
Abstract
Thermodynamic descriptions of the Fe-Mn-P system and its binary sub-system, Mn-P, are developed in the frame of a new Fe-X-P (X = Al, Cr, Cu, Mn, Mo, Nb, Ni, Si, Ti) database. The thermodynamic parameters of the binary sub-systems, Fe-Mn and Fe-P, are taken from the latest earlier assessments. Those of the Fe-Mn-P and Mn-P systems are optimized in this study using literature experimental thermodynamic and phase equilibrium data. The solution phases of the system are described with the substitutional solution model and the phosphides are treated as stoichiometric phases or semi-stoichiometric phases of the (A,B) p C q type described with the two-sublattice model. The formation of a miscibility gap in the liquid phase, at high P-contents is predicted.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The current work continues an earlier started project[1,2] for the development of a thermodynamic database of phosphorus containing Fe-X systems. In such a way a simple and compatible thermodynamic database for three-component steels is to be designed. It would provide important and practical input data for thermodynamic-kinetic models simulating their solidification.[3] The first two contributions concern the systems Fe-Cr-P[1] and Fe-Cu-P.[2]
The goal of the current work is to attain, for the first time, thermodynamic optimizations for the Fe-Mn-P and the Mn-P systems. The latter is based on the earlier Mn-P optimization of Miettinen.[4] The adjustable thermodynamic parameters used for the description of the Fe-Mn and the Fe-P phase diagrams are taken from Huang[5] and from Shim et al.,[6] respectively.
Phases, Modeling and Data
Detailed descriptions for the substitutional solution and sublattice models and their parameters are available from Ref 7 and in this journal by Ref 1. Thus, abbreviated descriptions of the phases and models are presented only (Table 1). The solution phases (liquid, bcc_A2, fcc_A1, cbcc_A12, cub_A13) are described with the substitutional solution model. Some phosphides are described with the sublattice model (Fe3P, Fe2P, Mn3P) and other phases are treated as stoichiometric (FeP, Mn3P2, MnP, white_P).
Complete mutual solubility was assumed between Fe2P and Mn2P, in agreement with the experimental data of Ref 8,9, though according to other authors,[10-12] an ordering reaction (at an undetermined temperature) yielding to an orthorhombic structure for compositions 0.31 < x < 0.62 of (Fe1−x ,Mn x )2P[13] was observed.
The experimental studies on the Fe-Mn-P system up to 1988 have been reviewed by Raghavan.[13] Table 2 shows the experimental information[9,14-22] selected in the current optimization for the Mn-P and Fe-Mn-P systems.
Results
The thermodynamic description of the Fe-Mn-P system is presented in Table 3. The parameters marked with a reference code were adopted from earlier assessments and those marked with O* or E* were, respectively, optimized or estimated in the current study. By O*, the parameter was optimized using literature experimental data (Table 2) and by E*, the parameter was estimated arbitrarily.
As previously reported,[1] the Gibbs energy expressions of Fe3P, Fe2P and FeP have been simplified by changing the reference states of Fe and P from HSER (used by Shim et al.[6]) to bcc-Fe and white P, respectively. In a similar way, the reference states for the Gibbs energy expression for Mn3P has been changed from cub-Mn and P2-gas (used by Zaitsev et al.[19]) to bcc-Fe and white P (Table 3). This change has no perceptible influence on the function values.
Further, calculated phase equilibria and thermodynamic quantities have been compared to original experimental data to in order to verify the results. All calculations were carried out with the ThermoCalc software.[24]
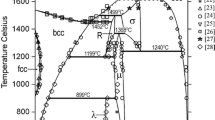
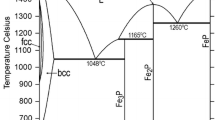
Figures 1 and 2 show the Fe-Mn and Fe-P phase diagrams calculated with the parameters of Huang[5] and of Miettinen and Vassilev,[1] respectively. Figure 3 shows the Mn-P phase diagram optimized in the current study. The agreement with the experimental data is good, as shown by Huang[5] for Fe-Mn, by[1,6] for Fe-P, and in Fig. 3 for Mn-P.

Calculated with the parameters of Huang[5] Fe-Mn phase diagram


Mn-P phase diagram, calculated with the parameters optimized in this work, together with experimental data points of Berak and Heumann[14]
Calculated Mn and P activities in liquid alloys at 1320 °C are shown in Fig. 4 and 5, while calculated Gibbs energies of formation of Mn3P and Mn2P are plotted in Fig. 6. The agreement with the experimental data of Ref 15-19 is reasonable.



The calculated liquidus projection of the Fe-Mn-P system, together with experimental data points of Vogel and Berak[9] is shown in Fig. 7. It is of worth noting that the manganese content in point U4 (see also Table 4) is inconsistent. This inconsistency was accepted since a stronger stabilization for the cub-phase would have lead to a too extensive primary surface for this phase (see the rounded form of the calculated cub-phase region in Fig. 7), whereas Vogel and Berak[9] presented it as a triangle starting from the binary Mn-P side and approaching point U4 as a very narrow region. The latter authors[9] suggested one more invariant point, U5, due to splitting of the fcc phase into Fe-rich and Mn-rich fcc phases. This point was ignored in the current work as such splitting has not been observed in the Fe-Mn system.[13]

Calculated liquidus projection of the Fe-Mn-P system, together with experimental data points of Vogel and Berak.[9] Shown also are the calculated liquidus isotherms between 1000 and 1500 °C (dotted lines)
Calculated vertical and isothermal sections are presented in Figs. 8, 9, 10, 11, 12, 13, 14, and 15, correspondingly. Good agreement of the current calculations with the experimental data of Vogel and Berak[9,20] is observed.

Calculated vertical section of the Fe-Mn-P system at mass ratio w Fe:w Mn = 19, together with experimental data points of Vogel and Berak[9] (Liquidus—liquidus points, 2. Arrest—thermal arrest data)

Calculated vertical section of the Fe-Mn-P system at mass ratio w Fe:w Mn = 9:1, together with experimental data points of Vogel and Berak[9] (Liquidus—liquidus points, 2. Arrest—thermal arrest data)

Calculated vertical section of the Fe-Mn-P system at mass ratio w Fe:w Mn = 5:5, together with experimental data points of Vogel and Berak[9] (Liquidus—liquidus points, 2. Arrest—thermal arrest data)

Calculated vertical section of the Fe-Mn-P system at mass ratio w Fe:w Mn = 1:9, together with experimental data points of Vogel and Berak[9] (Liquidus—liquidus points, 2. Arrest—thermal arrest data)

Calculated vertical section of the Fe-Mn-P system at 6 wt.% P, together with experimental data points of Vogel and Berak[9] (Liquidus—liquidus points, 2. Arrest—thermal arrest data)

Calculated vertical section of the Fe-Mn-P system at 12 wt.% P, together with experimental data points of Vogel and Berak[9] (Liquidus—liquidus points, 2. Arrest—thermal arrest data)

Calculated isotherm of the Fe-Mn-P system at 1000 °C, together with experimental data points of Kaneko et al.[20]

Calculated isotherm of the Fe-Mn-P system at 800 °C, together with experimental data points of Kaneko et al.[20]
In Fig. 16 the conformity between the calculated and experimental[21,22] P-activity coefficient (γ MnP ) data in liquid phase has been verified. The agreement is good, however, the temperature dependence of the calculated γ MnP values is slightly different from the experimental.

Finally, it is of worth noting that the formation of a liquid phase miscibility gap in the phosphorus-rich corner of the ternary system, at about 600 K, has been predicted. No experimental data is available to confirm the appearance of that gap but this was accepted in the present evaluation, in order to keep the liquid state ternary interaction parameters (Table 3) reasonably simple for the practical calculations at lower phosphorus contents. From theoretical view point[25] such a miscibility gap should be expected, because of the large difference between the atomic radii of the iron and manganese atoms, from one side, and that one of the phosphorus atoms, from other side. Moreover, there is a significant dissimilarity between the chemical character (e.g. the electronegativity) of the phosphorous and the metallic elements (Fe and Mn, in this case).
Summary
A thermodynamic description has been obtained for the ternary Fe-Mn-P system and its binary sub-system, Mn-P, making use of literature experimental thermodynamic and phase equilibrium data. In the current ternary phase diagram description, twelve phases, i.e., liquid, bcc, fcc, cbcc, cub, Fe3P (dissolving Mn), Fe2P (extending to Mn2P), Mn3P (dissolving Fe), FeP, Mn3P2, MnP and white-P, have been considered. Good or reasonable correlations between the calculated and the experimental thermodynamic and phase equilibrium data have been obtained.
The new Fe-Mn-P description is added as the next member of the thermodynamic database of Fe-X-P systems. No earlier thermodynamic optimization has been found in the open literature for this ternary system.
References
J. Miettinen and G. Vassilev, Thermodynamic Description of Ternary Fe-X-P Systems. Part 1: Fe-Cr-P, JPED, 2014, 35, p. xxx—under printing. doi:10.1007/s11669-014-0314-x.
J. Miettinen and G. Vassilev, Thermodynamic Description of Ternary Fe-X-P Systems. Part 2: Fe-Cu-P, JPED, 2014, 35, p. xxx—to be printed. doi:10.1007/s11669-014-0315-9.
J. Miettinen, S. Louhenkilpi, H. Kytönen, and J. Laine, IDS: Thermodynamic-Kinetic-Empirical Tool for Modeling of Solidification, Microstructure and Material Properties, Math. Comput. Simul., 2010, 80, p 1536-1550
J. Miettinen, Thermodynamic Solution Phase Data for Binary Mn-Based Systems, CALPHAD, 2001, 25, p 43-58
W. Huang, An Assessment of the Fe-Mn System, CALPHAD, 1989, 13, p 243-252
J.-H. Shim, C.-S. Oh, and D.N. Lee, Thermodynamic Properties and Calculation of Phase Diagram of the Fe-P System, J. Korean Inst. Met. Mater., 1996, 34, p 1385-1393
C. Qiu, Thermodynamic Study of Carbon and Nitrogen in Stainless Steels. PhD thesis, Royal Institute of Technology, Stockholm, 1993.
H. Nowotny and E. Henglein, Untersuchung ternärer Legierungen mit Phosphor (Study of Ternary Alloys Containing Phosphorus), Monatsch. Chem., 1948, 79, p 385-394, in German
R. Vogel and J. Berak, Über das System Mangan-Phosphor (The Iron-Phosphorus-Mangenese System), Arch. Eisenhüttenwes., 1952, 23, p 217-223, in German
R. Fruchart, A. Roger, and J.P. Senateur, Crystallographic and Magnetic Properties of Solid Solutions of the Phosphides M2P (M = Cr, Mn, Fe, Co and Ni, J. Appl. Phys., 1969, 40, p 1250-1257
A. Roger, J.P. Senateur, and R. Fruchart, Propriétés cristallographiques et magnétique des solutions solides parmis les phosphides Ni2P, Co2P, Fe2P, Mn2P et Cr2P (Crystallographic and Magnetic Properties of Solid Solutions among the Phosphides Ni2P, Co2P, Fe2P, Mn2P and Cr2P), Ann. Chim. (Paris), 1969, 4(2), p 79-91, in French
S. Nagase, H. Watanabe, and T. Shinohara, Magnetic Properties of the System Fe2P-Mn2P, J. Phys. Soc. Jpn., 1973, 34(4), p 911-916
V. Raghavan, Phase Diagrams for Ternary Iron Alloys, Part 3, Indian Institute of Metals, Calcutta, 1988, p 91-99
J. Berak and T. Heumann, Über das System Mangan-Phosphor, Z. Metallkd., 1950, 41(1), p 19-23, in German
M.T. Gasik, B.I. Yemlin, and V.F. Gorbachev, Metallurgy and Chemistry of Carbonite, Tekhnika Press, Kiev, 1972, p 3
G.I. Batalin, V.A. Stukalo and N.Ya. Neshchimenko, Tepмoдинимичecкaя aктивнocть фocфopa в жидкиx cплaвax мapгaнцa c фocфopa, (Phosphorous thermodynamic activity in liquid manganese phosphorous alloys) Укpaинcкий Xимичecкий Жypнaл (Ukr. Khim. Zh.), 1977, 43, p 602-604, in Russian
F. Granjean, D.W. Osborn, W.G. Lyon, and H.E. Flotow, Dimanganese Phosphide, Mn2P: Heat Capacity from 5 to 350 K, Magnetic Entropy, and Thermodynamic Functions to 1300 K, J. Chem. Thermodyn., 1977, 9, p 549-559
Y.E. Lee, Thermodynamics of the Mn-P System, Metall. Trans. B., 1986, 17, p 777-783
A.I. Zaitsev, A.D. Litvina, N.E. Shelkova, Z.V. Dobrokhotova, and B.M. Mogutnov, Thermodynamic Properties of Alloys of Phosphorus with Calcium, Barium, Chromium, Manganese and Iron, Z. Metallkd., 1997, 88, p 76-86
H. Kaneko, T. Nishizawa, K. Tamaki, and A. Tanifuji, Solubility of Phosphorus in α; and γ Iron, Nippon Kinzoku Gakkai-Si, 1965, 29, p 166-170, in Japanese
H. Schenck, E. Steinmetz, and H. Gitizad, Die Phosphoraktivität im flussigen Eisen und ihre Beeinflussung durch Nickel, Mangan and Chrom (The Influence of Nickel, Manganese and Chromium on the Phosphorous Activity in Liquid Iron), Arch. Eisenhüttenwes., 1969, 40, p 597-602, in German
S. Ban-Ya, N. Maruyama, and Y. Kawase, Effects of Ti, V, Cr, Mn Co, Ni, Cu, Nb, Mo and W on the Activity of Phosphorus in Liquid Iron, J. Iron Steel Inst. Jpn., 1984, 70, p 65-72, in Japanese
A.T. Dinsdale, SGTE Data for Pure Elements, CALPHAD, 1991, 15, p 317-425
J.-O. Andersson, T. Helander, L. Höglund, P. Shi, and B. Sundman, Thermo-Calc & DICTRA, Computational Tools for Materials Science, CALPHAD, 2002, 26, p 273-312
G.P. Vassilev, Systematic of Binary Phase Diagrams, Formed by Low-Melting Elements (Bi, Sn, Zn, In) and the Metals of IV-th and V-th Periods, JMM, 2005, 41B, p 79-93
Acknowledgment
Financial support of the Finnish Funding Agency for Technology and Innovation (TEKES) is gratefully acknowledged by Dr J. Miettinen. The research was carried out as part of the Finnish Metals and Engineering Competence Cluster (FIMECC)’s SIMP program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Miettinen, J., Vassilev, G. Thermodynamic Description of Ternary Fe-X-P Systems. Part 3: Fe-Mn-P. J. Phase Equilib. Diffus. 35, 587–594 (2014). https://doi.org/10.1007/s11669-014-0328-4
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11669-014-0328-4