
Abstract
ParE, a subunit of topoisomerase IV, is involved in the management of DNA topology and validated to be an attractive target for the development of antibacterial agents. Availability of the crystal structure of Francisella tularensis in combination with one of the pyrrolo [2,3-d] pyrimidine-2-thiolinhibitor facilitated us to emphasize the combined computational approach for discovering the presumed binding mode of selected inhibitors into the binding pocket of ParE (pdb. 4HY1). In the current study, pharmacophore modelling and 3D-QSAR studies were performed using 33 reported F. tularensis ParE inhibitors having pKi ranging from 5.06 to 9.00. The developed five featured pharmacophore model, DHHRR_1 was statistically validated with different parameters like Q2 (0.66), R2 (0.99) and F value (682) at four-component partial least squares factor. Enrichment analysis was performed to validate the generated pharmacophore model. Extra-precision molecular docking, binding free energy calculation using PRIME MM-GBSA were performed for the selected inhibitors. Induced fit docking was performed for the highest active inhibitor 16. The highest-ranked induced fit docked complex 16/4HY1 was used to run a 50 ns molecular dynamic simulation to validate the stability. Further, in silico High Throughput Virtual Screening was performed using 22 lakhs chemical database molecules to identify the potential virtual hits and predicted activity was found for the virtual hits. These results provide promising strategies for the development of novel molecules with better inhibitory activity against F. tularensis ParE.
Graphic abstract

Similar content being viewed by others
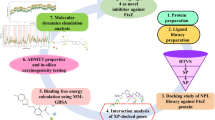
Avoid common mistakes on your manuscript.
Introduction
Antibacterial resistance is considered a global threat to mankind with a high rate of mortality and morbidity which needs dire attention (Levy and Marshall 2004). In addition, there is an increased demand for the novel types of antibacterials acting on different unexploited targets to overcome the prevailing resistance issue (Worthington and Melander 2013). The adenosine triphosphate (ATP)-dependent bacterial DNA gyrase (GyrA and GyrB) and topoisomerase IV (ParC and ParE) enzymes are heterotetrameric type II topoisomerases (Fisher et al. 2017). These enzymes are independently required to control the topology of DNA during transcription and replication and are functionally similar (Sugino et al. 1977). Both enzymes possess the ATPase active site at the N-terminal domain also known as ATP-operated clamp (Aldred et al. 2014). The absence of these enzymes in the host tissue and its high homology among nosocomial pathogens makes them attractive targets for the design and development of novel antibacterials (Fisher et al. 2017). GyrB plays a key role in the relaxation of DNA, introducing negative supercoils and removes positive supercoils by wrapping mechanism, whereas ParE involved in decatenating the daughter chromatids after the replication process behind the replication fork via a canonical mechanism (Sugino et al. 1977, Aldred et al. 2014). In contrast, ParE does not introduce any negative supercoils unlike GyrB (Levine et al. 1998).
Moreover, GyrB and ParE have structural similarities along with the ATP binding pocket across the species (Tourova et al. 2010). Similar to GyrB, ParE subunit comprises of two distinct domains: The N-terminal and C-terminal domain. N-terminal domain involved in the stabilization of dimer and acts as an ATP binding pocket, whereas at C-terminal the contacts are less extensive (Bellon et al. 2004). The ParE, ATP catalytic pocket of Streptococcus pneumonia, exhibited sequence similarity with that of GyrB ATP binding domains (43-kDa) of Escherichia coli (49.4% identity) and Thermus thermophilus (47% identity), demonstrating that ParE has the same fold as GyrB. In addition, S. pneumonia ParE consists of a highly conserved N-terminal binding domain and glycine-rich segment (G-loop), similar to the GyrB subunit (Sifaoui et al. 2003; Pan and Fisher 1999). However, there exists a difference in their structures, present in the longest α-helices at unconserved C-terminal end (Fernández-Moreira et al. 2006; Janoir et al. 1996). The E. coli GyrB is structurally heterogeneous with F. tularensis ParE due to differences in the sequence similarity. The crystal structure of F. tularensis ParE reveals that the N-terminal ATP-binding domain contains 39 kDa of residues which plays a vital role in the decatenating activity of DNA and stabilization of dimer (Tari et al. 2013). The asymmetric crystallographic unit (pdb.4HY1) contains two chains, A and B. These two chains were in lack of electron density for some of the residues and the C-terminal end contains histidine tag of the A chain. The B chain also includes a similar kind of disorder, but in addition, it possesses extra 218–381 residues (Trzoss et al. 2013).
In addition to aminocoumarins and fluoroquinolones, various scaffolds have been investigated as antibacterials targeting ParE enzyme. These include quinazolinediones (Huband et al. 2007, Pan et al. 2009), nonfluorinated quinolones (Jones et al. 2002), benzimidazoles (Charifson et al. 2008), ethylureas (Starr et al. 2009), azaindoles (Manchester et al. 2012), anilinopyridines (Uria-Nickelsen et al. 2013), pyrrolopyrimidines (Tari et al. 2013), [1,2,4]triazolo[1,5-a]pyridines (East et al. 2009), aminobenzimidazoles (Jeverica et al. 2014), and alcohol-containing benzothiazole urea. Inhibition of the ATPase activity of ParE has been explored for the development of novel synthetic scaffolds that are capable of inhibiting this enzyme, but potent activity against both Gram-negative and Gram-positive bacteria has not been accomplished. The high-resolution crystal structure of ParE investigated experimentally to develop potent antibacterial agents against ParE by stimulating the computational approaches (Tari et al. 2013; Trzoss et al. 2013; Basarab et al. 2013; Kale et al. 2014). In the current study, we performed pharmacophore modelling, 3D-QSAR study, extra-precision molecular docking, free energy calculation, molecular dynamic (MD) simulation studies to ascertain the critical interactions between the ligands and catalytic pocket responsible for ParE inhibitory activity. The generation of 3D-QSAR Pharmacophore model followed by its validation exhibited good predictive power for the experimental pKi values. The highest active compound from the selected data set was subjected to 50 ns MD simulation to validate the proposed binding mode.
Materials and methods
Data set
In the current study, we selected a total of 33 ParE inhibitors (Supplementary Table S1), sharing a common assay procedure (Tari et al. 2013; Trzoss et al. 2013). The Ki values of these inhibitors varied from 5.06 to 9.00 and were converted to their respective pKi. The 3D structures of these 33 inhibitors were prepared using the builder panel in Maestro 12.0 (Schrödinger 2019-2, LLC, New York, NY) (Bharadwaj et al. 2019) and optimized using the LigPrep module incorporated in Schrödinger 2019-2, LLC, New York NY. This procedure includes Van der Waals radii of the inhibitor non-polar atoms to 0.8 scaling factor with 0.15 as partial atomic charge cut-off. Ionization states of each ligand was produced (pH 7 ± 2.0) and energy minimization was performed with OPLS3e force field (Roos et al. 2019) till root mean square mean deviation (RMSD) of 0.01 Å was attained. The low energy conformations of each ligand were used for the molecular modelling studies.
Pharmacophore modelling and 3D-QSAR studies
In the current study, we used Phase module incorporated in Schrödinger 2019-2 to develop the 3D-QSAR pharmacophore models using the selected 33 F. tularensis ParE inhibitors. Ligands were geometrically refined and a maximum of 100 conformers were generated per structure using the conformational search engine, ConfGen. OPLS3e force field with distance-dependent dielectric constants was applied for the treatment of solvation system. The pharmacophore model was created by opting multiple ligands option followed by selecting entries, ignoring all other in search engines. The data set was generated by assigning the threshold of pKi > 8.6 as actives and pKi < 6.5 as inactives, whereas the remaining molecules were moderately active. Hypothesis settings include matching at least 50% of 8 actives mapped with pharmacophoric features like acceptor (A), donor (D), hydrophobic (H), negative ionic (N), positive ionic (P) and aromatic ring (R) for the generation of hypothesis. Then, we used a minimum of 200 target number of conformers to generate the model. Among the 33 inhibitors, 25 were selected as the training set based on the range of their pKi values (6.76–9.00) and diversity in the chemical structures of the respective inhibitors (Golbraikh and Tropsha 2000). The remaining eight compounds with pKi values ranging from 5.06 to 8.52 with diverse chemical structures were selected as test compounds. The existing six features were used to generate the pharmacophoric sites using the generated conformers of inhibitors. Based on the eight actives (Supplementary Table S1), pharmacophore hypotheses were developed keeping 1 Å for pharmacophore matching tolerance and 2 Å for the minimum intersite distance between two features. Among, the 20 generated models, five elements were found to be typical for all the actives. The best five hypothesized models were selected based on the vector, volume, sitescore, survival score, survival inactive and the number of matches (Table 1). Data set ligands were then aligned in accordance with the generated best five featured pharmacophore model DHHRR_1. The angles and distances between different pharmacophoric sites of the selected model are depicted in Fig. 1a, b, respectively. The van der Waals model of the training set chosen molecules was used to develop the atom-based 3D-QSAR model. The pharmacophore model which is statistically significant was generated based on the training set molecules for the selected hypothesis via partial least-square (PLS) regression. The increment of both predictivity and statistical significance was observed up to four PLS factors (Table 2), the further increase neither improve the statistics nor predictivity of the model. The best model DHHRR_1 was validated by predicting the activity of eight test set ligand, which were not used for the model generation. The fitness score of all the selected ligands were investigated by using this model (Supplementary Table S1). Further, to explore the structure–activity relationships of these ligands against ParE enzyme, contour plots (Fig. 3a–h) were generated and analysed.

a Pharmacophore model DHHRR_1 intersite angles in Å unit between the pharmacophoric points. b Pharmacophore model DHHRR_1 intersite distances in Å unit
Hypothesis validation
The best fitted five featured model DHHRR_1 exhibited an excellent predictive ability for the training set compounds, with a statistically significant coefficient of determination (R2) 0.99, SD (0.08) and F (682.2). The test set ligands exhibited a cross-validated correlation coefficient (Q2) of 0.66, RMSE (0.62), and Pearson R (0.93). The degree of correlation between the Phase predicted activity and experimental activity for the test and training set were computed and the results were depicted in Fig. 2a, b. From the scatter plots of both test and training sets, it is evident that there exist moderate differences between phase predicted and experimental activities for the test and training sets. Enrichment analysis was performed to validate the developed pharmacophore model, wherein the eight active ligands (pKi > 8.6) was mixed with three hundred decoy molecules retrieved from the Schrödinger database. To evaluate the ability of identification of actives from inactive and for the accurate ranking of inhibitors, robust initial enhancement and enrichment factors were calculated (Table 3 and Supplementary Figure S1).

a The plot of the correlation between the experimental and predicted activity (pKi) of pyrrolopyrimidine-based inhibitors using pharmacophore-based QSAR model of training set. b The plot of the correlation between the experimental and predicted activity (pKi) of pyrrolopyrimidine-based inhibitors using pharmacophore-based QSAR model of test set with best fit line y = 0.89 × 1.12 (R2 = 0.90)
Further, Y-randomization/scrambling test is performed to validate the generated QSAR model is reliable and not inferred by chance. This test is usually performed on the training set data as recommended by Tropsha et al. 2003. Firstly, the models are created by randomizing the activity data (dependent variable), while keeping the molecular descriptors unchanged (independent variable). After performing 10Y-randomization tests, the consequent models obtained are relied up on to have lowR2 and cross validated Q2 values, which confirm that the generated models are reliable (Supplementary Table S3).
Moreover, the presence of systematic errors in the developed model DHHRR_1 and its prediction quality were assessed by the mean absolute error (MAE)-based criteria method (https://dtclab.webs.com/sofware-tools). The MAE-based criteria are determined using the Xternal Validation plus 1.1 tool which was established by DTC software labs. Further, this tool is also able to compute all the external validation parameters using MAE-based criteria. Additionally, this criterion categorizes the prediction quality of model into ‘good’, ‘moderate’ and ‘bad’ based on their values of absolute error (AE) standard deviation and MAE. In turn, the absolute error standard deviation (σAE) are defined as follows: (1) good predictions (MAE + 3×σAE ≤ 0.2 × training set range and MAE ≤ 0.1 × training set range), (2) bad predictions (MAE + 3×σAE > 0.25 × training set range and MAE > 0.15 × training set range) and (3) moderate predictions (the predictions which do not fall under either of the above two criteria). Usually, MAE-based criterion is determined by eliminating 5% of compounds possessing high AE values in order to overcome the possibility of outlier predictions. To find out external test sets comprising of small number of badly predicted data, 95% of MAE-based criteria data is used, whereas 100% of MAE-based criteria data is used to penalize the predictivity of the generated model. Further, Xternal validation plus 1.1 tool by Roy et al. is also used to describe number of negative prediction errors (NNE), number of positive prediction errors (NPE), the mean of positive prediction errors (MPE), the absolute value of average prediction errors (AE), the average absolute prediction errors (AAE) and absolute value mean of negative prediction errors (MNE) in order to find out the presence of systematic error prediction (Roy et al. 2017 and Zhao et al. 2017). The results are said to be satisfied if any one or more of the prediction errors follow the rules defined by this method. The predictive ability of the developed model DHHRR_1 is observed to be good (Supplementary Table S4).
Molecular docking studies
X-ray crystal structure of F. tularensis ParE (pdb.4HY1, Resolution: 1.58 Å) was selected for the modelling study. The preparation of protein was performed using a Protein Preparation Wizard module of Schrödinger 2019-2 (Sidhu et al. 2019,; Sastry et al. 2013). The addition of hydrogens removed the crystallographic water molecules. Missing side chains were incorporated using Prime (Schrödinger 2019-2) (Jacobson et al. 2004). OPLS3e forcefield was employed for energy minimization keeping root mean square deviations (RMSDs) of heavy atoms at 0.30 Å (Roos et al. 2019) 10 Å radius around the bound ligand was used to define the active site and a grid box was generated keeping the co-crystallized ligand at the centre. The 33 ligands prepared using LigPrep were docked with extra-precision (XP) mode (Supplementary Table S5) using Glide (Friesner et al. 2006) keeping other options default. The selection of best-docked pose of each ligand was based on the Glide score, Glide energy and Glide Emodel energy values.
Binding free energy calculations
PRIME Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) approach (Schrödinger 2019-2) was used to calculate the binding free energy for all the protein–ligand complexes. OPLS3e force field with VSGB 2.0 solvation model was applied for the energy minimization. This method integrates a physics-based correction and optimized implicit solvation for hydrogen bonding, self-contact interactions, π–π interactions and hydrophobic interactions (Supplementary Table S6).
Molecular dynamics (MD) simulation
The docked complex of most active compound 16 (pKi = 9.00) in complex with 4HY1 was selected for performing the MD simulation with OPLS3e forcefield using the Desmond software (Schrödinger 2019-2). The molecular system was solvated with TIP4P water molecules with an approximate 10 Å buffering distance between the protein and the edges of the orthorhombic box. The solvated system comprises of 16,649 water molecules, 55,887 atoms, 1,174,189 Å3 final box volume. Appropriate counter Na+ ions were added to neutralize the system. Minimization of the solvated system by limited memory Broyden-Fletcher-Goldfarb-Shanno was achieved with a ten steepest descent steps and three vectors until a gradient threshold of 25 kcal/Mol/Å was reached (Guo et al. 2010). The short-range columbic interactions with 9.0 Å radius cut-off and for long-range electrostatic interactions smooth particle mesh Ewald method at a tolerance of 1e−09 was used (Essmann et al. 1995). The temperature of 300 k and a pressure of 1 bar was maintained by using Nose–Hoover thermostat and Martyna–Tobias–Klein barostat (Martyna et al. 1992, 1994), respectively. Under an isothermal-isobaric ensemble (NPT) conditions, the molecular system was simulated for 50 ns with a time step of 2 fs. A REPSA integration algorithm with multiple time steps was applied for bonded, far non-bonded, and near non-bonded interactions (2, 2 and 6 fs, respectively) and frames were composed for every 100 ps (Tuckerman et al. 1992). The analysis of trajectory poses and 3D structures were generated using the Maestro graphical interface.
Results and discussion
3D-QSAR study
To derive 3D-QSAR models, Phase (Schrödinger 2019-2) was employed. Based on the site score, volume, vector, matches, inactives, and survival score the best five hypotheses were selected. Among them, the best-fitted five featured models DHHRR_1 exhibited vector value (1.000), highest survival (5.891), selectivity score (2.103), survival inactive (2.412), volume score (0.885), site (1.000) scores (Table 1). This model consists of one hydrogen bond donor, two hydrophobic features and two ring features (Fig. 1a, b). The hypothesis DHHRR_1 alignment over inactive compounds (Fig. 3b, d) and active compounds (Fig. 3a, c) exposed that angles and distances are the key attributes (Supplementary Table S2(a) and (b)) for ParE inhibitory activity. The generated model DHHRR_1 is statistically significant with a high degree of confidence, implicated by the highest variance ratio (F = 682.2) and a smallest significance level of variance ratio (4.54e−21) (Table 2). In addition, the model exhibited a greater degree of confidence, based on the higher value of the QSAR model stability 0.522 on a maximum scale of 1, which is having a Pearson coefficient (r) of 0.9324, root-mean-square error (RMSE = 0.62) and a lower standard deviation (SD = 0.0867). The high coefficient of determinations (R2 = 0.9927) for training set molecules was used to predict the relevancy of the generating model. PLS factor four was used along with a higher cross-validated correlation coefficient (Q2 = 0.6616) exhibited the validity and model’s ability in predicting the experimental data of test set inhibitors (Supplementary Table S1). Among them, compounds 2 and 14 showed moderate deviations from the predicted QSAR model with residual values of 1.051 and 1.129, respectively.

a 3D QSAR contour plot visualization generated for the favourable and unfavourable hydrogen bond donor effects in active compounds. b 3D QSAR contour plot visualization generated for the favourable and unfavourable hydrogen bond donor effects in inactive compounds. c 3D QSAR contour plot visualization for the generated favourable and unfavourable hydrophobic interactions in active compounds. d 3D QSAR contour plot visualization for the generated favourable and unfavourable hydrophobic interactions in inactive compounds. e 3D QSAR contour plot visualization generated for the favourable and unfavourable hydrogen bond donor effects in compounds 10 and 16. f. 3D QSAR contour plot visualization generated for the favourable and unfavourable hydrogen bond donor effects in compounds 3 and 4. g 3D QSAR contour plot visualization for the generated favourable and unfavourable hydrophobic interactions in compounds 10 and 16. h 3D QSAR contour plot visualization for the generated favourable and unfavourable hydrophobic interactions in compounds 3 and 4
To validate the predictive ability of model DHHRR_1, enrichment analysis was performed (Supplementary Table S7–S9 and Figure S1). The top 5% of the database along with decent enrichment values, can be used to recover 50% of the actives. RIE was calculated for the generated models to estimate the contribution of actives ranking in the enrichment analysis. The hypothesized DHHRR_1 model exhibited higher RIE value 15.06, implicates the superiority ranking over random distribution. The area under curve (AUC) and ROC plots were generated to evaluate the performance of the pharmacophore models. The generated hypothesized model DHHRR_1 attained a better value of ROC (1.00) and AUC (0.99) (Table 3 and Supplementary Figure S1).
To examine the robustness and reliability of the model, Y-randomization test was employed. Y-scrambling test assures that the created model is reliable and not inferred by chance; and also validates the sufficiency of the training set molecules. This test compares the randomized scores generated with the scores of non-randomized data. If the predicted activity of the randomized model is identical to the original model, then the set of observation is not sufficient to support the generated model. After several repetitions, the new QSAR models were reported to have low R2 and Q2 values (Supplementary Table S3). The results obtained confirmed that the model DHHRR_1 is measurably noteworthy and vigorous. Further, these results are in agreement with all the statistical parameters such as RMSE, Pearson correlation coefficient.
The total 8 external test set compounds covering a response range of 0.852 logarithmic units and the prediction quality was found to be ‘good’ in accordance with MAE-based criteria (Supplementary Table S4). However, after elimination 5% data with predicted residuals MAE and MAE + 3 × σAE are exhibiting at 0.5310 and 1.5705, respectively. The threshold values utilized to judge the model predictions are 0.8526 (0.1 × training set range), 1.2789 (0.15 × training set range), 1.7052 (0.2 × training set range) and 2.1315 (0.25 × training set range). According to Roy et al. the generated QSAR model is reliable by the MAE-based criteria which further relied on the prediction errors. Additionally, to determine any possible systematic errors in the predicted model, it is very important to analyse the prediction errors of the compounds used as test set. In our study, the NPE/NNE (0.4286), NNE/NPE (2.333), │MPE/MNE│ (0.2043), │MNE/MPE│ (4.8938), AAE-│AE│ (0.0936) were calculated. The value of R2 (between observed values and predicted values) was found to be 0.8385.
Contour plots analysis
To identify the effect of the spatial arrangement of structural features of the ligands responsible for ParE inhibitory activity, contour maps were analysed. Blue cubes and Red cubes were used to show the favourable and unfavourable contributions for their inhibitory property against ParE. The best-fitted 3D-QSAR model DHHRR_1 of most active compounds 10 and 16 (pKi = 9.00 and 9.00 respectively) and the less active 3 and 4 (pKi = 5.69 and 5.92 respectively) was used for comparison. The favourable and unfavourable pharmacophoric features are designated in Fig. 3a–h. The hydrogen bond donating features was analysed for both active compounds 10 and 16 by visualizing contour plots and were represented in Fig. 3e and for inactive compounds 3 and 4 were depicted in Fig. 3f. The presence of blue cubes around position four of the pyrrolopyrimidine scaffold shows the favourable contribution of electron-donating groups at this position (Fig. 3a). Whereas the existence of electron-donating group (D5 pharmacophoric feature) at position two of pyrrolopyrimidine is unfavourable as it is showing a negative contribution to the inhibitory activity (Fig. 3b).The appearance of blue cubes at the NH group of pyrrole ring in the contour plot of 10 and 16 (Fig. 3e) shows the preference of hydrogen bond donating group at this position. While in the case of inactives 3 and 4, the presence of red cubes at position two of pyrrolopyrimidine indicates the non-preference of donating group with unfavourable contribution (Fig. 3f). The hydrophobic group in this hypothesized model is a significant feature influencing the inhibitory activity against ParE. 3D-QSAR contour plot visualization for the generated favourable and unfavourable hydrophobic features was represented for both actives (Fig. 3c) and inactives (Fig. 3d). Hydrophobicity or non-polar feature was analysed for both active compounds 10 and 16 (Fig. 3g). In this model, we emphasized the presence of two hydrophobic groups, namely H8 and H10. The presence of blue cubes at position five of pyrrolopyrimidine, indicates the preference of hydrophobic feature. Here, the presence of a chlorine atom at this position is significantly contributing to ParE inhibitory activity. Another hydrophobic feature at the position six of pyrrolopyrimidine molecule also necessitates significant activity. Moreover, ethyl group as a hydrophobic feature is found to be essential for the inhibitory activity as evident by the pKi (8.65–9.00) of highly active compounds. The appearance of blue cubes at position four of pyrrolopyrimidine moiety was observed to be indispensable for the inhibitory activity against F. tularensis ParE, indicating a positive contribution of the hydrophobic group at this position. However, for the hydrophobic contour map, the appearance of red cubes at the position two of pyrrolopyrimidine is observed to be non-crucial for the activity. Inactive compounds 3 and 4 contour maps showed the negative contribution of hydrophobic groups (red cubes) at position six of the pyrrolopyrimidine molecule (Fig. 3h). However, in these two compounds presence hydrophobic at position seven of pyrrolopyrimidine groups exhibited little positive contribution, which is occupied by a chlorine atom. Further, the presence of red cubes around the thiol group in compounds 3 and 4 indicated the unfavourable contribution of hydrophobic groups at position two of the pyrrolopyrimidine scaffold.
Molecular docking studies
The validation of the docking protocol has been achieved by redocking the co-crystal ligand (pdb.4HY1) into the binding pocket of the F. tularensis ParE enzyme. A similar conformational orientation of docked ligands with that of co-crystal (RMSD = 0.2643 Å) indicated the accuracy of the docking protocol (Supplementary Figure S2). All the ligands exhibited similar binding poses in the catalytic pocket, revealed by the comparative examination of docking results (Supplementary Table S5 and Figure S3 and S4). The hydrogen bonding and hydrophobic interactions with Asn41, Gly72, Asp68, Arg131 and Arg71 amino acid residues present in the C-terminal and N-terminal regions of the binding pocket played a crucial role in ligand binding. The binding poses of selected, highly active inhibitors 10 and 16 and low active inhibitors 3 and 4 were analysed and the results obtained were compared. From the docked poses of inhibitor 10 (Fig. 4a), the NH of pyrrole moiety, which is fused to the pyrimidine nucleus, formed hydrogen bond with the carbonyl oxygen of Asn41 (rNH···O=C, 1.75 Å). Further, the molecule was stabilized by charged interaction (π-cationic) between the ring nucleus of pyrrole and the protonated NH2 group of Lys7. In addition, this molecule also established hydrogen bonding interactions with amino acid residues Asp68, Thr162 Gly70 and Gly72. Another most active inhibitor, 16 (Fig. 4b), displayed four hydrogen-bonding interactions with 4HY1. The NH2 group present at position three of the pyrrolidine ring which is fused with pyrimidine nucleus established a hydrogen bonding interaction with the carbonyl oxygen of Asn41 (rNH2···O=C, 2.09 Å).

a Glide XP-docked poses of inhibitor 10, in the catalytic pocket of ParE (PDB ID: 4H1Y). b Glide XP-docked poses of inhibitor 16, in the catalytic pocket of ParE (PDB ID: 4H1Y)
Further, the NH group of the pyrrole nucleus established a hydrogen bonding interaction with the carboxyl oxygen of Asp68 (rNH···O=C, 1.85 Å). The oxygen of naphthyridine-1-oxide displayed two hydrogen bonds with the protonated NH2 group of Arg131 (rO···NH, 2.99 Å) and one more with the NH2 group of Arg131 (rO···NH, 2.09 Å). In addition, the 16/4HY1complex also showed one π-cationic interaction between the ring nucleus of pyrimidine and the protonated NH2 group of Arg71. The docking poses of these molecules revealed that the interactions are dominant in the N-terminal domain of the enzyme. This result is in agreement with the earlier report that ATPase activity is mainly confined to the N-terminal domain of the ParE enzyme, which is solely responsible for the bactericidal activity.
The lowest active ligand 3 exhibited three hydrogen bonding interactions between Met73 and nitrogen of pyrrolopyrimidine nucleus via water bridge. A water bridge interaction was also observed between nitrogen at position one of pyrrolopyrimidine moiety and Thr162 residue. Additionally, a hydrogen bonding interaction was observed between Asn41 and nitrogen of the pyrrolopyrimidine nucleus (Supplementary Figure S17). Another low active ligand 4 also displayed similar hydrogen bonding interaction with Met73. This inhibitor exhibited three hydrogen bonding interactions with Asp68, Gly72, Arg71 residues (Supplementary Figure S18). It is evident that the extensive hydrogen-bonding network is responsible for the stability of the high active ligands 10 and 16 within the binding pocket of the ParE enzyme.
Binding free energy of the ligands 1-33/4HY1 docked complexes was ranked using Prime MM-GBSA approach. The binding free energy (ΔGbind) values were found to be between − 54.27 and − 122.59 kcal/mol. It is evident from the results (Supplementary Table S6) that van der Waals energy (ΔGvdW − 46.85 to − 77.07 kcal/mol) is a major favourable contributor, while coulomb energy (ΔGCou 0.04 to − 73.77 kcal/mol) term is moderately favouring the inhibitor binding. In most of the active inhibitors lipophilic (ΔGlipo 14.60 to − 25.54 kcal/Mol) and solvation (ΔGsolv 7.7 to 49.55 kcal/mol) energy terms strongly disfavour the inhibitor binding.
MD simulation analysis
To investigate the stability and dynamic behaviour of 4HY1 bound with highly the high active ligand 16, the 16/4HY1 complex was subjected to a 50 ns molecular dynamics simulation using Desmond incorporated with Schrodinger suite 2019-2. From the thorough analysis of the trajectory frames, the metrics of root-mean-square deviation (RMSD) for Cα, backbone and heavy atoms of protein and ligand were depicted in Fig. 5a. The overall RMSD of Cα, backbone and heavy atoms were observed to be in the range 1.59–3.96, 1.44–-3.93 and 1.73–3.98 Å, respectively. During the equilibration phase, the RMSDs of Cα atoms, backbone atoms inhibitor complex increased till 14 ns and then converged after 30 ns phase. Fluctuations in the RMSD of Cα atoms were observed in the range of 1.94–3.96 Å during 14–20.5 ns. After the equilibration phase, the RMSD of Cα atoms stabilized in the range 3.96–2.82 Å during 20–50 ns. The root-mean-square fluctuations (RMSF) of the ParE protein residues obtained throughout the MD simulation study are depicted in Fig. 5b. The Cα, backbone and heavy atoms of loop residues (Lys98-Ser107) exhibited high RMSF values 2.35–4.69 Å, 2.45–4.82 Å and 2.94–5.28 Å, respectively. The RMSF plot revealed that the Cα atoms, backbone and heavy atoms of critical binding amino acid residues within the catalytic site showed a lower degree of fluctuations with RMSF values in the range of 0.53–1.56 Å, 0.56–1.51 Å and 0.58-1.23 Å, respectively. During MD simulation radius of gyration (rGyr) of Cα (red-diamond) and backbone (blue-circle) atoms of the specified protein-inhibitor complex (Supplementary Figure S5) was found to be stable, which indirectly indicates a low degree of flexibility in the protein structure. In addition, it is also implicated that the protein-inhibitor complex is in more relaxed conformations (stabilization), resulted from the effect of solvency. The RMSD of ligand (Fig. 5a) exhibited conformational variations from 14 ns and was then stabilized from 20 ns throughout the simulation time. Further, this plot revealed that the RMSD of binding pocket residues copes well with the inhibitor movement. Interactions like hydrogen bonding π-cationic and π–π stacking were observed with the stable key residues Ile38-Glu44, Val66-Asp76, Ile89-Ser107 and Val115 (Fig. 5b). However, no interactions were observed in the regions Leu12-Ile35, Leu47-Ser95-Gly114, Ser116-Gly161 and Phe166-Lys377. The other interacting residues Arg71, Arg131 and Ile38 also exhibited low RMSF values for Cα, backbone and heavy atoms in the range of 0.56–1.15, 0.06–1.19 and 0.71–1.26 Å, respectively.

a RMSD (Å) of the simulated positions of 4H1Y, Cα and backbone atoms from those in the initial structure. b Protein RMSF values of inhibitor 16/4H1Y complex during MD simulation. c It represents the interaction of ligand 16 with different residues in the catalytic pocket of 4H1Y during MD simulation
MD simulation trajectory analysis exhibited similar binding interactions as speculated by the XP docking interactions (Supplementary Figure S4). The NH pyrrole group of pyrrolopyrimidine scaffold of compound 16 showed two hydrogen bonds, one each with Asn41 (rNH2···O=C, 2.09 Å, 66%) and Asp68 (rNH···O=C, 1.85 Å, 100%). Two water-mediated hydrogen bonds between NH of pyridopyridine and Arg131 (CO···NH, 2.99 Å 67%) and NH2 of pyrrole andAsn41 (> C=O···NH, 2.09 Å, 39%), Asp68 also exhibited one more key interaction with NH of pyrrolopyrimidine scaffold (C=O···NH, 1.99 Å, 99% cross-bridged via water molecule). Asp68 is considered to be a critical interacting residue and is available throughout MD study (Supplementary Figure S6) and established two high-frequency hydrogen bonding interactions, one directly with the NH of pyrrole of pyrrolopyrimidine scaffold (100% of MD trajectory) and the other with NH of the pyrimidine group of pyrrolopyrimidine through a water molecule (99% of MD trajectory). This high-frequency hydrogen bonding interaction is due to the less conformational flexibility of Asp68 (ψ = 10°) (Supplementary Figure S7(a)). Hydrogen bonding interaction was also observed between the nitrogen of naphthyridine 1-oxide and carbonyl moiety of Arg131 via water molecule (67% of MD trajectory). This intermediate hydrogen bonding frequency is due to the moderate conformational flexibility of Arg131 (ψ = 50°) (Supplementary Figure S7(c)) side chain. In addition, oxygen of naphthyridine-1-oxide showed two hydrogen-bonding interactions with Arg131. The amino substituent of the pyrrolidine molecule exhibited a low-frequency hydrogen bonding interaction (39% of MD trajectory) with Asn41. This intermittent hydrogen bonding arises due to the moderate conformational flexibility of Asn41 (ψ = 20°) (Supplementary Figure S7(b)). The NH of pyrimidine moiety of pyrrolopyrimidine moiety established a water molecule mediated hydrogen bonding interaction with the side chain OH of Thr162 (–NH···H–O–H···OH, 43% of the MD trajectory). The moderate conformational flexibility of Thr162 (ψ = 40°) (Supplementary Figure S7(f)) is responsible for this moderate frequency hydrogen bonding interaction. One of the oxygen atoms which is associated with N-oxide formation formed a water-mediated hydrogen bonding interaction with Glu45 (35% of MD trajectory), while nitrogen of the pyridine ring established a strong frequency hydrogen bonding interaction with Glu10 (80% of MD trajectory). The moderate frequency hydrogen bonding interaction is due to comparatively less conformational flexibility of Glu45 (ψ = 30°) (Supplementary Figure S7(d)). The amino group present position three of pyrrolidine nucleus formed a moderate frequency water-mediated hydrogen bond with Asn41 (39% of MD trajectory). The nitrogen at position one of pyrrolopyrimidine scaffold established a water molecule mediated hydrogen bond with the carbonyl group of Gly72 (rNH···H–O–H···O = C<, 98% of MD trajectory). This higher frequency of hydrogen bonding is due to the less conformational ability of Gly72 (ψ = 18°) (Supplementary Figure S7(e)) during the simulation trajectory. The two pyridine rings of 1,5-naphthyridine moiety exhibited one π-cationic interaction each with the charged residue Arg71 (90% and 33% of MD trajectory). In addition, the nitrogen of N-oxide (side chain linked via sulphur) formed a strong salt bridge interaction (80% of MD trajectory) with Glu10 of the N-terminal domain. The results obtained in the MD simulation study are in good agreement with the docking studies.
In addition, 100 frames were extracted from simulation trajectory files; the clustered structure of 16/4HY1 was created with the trajectory clustering module (Schrödinger 2019-2). This generated structure of 16 unveiled two hydrogen-bonding interactions with Asn41, Asp68 residues (Supplementary Figure S8) (RMSD = 0.65 Å) which are in good agreement with the critical interactions visualized in the ligand interaction diagram of MD simulation (Supplementary Figure S9) and the results obtained in molecular docking studies. It is clearly understood that Asn41, Asp68 residues are critical for the inhibitor stabilization within the catalytic pocket. Further, inhibitor 16 showed an average radius of gyration (rGyr) 4.25 Å and an average RMSD 0.395 Å after 14 ns of MD study, representing less conformational changes and inhibitor stability during the simulation. The observed polar surface area (PSA) (104.10–152.34 Å2) and stable solvent accessible surface area (SASA) (83.24–153.7 Å2) demonstrated the inhibitor stabilization within the binding pocket. Fewer changes in the molecular surface area (MolSA) (364.2–368.5 Å2) during simulation indicated the stability of 16 during MD simulation (Supplementary Figure S6). Further, similar orientations was observed for conformations of 16 after MD study and XP-docking pose (RMSD: 1.811 Å) (Supplementary Figure S10(a)); conformations of 16 DHHRR_1 and XP-docking pose (RMSD: 2.752 Å) (Supplementary Figure S10(b)); conformations of 16 DHHRR_1 and MD pose (RMSD: 2.937 Å) (Supplementary Figure S10(c)).
Moreover, we computed thermal binding free energy for 102 trajectory frames for 50 ns using the module thermal_mmgbsa.py Script. It is evident that ΔGbind is constant throughout the MD simulation study, with an average of − 67.69 kcal/mol. However, the van der Waals energy (average ΔGvdW − 54.51 kcal/mol) and lipophilic energy (average ΔGlipo, − 18.115 kcal/mol) are observed to be the major contributors for the ligand binding, whereas the covalent energy (average 2.1325 kcal/mol) disfavours the binding. (Supplementary Figure S11). It is evident that the binding free energies obtained with the 16/4HY1 complex are in agreement with the binding free energy value calculated by the MM-GBSA approach.
In silico high throughput virtual screening
The generated pharmacophore model, DHHRR_1 was used as a query to screen approximately 22 lakhs molecules from the chemical database libraries (Timtec, Specs, Enamine etc.). The first 2000 molecules were separated based on fitness and phase screen score and clustering were performed using the module, Canvas in Schrödinger 2019-2. The Macro model atom types of overlapping volumes and ‘average’ linkage method was used to calculate the clusters. The Kelley index is a criterion to select an optimal number of clusters. The Kelley measure balances the normalized “spread” of the clusters at a particular level with the number of clusters at that level. The number of clusters to be formed was 25, and it is evident from the Kelley penalty curve that equal distribution of clusters was observed within the particular level (Supplementary Figure S12). The dendrogram for 2000 virtual hits provided the necessary information about the merging distance of the hits based on cluster indices (Supplementary Figure S13). Distance matrix or dissimilarity between the clusters (Supplementary Figure S14) based on the cluster order was calculated for the hits obtained through high throughput virtual screening (HTVS). Among 2000 virtual hits, 70 molecules were selected based on the virtual score. The extra-precision-docking was performed and the result is provided in the supplementary Table S10. These molecules were subjected to binding free energy calculation by the MM-GBSA approach. Ten molecules H1–H10 (Supplementary Figure S19) are selected based on the binding pose, glide score, glide emodel and glide energy values (Supplementary Table S10). In addition, the activity was predicted for these ten virtual l hits and was result is summarized in Supplementary Table S10. The virtual hit H1 (glide score − 5.811 kcal/mol, ΔGbind − 37.65 kcal/mol) fitted well within the catalytic pocket of 4HY1(Supplementary Figure S15). The thieno[2,3-d]pyrimidin-4(3H)-one moiety of the compound H1is buried between the charged residues and the carbonyl function accepted a hydrogen bond from the protonated NH2 of Arg131. The NH of thieno[2,3-d]pyrimidin-4(3H)-one scaffold formed hydrogen bonding interaction with Gly72 residue of the N-terminal domain. The -NH of H1 also exhibited two hydrogen-bonding interactions one each with Asn41 and Gly72 via water molecules. This compound also showed a π-cationic interaction with the thieno [2,3-d] pyrimidin-4 (3H)-one moiety and Arg71 (Supplementary Figure S15). The thieno[2,3-d]pyrimidin-4 (3H)-one scaffold present in compound H2 (∆Gbind-59.58 kcal/mol, Gscore-5.709 kcal/mol)is surrounded by the polar residues (Supplementary Figure S16). The substituent 4-fluorobenzene is embedded with the hydrophobic groups of the binding pocket. This moiety is also involved in the hydrogen bonding interaction with Val115 and Asn41. Further, 4-fluorobenzene also exhibited a π-π interaction with the His94 residue.
Conclusion
In the current study, pyrrolopyrimidine based analogues were subjected to a combined computational approach to investigate the structural relationship and molecular mechanism responsible for ParE inhibitory activity. The hypothesized model DHHRR_1 established a cross-validation coefficient (Q2 = 0.66) and also a high coefficient of determination (R2= 0.99) validated by using decoy data set and enrichment study. Contour plot analysis exposed essential pharmacophoric features necessary for ParE inhibition. The results demonstrated that the ligands 1–33 occupied the N-terminal domain of binding pocket and showed hydrophobic, electrostatic, and hydrogen bonding interactions with Asp68, Asn41, Arg131, Thr162, Gly72 and Glu45 residues of the ATP-binding pocket. Further, from the binding free energy calculation, Van der Waals energy is observed to be the major contributor, whereas coulombic energy moderately favours the binding. From the 50 ns MD simulation trajectory analysis, the pose of 16/4HY1 complex exhibited similar modes of interactions which are in well agreement with XP-docking results. Hydrogen bonding, hydrophobic, π-cationic interactions played an essential role in the ligand stabilization during MD study. Trajectory clustering displayed low (RMSD = 0.65Å) with the conformational structure of 16 after MD simulation study. Model DHHRR_1 was used for in silico high throughput virtual screening against chemical database library. Further, these hits were subjected to extra-precision docking and binding free energy calculation by the MM-GBSA approach. Ten virtual hits H1–H10 with high binding affinity against 4HY1 are identified.
References
Aldred KJ, Kerns RJ, Osheroff N (2014) Mechanism of quinolone action and resistance. Biochemistry 53(10):1565–1574. https://doi.org/10.1021/bi5000564
Basarab GS, Manchester JI, Bist S, Boriack-Sjodin PA, Dangel B, Illingworth R et al (2013) Fragment-to-hit-to-lead discovery of a novel pyridylurea scaffold of ATP competitive dual targeting type II topoisomerase inhibiting antibacterial agents. J Med Chem 56(21):8712–8735. https://doi.org/10.1021/jm401208b
Bellon S, Parsons JD, Wei Y, Hayakawa K, Swenson LL, Charifson PS et al (2004) Crystal structures of Escherichia coli topoisomerase IV ParE subunit (24 and 43 kilodaltons): a single residue dictates differences in novobiocin potency against topoisomerase IV and DNA gyrase. Antimicrob Agents Chemother 48(5):1856–1864. https://doi.org/10.1128/aac.48.5.1856-1864.2004
Bharadwaj S, Lee KE, Dwivedi VD, Yadava U, Kang SG (2019) Computational aided mechanistic understanding of Camellia sinensis bioactive compounds against co-chaperone p23 as potential anticancer agent. J Cell Biochem 120(11):19064–19075. https://doi.org/10.1002/jcb.29229
Charifson PS, Grillot A-L, Grossman TH, Parsons JD, Badia M, Bellon S et al (2008) Novel dual-targeting benzimidazole urea inhibitors of DNA gyrase and topoisomerase IV possessing potent antibacterial activity: intelligent design and evolution through the judicious use of structure-guided design and structure-activity relationships. J Med Chem 51(17):5243–5263. https://doi.org/10.1021/jm800318d
East SP, White CB, Barker O, Barker S, Bennett J, Brown D, Boyd EA, Brennan C, Chowdhury C, Collins I, Convers-Reignier E (2009) DNA gyrase (GyrB)/topoisomerase IV (ParE) inhibitors: synthesis and antibacterial activity. Bioorg Med Chem Lett 19(3):894–899. https://doi.org/10.1016/j.bmcl.2008.11.102
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103(19):8577–8593. https://doi.org/10.1063/1.470117
Fernández-Moreira ES, Balas D, González I, De La Campa AG (2006) Fluoroquinolones inhibit preferentially Streptococcus pneumoniae DNA topoisomerase IV than DNA gyrase native proteins. Microb Drug Resist 6(4):259–267. https://doi.org/10.1089/mdr.2000.6.259
Fisher RA, Gollan B, Helaine S (2017) Persistent bacterial infections and persister cells. Nat Rev Microbiol 15(8):453. https://doi.org/10.1038/nrmicro.2017.42
Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT (2006) Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J Med Chem 49(21):6177–6196. https://doi.org/10.1021/jm051256o
Golbraikh A, Tropsha A (2000) Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. Mol Divers 5(4):231–243. https://doi.org/10.1023/a:1021372108686
Guo Z, Mohanty U, Noehre J, Sawyer TK, Sherman W, Krilov G (2010) Probing the α-helical structural stability of stapled p53 peptides: molecular dynamics simulations and analysis. Chem Biol Drug Des 75(4):348–359. https://doi.org/10.1111/j.1747-0285.2010.00951.x
Huband MD, Cohen MA, Zurack M, Hanna DL, Skerlos LA, Sulavik MC, Gibson GW, Gage JW, Ellsworth E, Stier MA, Gracheck SJ (2007) In vitro and in vivo activities of PD 0305970 and PD 0326448, new bacterial gyrase/topoisomerase inhibitors with potent antibacterial activities versus multidrug-resistant gram-positive and fastidious organism groups. Antimicrob Agents Chemother 51(4):1191–1201. https://doi.org/10.1128/AAC.01321-06
Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA (2004) A hierarchical approach to all-atom protein loop prediction. Proteins 55(2):351–367. https://doi.org/10.1002/prot.10613
Janoir C, Zeller V, Kitzis MD, Moreau NJ, Gutmann L (1996) High-level fluoroquinolone resistance in Streptococcus pneumoniae requires mutations in parC and gyrA. Antimicrob Agents Chemother 40(12):2760–2764. https://doi.org/10.1128/AAC.40.12.2760
Jeverica S, Golparian D, Hanzelka B, Fowlie AJ, Matičič M, Unemo M (2014) High in vitro activity of a novel dual bacterial topoisomerase inhibitor of the ATPase activities of GyrB and ParE (VT12-008911) against Neisseria gonorrhoeae isolates with various high-level antimicrobial resistance and multidrug resistance. J Antimicrob 69(7):1866–1872. https://doi.org/10.1093/jac/dku073
Jones ME, Critchley IA, Karlowsky JA, Blosser-Middleton RS, Schmitz F-J, Thornsberry C et al (2002) In vitro activities of novel nonfluorinated quinolones PGE 9262932 and PGE 9509924 against clinical isolates of Staphylococcus aureus and Streptococcus pneumoniae with defined mutations in DNA gyrase and topoisomerase IV. Antimicrob Agents Chemother 46(6):1651–1657. https://doi.org/10.1128/AAC.46.6.1651-1657.2002
Kale RR, Kale MG, Waterson D, Raichurkar A, Hameed SP, Manjunatha M et al (2014) Thiazolopyridoneureas as DNA gyrase B inhibitors: optimization of antitubercular activity and efficacy. Bioorg Med Chem Lett 24(3):870–879. https://doi.org/10.1016/j.bmcl.2013.12.080
Levine C, Hiasa H, Marians KJ (1998) DNA gyrase and topoisomerase IV: biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim Biophys Acta 1400(1–3):29–43. https://doi.org/10.1016/S0167-4781(98)00126-2
Levy SB, Marshall B (2004) Antibacterial resistance worldwide: causes, challenges and responses. Nat Med 10(12):S122–S129. https://doi.org/10.1038/nm1145
Manchester JI, Dussault DD, Rose JA, Boriack-Sjodin PA, Uria-Nickelsen M, Ioannidis G, Bist S, Fleming P, Hull KG (2012) Discovery of a novel azaindole class of antibacterial agents targeting the ATPase domains of DNA gyrase and topoisomerase IV. Bioorg Med Chem Lett 22(15):5150–5156. https://doi.org/10.1016/j.bmcl.2012.05.128
Martyna GJ, Klein ML, Tuckerman M (1992) Nosé-Hoover chains: the canonical ensemble via continuous dynamics. J Chem Phys. 97(4):2635–2643. https://doi.org/10.1063/1.463940
Martyna GJ, Tobias DJ, Klein ML (1994) Constant pressure molecular dynamics algorithms. J Chem Phys 101(5):4177–4189. https://doi.org/10.1063/1.467468
Pan XS, Fisher LM (1999) Streptococcus pneumoniae DNA gyrase and topoisomerase IV: overexpression, purification, and differential inhibition by fluoroquinolones. Antimicrob Agents Chemother 43(5):1129–1136. https://doi.org/10.1128/AAC.43.5.1129
Pan XS, Gould KA, Fisher LM (2009) Probing the differential interactions of quinazolinedione PD 0305970 and quinolones with gyrase and topoisomerase IV. Antimicrob Agents Chemother 53(9):3822–3831. https://doi.org/10.1128/AAC.00113-09
Roos K, Wu C, Damm W, Reboul M, Stevenson JM, Lu C, Dahlgren MK, Mondal S, Chen W, Wang L, Abel R (2019) OPLS3e: extending force field coverage for drug-like small molecules. J Chem 15(3):1863–1874. https://doi.org/10.1021/acs.jctc.8b01026
Roy K, Ambure P, Aher RB (2017) How important is to detect systematic error in predictions and understand statistical applicability domain of QSAR models? Chemom Intell Lab Syst 162:44–54. https://doi.org/10.1016/j.chemolab.2017.01.010
Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aid Mol Des 27(3):221–234. https://doi.org/10.1007/s10822-013-9644-8
Sidhu JS, Sharma S, Singh A, Garg N, Kaur N, Singh N (2019) A naphthalimide-based novel “Turn-On” fluorescence approach for the determination of uric acid and monitoring of xanthine oxidase activity. Anal Methods UK 11(32):4190–4196. https://doi.org/10.1039/c9ay01464k
Sifaoui F, Lamour V, Varon E, Moras D, Gutmann L (2003) ATP-bound conformation of topoisomerase IV: a possible target for quinolones in Streptococcus pneumoniae. J Bacteriol Res 185(20):6137–6146. https://doi.org/10.1128/JB.185.20.6137-6146.2003
Starr JT, Sciotti RJ, Hanna DL, Huband MD, Mullins LM, Cai H, Gage JW, Lockard M, Rauckhorst MR, Owen RM, Lall MS (2009) 5-(2-Pyrimidinyl)-imidazo [1, 2-a] pyridines are antibacterial agents targeting the ATPase domains of DNA gyrase and topoisomerase IV. Bioorg Med Chem Lett 19(18):5302–5306. https://doi.org/10.1016/j.bmcl.2009.07.141
Sugino A, Peebles CL, Kreuzer KN, Cozzarelli NR (1977) Mechanism of action of nalidixic acid: purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proc Natl Acad Sci 74(11):4767–4771. https://doi.org/10.1073/pnas.74.11.4767
Tari LW, Trzoss M, Bensen DC, Li X, Chen Z, Lam T, Zhang J, Creighton CJ, Cunningham ML, Kwan B, Stidham M (2013) Pyrrolopyrimidine inhibitors of DNA gyrase B (GyrB) and topoisomerase IV (ParE). Part I: Structure guided discovery and optimization of dual targeting agents with potent, broad-spectrum enzymatic activity. Bioorg Med Chem Lett 23(5):1529–1536. https://doi.org/10.1016/j.bmcl.2012.11.032
Tourova TP, Korshunova AV, Mikhailova EM, Sokolova DS, Poltaraus AB, Nazina TN (2010) Application of gyrB and parE sequence similarity analyses for differentiation of species within the genus Geobacillus. Mol 79(3):356–369. https://doi.org/10.1134/S0026261710030124
Tropsha A, Gramatica P, Gombar VK (2003) The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb Sci 1:69–77. https://doi.org/10.1002/qsar.200390007
Trzoss M, Bensen DC, Li X, Chen Z, Lam T, Zhang J, Creighton CJ, Cunningham ML, Kwan B, Stidham M, Nelson K (2013) Pyrrolopyrimidine inhibitors of DNA gyrase B (GyrB) and topoisomerase IV (ParE), Part II: development of inhibitors with broad spectrum, Gram-negative antibacterial activity. Bioorg Med Chem Lett 23(5):1537–1543. https://doi.org/10.1016/j.bmcl.2012.11.073
Tuckerman MB, Berne BJ, Martyna GJ (1992) Reversible multiple time scale molecular dynamics. J Chem Phys 97(3):1990–2001. https://doi.org/10.1063/1.463137
Uria-Nickelsen M, Neckermann G, Sriram S, Andrews B, Manchester JI, Carcanague D, Stokes S, Hull KG (2013) Novel topoisomerase inhibitors: microbiological characterisation and in vivo efficacy of pyrimidines. Int J Antimicrob Agents 41(4):363–371. https://doi.org/10.1016/j.ijantimicag.2012.12.001
Worthington RJ, Melander C (2013) Combination approaches to combat multidrug-resistant bacteria. Trends Biotechnol 31(3):177–184. https://doi.org/10.1016/j.tibtech.2012.12.006
Zhao M, Wang L, Zheng L, Zhang M, Qiu C, Zhang Y, Niu B (2017) 2D-QSAR and 3D-QSAR analyses for EGFR inhibitors. Biomed Res Int. https://doi.org/10.1155/2017/4649191
Acknowledgements
The authors much acknowledge the All India Council of Technical Education (AICTE), National doctoral fellowship (NDF-2018), Application number—56149 for providing the funding support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest in this study. The authors alone are responsible for the content and writing of the paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yele, V., Azam, M.A. & Jupudi, S. Ligand-based pharmacophore modelling, in silico virtual screening, molecular docking and molecular dynamic simulation study to identify novel Francisella tularensis ParE inhibitors. Chem. Pap. 74, 4567–4580 (2020). https://doi.org/10.1007/s11696-020-01274-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-020-01274-3