ = 3.34 (s, 6H, N–(CH 3)2), δ = 3.41–3.44 (t, 2H, –CH 2–N), δ = 3.55–3.56 (t, 2H, C11–CH 2–O), δ = 3.60–3.61 (t, 2H, O–CH 2–CH2N), δ = 3.62–3.65 (m, 4H, C12–O–C2 H 4–O), δ = 3.88 (s, 2H, N–CH 2COO), δ = 3.93–3.96 (m, 4H, O–C2 H 4–OC2H4N). C 12 EO 3 SBe 1H NMR [400 MHz, (CDCl3)] δ = 0.86–0.90 (t, 3H, –CH 3), δ = 1.29 (m, 18H, –(CH 2)9–), δ = 1.52–1.57 (m, 2H, C10H21–CH 2–CH2–O), δ = 2.27 (m, 2H, –CH2–CH 2–CH2–S), δ = 2.90 (t, 2H, –CH 2–CH2–CH2–S), δ = 3.28 (s, 6H, N–(CH 3)2), δ = 3.43–3.45 (t, 2H, –CH 2–N), δ = 3.50–3.55 (t, 2H, C11–CH 2–O), δ = 3.57–3.67 (m, 8H, C12H25–(OC2 H 4)2–O), δ = 3.79 (t, 2H, O–CH 2–CH2N), δ = 3.95 (t, 2H, –CH 2–S).
The Surface Activity Parameters of C12EO3AO, C12EO3Be and C12EO3SBe
As shown in Fig. 3, the γ of the solution (3.0 × 10−5 mol L−1) decreases with the drop detachment time increasing with breakpoints at t ≈ 3 min (for C12EO3Be) and t ≈ 6 min (for C12EO3AO), respectively. The result of C12EO3SBe is similar to that of C12EO3Be. The change of γ is <±0.1 mN m−1 in the region of t > 10 min. Under this condition, the γ could be considered as an equilibrium surface tension. The lower concentration sample solution needs longer aging time for the drop surface to reach the equilibrium state. Thus, in order to make the drop surfaces reach the equilibrium state, all the drops of the surfactant solutions were aged for at least 10 min.
The plots of the γ versus logc for the hybrid surfactants in this work and the corresponding traditional surfactants are shown in Fig. 4. One can see that the γ gradually decreases to a plateau region with the surfactant concentration increasing. A decrease in surface tension indicates that the surfactant molecules adsorbed at the air-solution interfaces. The break point appearing in the γ-logc curves suggests the formation of micelles in aqueous solutions. Here, it is worth mentioning that no evidence of surface tension minima appears in the γ-logc curves [11]. The surface activity parameters, such as CMC, γ CMC, pC 20, Π CMC, Γ m and A min were obtained and/or calculated [10] from the γ-logc curves and are listed in Table 1.
The CMC for N,N-dimethyl-N-dodecyl amine oxide (C12AO) is 1.18 × 10−3 mol L−1 (ring method) [12], for N,N-dimethyl-N-dodecyl betaine (C12Be) 2.00 × 10−3 mol L−1 (ring method) [13], and for N,N-dimethyl-N-dodecyl sulfobetaine (C12SBe) is 2.20 × 10−3 mol L−1 (maximum bubble pressure method) [14], respectively. As can be seen in Table 1, the CMC values of C12AO, C12Be and C12SBe in this work match well with those in the literature [12–14] despite the difference in the γ determination methods. All the CMC values of the hybrid surfactants in this work fall within the 10−4 mol L−1 range, which is one order of magnitude smaller than those of structure related traditional surfactants. The similar relations hold true for sodium dodecyl sulfate and dodecyl polyoxyethylene sulfates [2, 15]. It could be attributed to the hydrophilicity of the hybrid surfactants in this work ‘continuously’ increasing from hydrophobic (C12) to less hydrophobic/less hydrophilic (EO3) then to hydrophilic (AO, Be, and SBe). As regards the change in the hydrophilicity at the water–air interface, one can argue that it changes from hydrophobic (air) over a less hydrophobic/less hydrophilic interfacial zone (EO3) between the two bulk phases to hydrophilic (water) [1]. However, the hydrophilicity of the structure-related counterparts changes ‘discontinuously’ from hydrophobic tails to hydrophilic head groups without a less hydrophobic/less hydrophilic transition zone (EO3), which, in turn, explains the higher surface activity of the hybrid surfactants in this work.
If one looks at the structure difference among the hybrid surfactants in this work, one sees the main structure difference lies in the head groups of AO, Be and SBe. Due to AO being a totally non-ionic head group, it has the smallest hydrophilicity among these three head groups. Be and SBe are zwitterionic head groups, possessing higher hydrophilicity than that of AO. However, the head group of Be is more hydrophilic than the SBe ones [14]. The hydrophilic difference of the head groups leads to the difference in the CMC values. The order of the CMC values for the hybrid surfactants in this work is CMCAO < CMCSBe < CMCBe, a similar relation can be observed in structure related un-ethoxylated ones. It could be attributed to the hydrophilic difference of the head groups [14].
References
Catanoiu G, Blunk D, Stubenrauch C (2012) Novel ethoxylated inositol derivatives—hybrid carbohydrate/oligoethylene oxide surfactants. J Colloid Interface Sci 371:82–88
Wang X, Yan F, Li Z, Zhang L, Zhao S, An J, Yu J (2007) Synthesis and surface properties of several nonionic–anionic surfactants with straight chain alkyl-benzyl hydrophobic group. Colloids Surf A 302:532–539
Tokiwa F, Ohki K (1967) Micellar properties of a series of sodium dodecylpolyoxyethylene sulfates from hydrodynamic data. J Phys Chem 71(5):1343–1348
Alargova RG, Ivanova VP, Kralchevsky PA, Mehreteab A, Broze G (1998) Growth of rod-like micelles in anionic surfactant solutions in the presence of Ca2+ counterions. Colloids Surf A 142:201–218
Gong R, Han L, Gao C, Shu M, Che S (2009) Molecular design of AEC tri-block anionic surfactant towards rational synthesis of targeted thick-walled mesoporous silica. J Mater Chem 19:3404–3411
Koebner A, Potts HA (1965) N-dialkyl-alkyl- and alkaryl-oxyalkyl-amine oxides. US Patent 3,206,512
Yoshinobu N (1985) Ampholytic surface-active betaine compound and production thereof. J.P. Patent 60,089,458
Lange F, Meffert A (1987) Process for preparing tertiary ether amines. US 4,650,865
Harkins WD, Brown F (1919) The determination of surface tension (free surface energy), and the weight of falling drops: the surface tension of water and benzene by the capillary height method. J Am Chem Soc 41:499–524
Liu XF, Hu J, Huang YJ, Fang Y (2013) Aggregation behavior of surface active dialkylimidazolium ionic liquids [C12Cnim]Br (n = 1–4) in aqueous solutions. J Surfactants Deterg 16:539–546
Mohamed A, Trickett K, Chin SY, Cummings S, Sagisaka M, Hudson L, Nave S, Dyer R, Rogers SE, Heenan RK, Eastoe J (2010) Universal surfactant for water, oils, and CO2. Langmuir 26:13861–13866
Goracci L, Germani R, Rathman JF, Savelli G (2007) Anomalous behavior of amine oxide surfactants at the air/water interface. Langmuir 23:10525–10532
Chevalier Y, Storet Y, Pourchet S, Perchec PL (1991) Tensioactive properties of zwitterionic carboxybetaine amphiphiles. Langmuir 7:848–853
Weers JG, Rathman JF, Axe FU, Crichlow CA, Foland LD, Scheuing DR, Wiersema RJ, Zielske AG (1991) Effect of the intramolecular charge separation distance on the solution properties of betaines and sulfobetaines. Langmuir 7:854–867
Rosen MJ (2004) Surfactants and interfacial phenomena, 3rd edn. Wiley, New Jersey, pp 129–135
Acknowledgments
This work was jointly supported by the National Natural Science Foundation of China under Grant No. 21071065, the Scientific Research Foundation for the Returned Overseas Chinese Scholars (2011), State Education Ministry, and Qinlan Project (2010) of Jiangsu Province.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article
Cite this article
Hou, L., Zhang, H., Chen, H. et al. Synthesis and Surface Properties of N,N-Dimethyl-N-dodecyl Polyoxyethylene Amine-Based Surfactants: Amine Oxide, Betaine and Sulfobetaine. J Surfact Deterg 17, 403–408 (2014). https://doi.org/10.1007/s11743-013-1540-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11743-013-1540-7