
Abstract
Salt in saline land is regarded as a kind of abiotic stress that limits the productivity of plants and their geographical distribution. To understand the mechanism of how shrub willow clones seedling respond to salt stress at the proteomic level, proteins extracted from seedling leaves of salt sensitive cultivar JW9-6 and salt tolerant cultivar JW2372 were tested under salt stress for the different durations, including 2, 12 and 72 h, using 2-D electrophoresis. Totally, 83 differentially expressed proteins were found using MALDI-TOF/TOF MS. These proteins were divided into 11 classes. The primary findings from this study are: (1) enhanced ROS scavenging capacity leads to increased salt tolerance for the shrub willow that protects redox homeostasis system from being damaged; (2) different measures, e.g., the inhibition of protein synthesis, protein folding and assembly, and enhancing protein proteolysis, were essential for shrub willow seedlings to respond to salt stress; (3) salt stress could affect the pathways of photosynthesis, carbohydrate metabolism, energy supply, and metabolism for amino acid and nitrogen. (4) JW2372 are more salt tolerant than that of cultivar JW9-6 due to overall performance of the above pathways.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Shrub willow (Salix spp.), one of the most widely distributed tree species in the world, has its unique characteristics, e.g., grows apace and strong adaptation to different soil types and areas. The number of willow species is more than 190 and some of them are salt-tolerant to some extent, including S.integra Thrnb, salix mongolica, Salix triandra and JW 2372 (Sui et al. 2011).
Salt in saline and alkaline land is a major type of abiotic stress that limits the productivity of plants and their geographical distribution (Wiebe et al. 2007). Nearly 9,900 million km2 lands are affected by salinity in China (Askari et al. 2006). The salinization of arable land is anticipated to be increased by 30 % in the next 20 years, and it could be further increased by 50 % till 2050 (Wang et al. 2003). These saline and alkaline lands could lead to the reduction of forest area and deterioration of ecological environment. As a result, the development of agriculture and forestry using saline and alkaline lands has become a priority for modern biotechnology (Hoshida et al. 2000). Therefore, it is important to pursue a solid understanding of how plants respond to the salt stress. There are several different molecular mechanisms for salt stress tolerance, including detoxification of ROS, salt uptake/exclusion and compartmentalization, carbohydrate and energy metabolism, etc. (Zhao et al. 2013).
To study the mechanisms of shrub willow clones in response to salt stress, Jiangsu academy of forestry established an index to evaluate salt tolerance for shrub willow clones at the seeding stage (Sui et al. 2011). Results showed that different factors including plant height, relative water content and transpiration could be used as salt-tolerance evaluation indices. Additionally, clone JW 2372 was found better than clone JW9-6 in terms of salinity tolerance (Sui et al. 2011). Since there are few studies on the effects of salts on physical and physiological processes in willow, the mechanism of how shrub willow seedling leaves respond to salinity stress is not quite clear. Hence, it is necessary to use a proteomic strategy which has distinct advantages in molecular level to study this problem.
In this paper, a proteomic method was used to analyze leaves of the two shrub willow clones cultivars, JW9-6 (salt-sensitive) and JW2372 (salt-tolerant) to: (1) study expression pattern of the proteome; (2) examine the difference of the expressed proteins in responding to salt stress; and (3) understand how these proteins involved in pathways in the shrub willow clones seedling leaves. The mechanism of how shrub willow clones seedling leaves respond to salinity stress was discussed.
Materials and methods
Plant materials
Two Shrub willow clones (salt-sensitive type JW9-6 and salt-tolerant type JW2372) (17 cm) were cultivated in nutrient solution containing 1/4 Hoagland. It was replaced by fresh solution every 7 days. The simulation of salt stress was modified based on Sui et al. (2011). The seedlings were planted in a growing chamber under 20–30 °C temperature. Six-week-old seedlings were grown in each ampulla (350 ml) under 1/4 Hoagland nutrient solution that included 3 % NaCl. The control were grown in the 1/4 Hoagland nutrient solution. Leaves of Shrub willow clones collected at different salt stress exposure durations (0, 2, 12 and 72 h) were used immediately; or frozen in liquid nitrogen and stored under the temperature of −70 °C. Leaves of Shrub willow clones which were unstressed, were sampled at 2, 12 and 72 h, respectively, and used as control.
Protein sample preparation
For quality control, three biological repeats were carried out for each sample analysis. Willow leaves were extracted using acetone/TCA precipitation as described in Parker et al. (2006) with slight modifications. The powder of leaf samples was put in the solution of 10 % w/v trichloroacetic acid/acetone with 1 % (w/v) DTT at −20 °C for 2 h. After centrifugation and rinse, the size of the sample was 595 nm using the method described in Bradford (BioRad, Bradford), where bovine serum albumin was used as the standard.
2-D electrophoresis, gel staining, and image analysis
First-dimensional electrophoresis, in this study, was performed on an IPG-phor IEF system (Bio-Rad products from USA). About 1,200 μg protein was diluted in a two-dimensional rehydration buffer (4 % w/v CHAPS, 0.5 % v/v IPG buffer, 2 M thiourea, and 7 M urea) and added to each IPG strip (24 cm non-linear, pH 4–7). It was then rehydrated at 50 V and 20 °C for 13 h. Followed by this, it was treated on the IPG phor apparatus under the following conditions: 200 V for 1 h, 500 V for 1 h, 1,000 V for 2 h, 8,000 V for 4 h, and 8,000 V to reach 110,000 VH. Before using SDS-PAGE, the strips were equilibrated for 15 min in 10 ml of the equilibration buffer [6 M urea, 0.375 M Tris–HCl (pH 8.8), 2 % (w/V) SDS, 20 % glycerol (V/V), and 2 % (w/V) DTT] and then kept for another 15 min in alkylating equilibration buffer containing 2.5 % (w/V) iodoacetamide instead of 2 % DTT.
The second electrophoretic dimension was treated by 12 % SDS-PAGE. Gel electrophoresis was conducted at 16 °C with a 1.0 W/gel for 1 h and then mixed with 10 W/gel until the dye front at about 1 cm above the bottom of the gel. The signal was visualised using the equipment CBB G-250. Gel images were first digitalized with a Bio-Rad FluorS system and further analyzed using the software PDQuest (Version 7.2.0 of the software from BioRad). Protein spots were first identified and then matched automatically based on total density of gels. For each of the identified spot, the mean relative volume was assumed to be equal to its expression level at each stage. The spots with a mean relative volume that has a magnitude of change over 15 % or smaller than 6.6 % (P < 0.05) were treated as differentially expressed protein spots.
Protein in-gel digestion, protein identification, and database search
The differentially expressed proteins were manually identified and extracted from gels, by washing with double-distilled water and destaining twice with 50 mM NH4HCO3 in 50 % acetonitrile (ACN) for CBB G-250 staining spots. The proteins were then mixed with DTT of 10 mM in NH4HCO3 of 50 mM; and then alkylated in iodoacetamide of 40 mM and NH4HCO3 of 50 mM for 1 h at room temperature. The gel was first dried with 100 % ACN; and then digested overnight at 37 °C with an addition of 15 µL of trypsin (Promega, USA, 1:50, enzyme to protein) in 50 mM NH4HCO3. The resulting peptides were extracted twice with 0.1 % TFA in 50 % ACN. The samples were air-dried and analyzed with a 4800 MALDI-TOF/TOF Proteomics Analyzer (Applied Biosystems, USA).
All spectra of proteins were searched using a Mascot search engine (http://www.matrixscience.com) in the NCBInr databases. The following searching parameters were used: taxonomic category restrictions to Viridiplantae (Green plants). Mass tolerance for peptides is 100 ppm and mass tolerance of TOF/TOF fragments is 0.5 Da using cysteines carbamidomethylation for a fixed modification. At the same time, methionine oxidation was used as a variable modification. The confidence in the peptide mass fingerprinting matches (P < 0.05) was determined based on the MOWSE score. This was confirmed if an accurate overlapping of the matched peptides with the major peaks of the mass spectrum was found. Only significant hits (P < 0.05) based on the MASCOT probability analysis were accepted.
Results
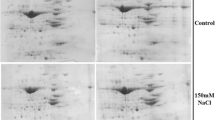
To investigate the protein profiles changes (0, 2, 12, and 72 h) for the two cultivars under salt stress, 2-D electrophoresis maps were obtained using IEF on 24 cm pH4–7 nonlinear IPG gels. Results showed that more than 800 protein spots could be detected in the range of pH 4–7 (Fig. 1). The reproducibility of 2-DE gels, for different samples, could be reflected by the data shown in Supplemental 1. Quantitative analysis of the three replicates indicated that there were 100 protein spots showed a more than 1.5-fold or less than 0.66-fold difference (P < 0.05) in expression values in at least one sample compared to the control. Quantitative analysis using PDQuest were shown in Supplemental material 2. Pictures of 2-D gels from biological triplicate analysis were shown in Supplemental material 3. The 100 spots, identified in 2-D electrophoresis gels, were excised and digested and MALDI-TOF-TOF MS analysis. The 83 proteins, shown in Fig. 1, were identified based on the NCBI database (Table 1).

The position of 83 leaf protein spots from cultivars JW9-6 (salt-sensitive) and JW2372 (salt-tolerance) under salt treatment for 2, 12 and 72 h were shown in 2-DE pictures. The numbers with arrows indicate the identified protein spots. And 2-D gel picture of each time point have been shown
The 83 identified proteins, which exhibited more than 15 % (P < 0.05) or less than 6.6 % (P < 0.05) differences, were sub-grouped into 11 categories according to KEGG. Various metabolic pathways and processes were found associated with the identified proteins. Such pathways included redox homeostasis (7 %), protein synthesis (5 %), proteolytic proteins (5 %), protein folding and assembly (5 %), carbohydrate metabolism (7 %), amino acid and nitrogen metabolism (7 %), cellular processes (3 %), photosynthesis (40 %), nucleotide metabolism (2 %), energy metabolism (13 %) and unclassified (6 %). The ‘unclassified’ class denotes those matched with unknown functions in the database. The largest function group (40 %) was proteins involved in photosynthesis that were heavily affected by salt stress (Fig. 2).

Functional groups of differentially expressed proteins identified from control and salt treated shrub willow clones (cultivars JW9-6 and JW2372) seedling leaves for different stress hours (2, 12 and 72 h). This classification is based on KEGG (http://www.kegg.jp/kegg/pathway.html) and literature
The change patterns in the proteins were quite different as salt stress progressed (Table 1). Take JW9-6 as an example. Some proteins, i.e., spots 1, 7, 48 and 53 were found to be strongly increased in the first 2 h, and then decreased to a constant lower level even when it was exposed to a long duration of stress (12 or 72 h). Some proteins, i.e., spots 20, 32 and 74, were found to be strongly decreased in the first 2 h, and then increased to a constant higher level even when it was exposed to a longer period of stress (12 or 72 h). Some proteins, i.e., spots 31, 36, 39, 41, 51 and 75 increased steadily, whereas other proteins, spot 55 showed a peak at 2 and 12 h stage, followed by a decrease at the subsequent stage (72 h). These results implied that shrub willow clones seedling leaves responded to different periods of salt stress and modulated the corresponding protein expression to minimize potential damage.
Discussion
Changes in two shrub willow clones seedling leaves during salt stress were associated with the disruption of various aspects. However, there is little information regarding the proteome in the biological processes. Hence, a comprehensive proteomics analysis between two different salt-tolerant shrub willow clones is important to understand the molecular mechanisms in plant under salt stress.
Proteins related to redox homeostasis
In the literature, it is found that proteins that are related to reactive ROS in the processes of plant growth are usually regulated by salt stress, including GST, ascorbate peroxidase (APX) (Csiszár et al. 2011). In this paper, four identifiers were found to be related to redox homeostasis in shrub willow clones seedling leaves. Ascorbate peroxidase (APX) (spot 1), was up-regulated significantly in the leaves of the cultivar of JW2372, but down-regulated at the 72 h of JW9-6. The protein is a major ROS-scavenging protein (Dixon et al. 2002), and its expressions were significantly up-regulated by stresses (Wan and Liu 2008). At the same time, ATP-sulfurylase (ATPS) precursor (spot 4) was up-regulated significantly (P < 0.05) in cultivar JW9-6’s leaves by salt stress. It was, however, significantly (P < 0.05) down-regulated in cultivar JW2372’s seedling leaves at 72 h. ATPS plays in bio-transportation of sulfate, respectively as the key protein and enzyme in sulfate uptake by the roots and the assimilation of sulfate in plants (Zhu et al. 2007). Results indicated that the antioxidative defense system was enhanced in the leaves of JW2372, and decreased in the seedling leaves of JW 9-6 after salt stress. Unexpectedly, there were two protein spots, NADPH-dependent thioredoxin reductase isoform 2 (spot 2) and lactoylglutathione lyase-like (spot 3), that were down-regulated significantly (P < 0.05) due to salt treatment, the later remained unchanged (P > 0.05) in cultivar JW2372’s seedling leaves, under both non-salt and salt stresses (Table 1). Thioredoxin is a ubiquitous small protein disulfide reductase, involved in cellular redox regulation (Laloi et al. 2004). Lactoylglutathione lyase is also known as glyoxalase, which can convert toxic 2-oxoaldehydes into less reactive 2-hydroxyacids using glutathione as a co-factor (Espartero et al. 1995).
Overall, higher expression of two proteins (APX and ATPS) might imply that their hydrogen peroxide-scavenging capacity was enhanced at the beginning of salt treatment. The two down-regulated proteins (thioredoxin and lactoylglutathione lyase) implied that antioxidative systems of the two cultivars were impaired under salt stress, and more down-regulated proteins in cultivar JW9-6 under salt stress at the late stage of salt treatment denoted that cultivar JW2372 is of higher hydrogen peroxide-scavenging capacity, comparing to cultivar JW9-6. This may be due to the fact that cultivar JW2372 is of better salt tolerance than cultivar JW9-6. Results also revealed that shrub willow increased salt tolerance through increasing scavenging capacity of the hydrogen peroxide and protecting the redox homeostasis system from damaged.
Proteins related to protein biosynthesis, protein folding and assembly, and protein proteolytic
A total of 12 identifier were found to be involved in protein biosynthesis, protein folding and assembly, and protein proteolytic (Table 1). There were four proteins that were identified in protein biosynthesis. Among them, chloroplast elongation factor Tu A (EF-TuA) (spot 6) was up-regulated, and eukaryotic translation elongation factor (spot 8) was down-regulated in cultivar JW9-6’s leaves. However, these two proteins were not affected in cultivar JW2372’s leaves by salt stress. These proteins were directly involved in initiation and elongation of the newly growing peptide chains (Mittler et al. 2004). The 30S ribosomal protein S1 (spot 7) was up-regulated in JW9-6’s seedling leaves, but it was down-regulated at the early phase of stress exposure (2 h) and then up-regulated at the late phase of stress exposure time (72 h) in cultivar JW2372’s seedling leaves. Ribosomal proteins are believed to take part in the regulation of protein synthesis at the level of the protein elongation step (Marek 2002). The differently express of these proteins demonstrated that proteins biosynthesis was increased in cultivar JW9-6 while decreased in cultivar JW2372.
Four identified proteins were identified to be associated with protein folding and assembly (spot 13, 14, 15 and 16). On the one hand, two proteins (spots 14 and 15) were up-regulated in cultivar JW9-6, but no significant changes in cultivar JW2372. They were identified as 40 kDa thylakoid lumen PPIase and cyclophilin-like protein. PPIase catalyses the slow cis–trans isomerization of proline peptide bonds in oligopeptides and accelerates slow, rate-limiting steps in the folding of several proteins (Fischer and Bang 1985; Lang et al. 1987). Cyclophilin, a family of proteins, usually bind with the immunosuppressant cyclosporin A, and assist in protein folding (Fischer et al. 1989; Schreiber 1991). The chaperones and Hsp were found to be related to protein folding and assembly (Scarpeci et al. 2008; Razavizadeh et al. 2009). One heat shock protein, cognate 1 at spot 13, was up-regulated in cultivar JW2372 and the other proteins (spot 16, cp10-like protein) were down-regulated in cultivar JW9-6. The up-regulation of these proteins showed that protein folding and assembly were increased under salt stress.
Protein proteolytic pathways play a dynamic and vital role in the regulating different metabolic processes and in the cell’s response to external environmental conditions. It is capable of removing irreversibly damaged polypeptides that may interfere with these pathways (Joanna et al. 1999). There were four identities that were found to be associated with protein proteolytic pathways (Table 1). Among them, papain-like cysteine proteinase (spot 9) was up-regulated in the two cultivars. It was, however, down-regulated at the late stress stage in the cultivar JW9-6. Putative cysteine proteinase precursor (spot 10) was significantly up-regulated in cultivar JW9-6’s seedling leaves (P < 0.05) by salt treatment but down-regulated significantly in cultivar JW2372 at the late treatment. Cysteine proteinases are also referred to as thiol proteases and play an essential role in proteins proteolytic pathway in various cellular compartments. They also participate in plant growth, senescence and programmed cell death (Malgorzata and Barbara 2004). The 26S protease regulatory subunit 6 identified at spot 11 was down-regulated significantly (P < 0.05) in cultivar JW9-6’s leaves at the late stress treatment. They were not changed in expression in cultivar JW2372. Proteasomes are parts of a major mechanism by which cells may regulate the concentration of some proteins and degrade misfolded proteins (Lodish et al. 2004). At last, one protein (ATP-dependent Clp protease ATP-binding subunit ClpC, spot 12) was down-regulated in the two cultivars of willow leaves under salt stress. ClpC belongs to the clpA/clpB family, ATP-dependent specificity component of the ClpAP protease. The primary function of the ClpA-ClpP complex appears to be the degradation of unfolded or abnormal proteins. Thus, the pattern of this protein showed that protein proteolysis was decreased.
Overall, regulated expression response patterns of above-mentioned three groups showed that decreasing of protein biosynthesis, protein folding and assembly, and increasing protein proteolysis were required for shrub willow seedlings to survive under salt treatment. Protein synthesis in cultivar JW2372 was opposition to cultivar JW9-6, which indicated that shrub willow clones need more proteins to resist the salt treatment.
The relationship between differentially expressed proteins and photosynthesis
Photosynthesis is the process of using energy from sunlight to split water to liberate O2 and converts CO2 into organic compounds, e.g., sugars (Bryant and Frigaard 2006). In this study, a total of 34 differentially expressed identities were found to be associated with the photosynthesis processes (Table 1). Six proteins (spots 35, 42, 59, 60, 61 and 62) were associated with light-harvesting reaction. Both spots 35 and 42 were found as type III chlorophyll a/b binding protein. Spot 42 was up-regulated at 0–12 h in the seedling leaves. Spot 36 was down-regulated in the seedling leaves of the cultivar JW9-6, and later up-regulated in cultivar JW2372 (Table 1). Type III chlorophyll a/b binding protein, a component of light-harvesting complex in plants, facilitates light absorption and transfers the excitation energy to reaction centers due to reduced amount of NADP+ to NADPH (Wan and Liu 2008). The protein at spot 59, identified as cytochrome b6-f complex iron-sulfur subunit, was up-regulated in cultivar JW9-6 and later down-regulated in cultivar JW2372 at the stress stage 12 and 72 h. Three proteins (spots 60, 61 and 62) were identified as coproporphyrinogen oxidase, ferredoxin–NADP reductase, leaf isozyme, chloroplastic-like isoform 1 and ATP-dependent zinc metalloprotease FTSH 2, respectively. Coproporphyrinogen oxidase and ferredoxin–NADP reductase, leaf isozyme, chloroplastic-like isoform 1 were all significantly (P < 0.05) down-regulated in cultivar JW9-6’s seedling leaves, but ferredoxin–NADP reductase, leaf isozyme, chloroplastic-like isoform 1 was up-regulated in cultivar JW2372’s seedling leaves. Coproporphyrinogen oxidase catalyzes chlorophyll biosynthesis pathway(s) (Ole et al. 1993). ATP-dependent zinc metalloprotease FtsH was related to photosystem II, preventing cell death under high-intensity light conditions (Ge et al. 2007; Hui et al. 2004). Overall, the decrease in expression of the proteins (Type III chlorophyll a/b binding protein, Cytochrome b6-f complex iron-sulfur subunit and ferredoxin–NADP reductase, leaf isozyme, chloroplastic-like isoform is associated with PS I, which could reduce light absorption and electron transfer in the photosynthetic pathways.
Oxygen-evolving enhancer protein 2 (OEEP2), was related to oxygen-evolving of photosystem II. OEEP2 was significant up-regulated in cultivar JW9-6, but down-regulated at 12 h in cultivar JW2372. Results revealed that oxygen-evolving of PS II was enhanced in cultivar JW9-6 and decreased in cultivar JW2372.
Additionally, there were 26 identities in CO2 assimilation, including two ribulose 1,5-bisphosphate carboxylase, rubisco (spots 41 and 54), seven ribulose 1, 5-bisphosphate carboxylase large subunits, RLSs (spots 37, 39, 40, 46, 47, 49 and 57), nine glyceraldehyde 3-phosphate dehydrogenase, G3PDH (spots 33, 34, 38, 48, 51, 53, 55, 56 and 63), five transketolases, Trans (spots 43, 44, 45, 50 and 52), one phosphoribulokinase (spot 58), one sedoheptulose-1, 7-bisphosphatase, chloroplast, SBPase (spot 64) and one carbonic anhydrase, CA (spot 65) (Table 1). Rubisco often combines carbon dioxide with ribulose-1, 5-bisphosphate to generate 3-phosphoglycerate (Makino et al. 2000). G3PDH could remove hydrogen from NADPH and added it to the 1, 3-bisphosphoglycerate, where glyceraldehyde-3-phosphate could be generated in Calvin cycle (Maberly et al. 2010). Transketolase could combine sedoheptulose-7-mitochondrial phosphate with glyceraldehyde-3-phosphate to assemble ten carbons in a Calvin cycle (Van et al. 1996). Phosphoribulokinase was also involved in the Calvin cycle, catalyzing the ATP-dependent phosphorylation of ribulose 5-phosphate to ribulose 1, 5-bisphosphate (Avilan et al. 2000). SBPase is a unique enzyme to photosynthetic organisms and plays an important role in regulating the photosynthetic (Dunford et al. 1998). Of these 26 identities, seven proteins (spots 34, 37, 40, 50, 52, 56 and 65) were down-regulated in only one cultivar or in both cultivar seedling leaves. Totally, 15 proteins (spots 33, 38, 39, 41, 43, 44, 46, 47, 49, 51, 54, 57, 58, 63 and 64) were found to show the increasing abundance in only one cultivar or in both cultivar seedling leaves under salt stress. However, three proteins at spots 48, 53 and 55 were up-regulated at the early stress stage and then down-regulated at the 72 h stress time in cultivar JW9-6’s seedling leaves. Only one protein (spot 45) was significantly up-regulated in cultivar JW9-6’s but significantly down-regulated in cultivar JW2372 at the early stress stage. The data presented in this section indicated that Calvin cycle in willow seedling leaves was enhanced by salt stress and salt-tolerance cultivar JW2372 had the stronger photosynthesis than salt-sensitive cultivar JW9-6 by salt treatment.
Further, one protein (spot 32) engaged in rubisco activation was rubisco activase. It was down-regulated in cultivar JW9-6 at the early stress stage and up-regulated later at the late stress stage (72 h) by salt stress. It was later significantly up-regulated by cultivar JW2372. Results indicated that rubisco activation was significantly affected by salt stress in willow seedling leaves.
Identified proteins related to carbohydrate metabolism
There were six identities (spots 17, 18, 19, 20, 21 and 22) associated with carbohydrate metabolism. Two proteins (spots 17 and 18, Malate dehydrogenase, (MDH)) of them were up-regulated in cultivar JW9-6 by salt stress in the seedling leaves. MDH is an enzyme which was involved in tricarboxylic acid cycle and the generation of oxaloacetate from malate (Wang et al. 2007). Aldolase (spots 19 and 20), ribulose-phosphate 3-epimerase (spot 21), fructose-1, 6-bisphosphatase, FBPase (spot 22), were found down-regulated in only one cultivar or in both cultivars seedling leaves under salt stress. Ribulose-phosphate-3-epimerase could catalyze bidirectional conversion of ribulose-5-phosphate to xylulose-5-phosphate (Rogers et al. 2007). FBPase could catalyze the break-down of fructose-1, 6-bisphosphate to fructose-6-phosphate (Ge et al. 2007). The down-regulation of proteins indicated that carbohydrate metabolism was decreased in willow leaves under salt stress.
Proteins are associated with energy metabolism
Under salt stress, low energy metabolism rates were found in plants to conserve energy and limit generation of ROS (Moller 2001). There were 11 identities (spots 66, 67, 68, 69, 70, 71, 72, 73, 74, 75 and 76) associated with energy metabolism in this study. Two proteins (spots 67 and 71) of them were significantly (P < 0.05) down-regulated in cultivar JW9-6 but up-regulated significantly (P < 0.05) in cultivar JW2372’s leaves. These two proteins were identified as ATP synthase beta subunit. ATP synthases have been found to be responsive to NaCl stimulus (Jiang et al. 2007). Five proteins (spot 66, 68, 70, 73 and 76) were found significantly down-regulated in only one cultivar or in both cultivars seedling leaves under salt stress, including formate dehydrogenase (FDH), ATP synthase beta subunit and phosphoglycerate kinase (PGK), putative FDH plays an significant role in energy supply and in response to stresses in other plants (Tishkov and Popov 2004). PGK could catalyze the reversible transfer of a phosphate group from 1, 3-bisphosphoglycerate to ADP, producing 3-phosphoglycerate and ATP. It acts as a major enzyme in the first ATP-generating step of the glycolytic pathway. Spot 69 was up-regulated in cultivar JW2372 and spot 72 was up-regulated in cultivar JW9-6 seedling leaves by salt treatment. The two spots were detected as ATP synthase beta subunit. ATP synthase CF1 alpha subunit identified at spot 74 was down-regulated at the early stress and up-regulated at the stress time (72 h) in cultivar JW9-6, while significantly up-regulated in cultivar JW2372. The differently express pattern of these proteins indicated that energy supply was inhibited in both cultivars after stresses, but more seriously in cultivar JW9-6 than in cultivar JW2372. This may imply that sufficient energy supply is required for shrub willow seedling to handle salt stress. At the same time, energy in the seedling leaves of genotype cultivar JW9-6 is more abundant than in cultivar JW2372. This may explain the fact that cultivar JW2372 is more salt-tolerant than cultivar JW9-6.
Proteins related to other metabolisms and pathways in shrub willow seedling leaves
In this study, 11 proteins were associated with the primary metabolisms and its pathways, including metabolisms of nucleotide, metabolism and cellular processes for amino acid and nitrogen. Six proteins (spots 23, 24, 25, 26, 27 and 28), which were involved with amino acid and nitrogen metabolism, were found to be differentially expressed in abundance under salt treatment (Table 1). One protein (spot 24) of them was down-regulated and two proteins (spots 25 and 26) were up-regulated in cultivar JW9-6, but no change identified in abundance in cultivar JW2372. These were identified as s-adenosylmethionine synthase 4-like isoform 1 (SAM synthase), cysteine synthase1 and Gln synthetase (GS), respectively. SAM synthase could catalyze the biosynthesis of SAM from ATP and l-methionine and involved in ethylene biosynthesis under salt stress (Ma et al. 2012). SAM often serves as a methyl group donor in transmethylation reactions (Van et al. 1994). Here, SAM synthase decreased in abundance under NaCl treatment that is consistent with previous results (Jiang et al. 2007). Cysteine synthase is responsible for the last step in biosynthesis of Cys (Masaaki et al. 2001). GS is an important enzyme related with assimilating inorganic nitrogen into organic forms. It also plays a key role in the nitrogen metabolism by catalyzing condensation of glutamate and ammonia to form glutamine (Hoelzle et al. 1992). It also plays a vital role in improving rice tolerance to salt (Hoshida et al. 2000). The other three proteins (spot 23, glutamate-1-semialdehyde aminotransferase (GSA-AT), spot 27, glutamate–ammonia ligase, cytosolic, spot 28, AlaT1) were down-regulated in cultivar JW2372 at the late stress stage. GSA-AT catalyzes the transamination of GSA to form ALA (Sandra et al. 1992). AlaT1, known as glutamate-pyruvate transaminase 1, can generate pyruvate and glutamate by catalyzing the reversible transamination between 2-oxoglutarate and alanine. Therefore, it plays an important role in the metabolism of amino acids and glucose (Pollard and Cooper 2009). Our analysis indicated that the nitrogen metabolism and amino acid are increased in cultivar JW9-6, but inhibited in cultivar JW2372 under salt stress.
Additionally, three proteins (spot 29, actin-3-like, spot 30, actin-depolymerizing factor and spot 31, alpha-tubulin) were found to be related to cellular processes. Two of them (spots 29 and 31) were up-regulated in the seedling leaves of the two cultivars under salt stress, while the third one was up-regulated only in cultivar JW9-6. While one protein (spot 30) down-regulated only in the cultivar JW9-6 related to cellular processes. Actin is a component in cytoskeletal system that enables cell movements and cellular processes (Pollard and Cooper 2009). Tubulin is one of several members of a small family of globular proteins and a component of the cytoskeleton. Both actin and tubulin dynamics are of importance to cellular homeostasis. Up-regulation of these proteins revealed that cellular processes are improved by salt stress in shrub willow seedling leaves.
We further found, for two proteins (spot 77 and 78), the mechanism of their responding to salt was related to nucleotide metabolism. One protein (spot 77, UvrB/UvrC protein : AAA ATPase) was down-regulated but the other protein (spot 78, Adenine phosphoribosyltransferase 1, (APRTase)) was up-regulated in cultivar JW9-6. At the same time, it was not affected (P > 0.05) by the expression in cultivar JW2372 under both salt and non-salt stresses. APRTase is a kind of enzyme that is involved in the pathway for purine nucleotide salvage. It is a catalyst in the reaction between phosphoribosyl pyrophosphate (PRPP) and adenine to form AMP (Sancar and Hearst 1993). Nucleotide excision repair is initiated by the excinuclease which excises a wide variety of DNA damages in a dodecanucleotide (Grossman 1993). This is due to fact that the coordinated reactions among UvrA, UvrB, and UvrC proteins. Our results indicated that under salt stress nucleotide metabolism in shrub willow seedling leaves is affected.
Conclusions
In this study, comparative analysis of seedling leaves of two shrub willow clones (salt-tolerant cultivar JW2372 and salt-sensitive cultivar JW9-6) under salt stress were conducted to investigate changes of total proteins under salt stress. Totally, 83 differentially expressed proteins were successfully identified using MALDI-TOF-TOF MS (Table 1).
Major findings from this study are listed as follows: (1) shrub willow increases salt tolerance by enhancing its ROS scavenging capacity and protecting redox homeostasis system from damage; (2) the inhibition of protein synthesis as well as protein folding and assembly, and enhancing of protein proteolysis were required by shrub willow seedlings to survive in salt stress; (3) salt stress might affect the pathways of photosynthesis, carbohydrate metabolism, energy supply, and nitrogen and amino acid metabolism. (4) cultivar JW2372 more salt tolerant than that of cultivar JW9-6 due to comprehensive performance of all the pathways.
Investigating the mechanism at proteomic level allows us to further our understanding and possibly develop management strategies of cellular activities in salt-treated shrub willow clones, providing new insights on the responses of shrub willow leaves to salt stress.
Author contribution statement
Dezong Sui designed and performed research, analyzed data and wrote the manuscript. Shizheng Shi and Xudong He participated in the design of the study. Baosong Wang designed research and revised the manuscript. All the authors have read and approved the final manuscript.
References
Askari H, Edqvist J, Hajheidari M, Kafi M, Salekdeh GH (2006) Effects of salinity levels on proteome of Suaeda aegyptiaca leaves. Proteomics 6:2542–2554
Avilan L, Lebreton S, Gontero B (2000) Thioredoxin activation of phosphoribulokinase in a bi-enzyme complex from Chlamydomonas reinhardtii chloroplasts. J Biol Chem 275:9447–9451
Bryant DA, Frigaard NU (2006) Prokaryotic photosynthesis and phototrophy illuminated. Trends Microbiol 14:488–496
Csiszár J, Váry Z, Horváth E, Gallé Á, Tari L (2011) Role of glutathione transferases in the improved acclimation to salt stress in salicylic acid-hardened tomato. Acta Biologica Szegediensis 55:67–68
Dixon DP, Lapthorn A, Edwards R (2002) Plant glutathione transferases. Genome Biol 3:1–10
Dunford RP, Catley MA, Raines CA, Lloyd JC, Dyer TA (1998) Purification of active chloroplast sedoheptulose-1,7-bisphosphatase expressed in Escherichia coli. Protein Expr Purif 14:139–145
Espartero J, Sanchez-Aguayo I, Pardo JM (1995) Molecular characterization of glyoxalase-I from a higher plant; upregulation by stress. Plant Mol Biol 29:1223–1233
Fischer G, Bang H (1985) The refolding of urea-denatured ribonuclease A is catalyzed by peptidyl-prolyl cis-trans isomerase. Biochim Biophys Acta 828:39–42
Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX (1989) Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 337:476–478
Ge J, Shi L, Gu WB (2007) Photosynthetic characteristics and the salt-stressed sweet sorghum seedlings. Acta Agron Sin 33:1272–1278
Grossman LS (1993) Thiagalingam nucleotide excision repair, a tracking mechanism in search of damage. J Biol Chem 268:16871–16874
Hoelzle I, Finer JJ, McMullen MD, Streeter JG (1992) Induction of glutamine synthetase activity in nonnodulated roots of glycine max, phaseolus vulgaris, and pisum sativum. Plant Physiol 100:525–528
Hoshida H, Tanaka Y, Hibino T, Hayashi Y, Tanaka A, Takabe T (2000) Enhanced tolerance to salt stress in transgenic rice that overexpresses chloroplast glutamine synthetase. Plant Mol Biol 43:103–111
Hui HX, Xu X, Li ShM (2004) Possible mechanism of inhibition on photosynthesis of Lyeium barbarum under salt stress.Chin. J Ecol 23:5–9
Jiang YQ, Yang B, Neil S, Harris MK (2007) Deyholos. Comparative proteomic analysis of NaCl stress-responsive proteins in Arabidopsis roots. J Exp Bot 58:3591–3607
Joanna P, Wang JM, Adrian K (1999) New insights into the ATP-dependent Clp protease: Escherichia coli and beyond. Mol Microbiol 32:449–458
Laloi C, Mestres-Ortega D, Marco Y, Meyer Y, Reichheld JP (2004) The Arabidopsis cytosolic thioredoxin h5 gene induction by oxidative stress and its W-box-mediated response to pathogen elicitor. Plant Physiol 134:1006–1016
Lang K, Schmid FX, Fischer G (1987) Catalysis of protein folding by prolyl isomerase. Nature 329:268–270
Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, Darnell J (2004) “3” Molecular cell biology (5th ed.). W.H. Freeman, New York, pp 66–72
Ma HY, Song LR, Shu YJ, Wang S, Niu J, Wang ZK, Yu T, Gu WH, Ma H (2012) Comparative proteomic analysis of seedling leaves of different salt tolerant soybean genotypes. J Proteomics 75:1529–1546
Maberly SC, Courcelle C, Groben R, Gontero B (2010) Phylogenetically-based variation in the regulation of the Calvin cycle enzymes, phosphoribulokinase and glyceraldehydes-3-phosphate dehydrogenase, in algae. J Exp Bot 61:735–745
Makino A, Nakano H, Mae T, Shimada T, Yamamoto N (2000) Photosynthesis, plant growth and N allocation in transgenic rice plants with decreased Rubisco under CO2 enrichment. J Exp Bot 51:383–389
Małgorzata G, Barbara Z (2004) Multifunctional role of plant cysteine proteinases. Acta Biochim Pol 51:609–624
Marek T (2002) Molecules in focus the acidic ribosomal P proteins. Int J Biochem Cell Biol 34:911–915
Masaaki N, Maiko S, Michimi N, Mitsuko A, Hikaru S, Kazuki S (2001) Cysteine synthase overexpression in tobacco confers tolerance to sulfur-containing environmental pollutants. Plant Physiol 126:973–980
Mittler R, Vanderauwera S, Gollery M, Breusegem FV (2004) Reactive oxygen gene network of plants. Trends Plant Sci 9:490–498
Moller IM (2001) Plant mitochondria and oxidative stress: electron transport, NADPH turnover, and metabolism of reactive oxygen species. Annu Rev Plant Physiol Plant Mol Biol 52:561–591
Ole M, Lene S, Niels NS, Kjeld AM (1993) A soybean coproporphyrinogen oxidase gene is highly expressed in root Nodules. Plant Mol Biol 23:35–43
Parker R, Flowers TJ, Moore AL, Harpham NVJ (2006) An accurate and reproducible method for proteome profiling of the effects of salt stress in the rice leaf lamina. J Exp Bot 57:1109–1118
Pollard TD, Cooper JA (2009) Actin, a central player in cell shape and movement. Science 326:1208–1212
Razavizadeh R, Ehsanpour AA, Ahsan N, Komatsu S (2009) Proteome analysis of tobacco leaves under salt stress. Peptides 30:1651–1665
Rogers MB, Watkins RF, Harper JT, Durnford DG, Gray MW, Keeling PJ (2007) A complex and punctate distribution of three eukaryotic genes derived by lateral gene transfer. BMC Evol Biol 7:89–102
Sancar A, Hearst JE (1993) Molecular Matchmakers. Science 259:1415–1420
Sandra L, Berry L, Bernhard G, Marvin A, Smith C, Gamini K (1992) Purification and characterization of glutamate 1-semialdehyde aminotransferase from barley expressed in escherichia coli. Plant Physiol 99:1597–1603
Scarpeci TE, Zanor MI, Valle EM (2008) Investigating the role of plant heat shock proteins during oxidative stress. Plant Signal Behav 3:856–857
Schreiber SL (1991) Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science 251:283–287
Sui DZ, Wang BS, Shi SZ, Jiao ZY (2011) Selection of identification index and comprehensive evaluation of salt tolerance at seeding stage of shrub willow clones. J Northwest For Univ 26:61–64
Tishkov VI, Popov VO (2004) Catalytic mechanism and application of formate dehydrogenase. Biochemistry 69:1252–1267
Van BF, Dekeyser R, Gielen J, Van MM (1994) Characterization of a s-adenosylmethionine synthase gene in rice. Plant Physiol 105:1463–1464
Van D, Bergh ER, Baker SC, Raggers RJ, Terpstra P, Woudstra EC, Dijkhuizen L (1996) Primary structure and phylogeny of the Calvin cycle enzymes transketolase and fructosebisphosphate aldolase of Xanthobacter flavu. J Bacteriol 178:888–893
Wan XY, Liu JY (2008) Comparative proteomics analysis reveals an intimate protein network provoked by hydrogen peroxide stress in rice seedling leaves. Mol Cell Proteomics 7:1469–1488
Wang W, Vinocur B, Altman A (2003) Plant responses to drought, salinity and extreme temperatures: towards genetic engineering for stress tolerance. Planta 218:1–14
Wang W, Huang SL, Ding GJ (2007) Morphological and physiological changes accompanying the induction of salt tolerance in Neolitsea sericea seedlings. J Zhejiang For Coll 24:168–172
Wiebe BH, Eilers RG, Eilers WD, Brierley JA (2007) Application of a risk indicator for assessing trends in dryland salinization risk on the Canadian prairies. Can J Soil Sci 87:213–224
Zhao Q, Zhang H, Wang T, Chen SX, Dai SJ (2013) Proteomics-based investigation of salt-responsive mechanisms in plant roots. J Proteomics 82:230–253
Zhu Ch, Wang BL, Qu D (2007) Cloning and sequence analysis of partial cDNAs of ST and ATPS from Maize. Acta Botanica Boreali Occidentalia Sinica 27:1742–1746
Acknowledgments
This work was supported by the Natural Science Foundation of Jiangsu Province (Grant No. BK2011873) and Jiangsu provincial science and technology support program (Grant No. BE2013449).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Hajduch.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sui, D., Wang, B., Shi, S. et al. Changes of protein expression during leaves of shrub willow clones in response to salt stress. Acta Physiol Plant 37, 51 (2015). https://doi.org/10.1007/s11738-015-1811-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-015-1811-1