
Abstract
Thermodynamic descriptions of the Fe-Mo-P system and its binary sub-system, Mo-P, are developed in the frame of a new Fe-X-P (X = Al, Cr, Cu, Mn, Mo, Nb, Ni, Si, Ti) database. The thermodynamic parameters of the binary sub-systems, Fe-Mo and Fe-P, are taken from earlier assessments (modifying slightly the Fe-Mo description) and those of the Fe-Mo-P and Mo-P systems are optimized in this study using experimental thermodynamic and phase equilibrium data available in the literature. The solution phases (i.e., bcc_A2, fcc_A1, Liquid) are described with the substitutional solution model. The binary compounds: Fe2Mo, FeP, Mo3P and MoP, are treated as stoichiometric phases, while for Fe3P and Fe2P two-sublattice models accounting for the dissolution of Mo were applied. Some intermetallic compounds (Mu, R and Sigma) are described with three-sublattice models.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The current paper continues an earlier started project[1–3] for the development of a Fe-X-P database. The aim is to design a simple and compatible thermodynamic database, which provides important and practical input data for thermodynamic-kinetic models simulating the solidification of phosphorous steels.[4]
The binary thermodynamic data retained from the literature are these of Andersson[5] (mainly) for the Fe-Mo system, and from Shim et al.[6] and Miettinen and Vassilev[1]—for the Fe-P system. We would like to emphasize that the latter optimization has been implemented by Shim et al.[7] to the very important system Fe-P-C as well. Thus, the current database would be compatible with the a.m. assessment.
The Fe-P system was optimized by Ohtani et al.[8] as well, using the first principle calculation of the enthalpy of formation of the Fe-P intermetallics (the Fe-P optimization of Shim et al.[6] (already published at that time) was not mentioned neither discussed by those authors[8]). However, Changjun et al.[9] reported that the thermodynamic parameters of Ohtani et al.[8] “may underestimate the Gibbs energy of the Fe3P phase while may over-estimate that of the Fe2P phase”. For this reason, these authors[9] have retained the Fe-P parameters of Gustafsson[10] used in the optimization of the Fe-Mo-P system. Anyhow, that description could not be directly adapted for the needs of the current database.
Thus, the goal of this work is to achieve new thermodynamic optimizations of the ternary Fe-Mo-P system and its binary sub-system Mo-P. The thermodynamic description of the Fe-Mo system had to be partially modified as well, in order to get a better fit for some solid-liquid equilibria in this as well as in other ternary systems (Fe-Cr-Mo, Fe-Mo-Ni and Fe-Mo-Si).
Phases, Modeling and Data
Detailed descriptions of the substitutional solutions and sublattice models and their parameters are available from Lukas et al.[11] Thus, a summary of the phases and the models used in this work is presented in Table 1.
The solution phases (i.e. bcc_A2, fcc_A1, Liquid) are modeled with the substitutional solution model (Table 1). Most of the binary compounds: Fe2Mo, FeP, Mo3P and MoP, are treated as stoichiometric phases, while for Fe3P and Fe2P two-sublattice models accounting for the dissolution of Mo were applied.
Some intermetallic compounds (Mu (μ), R and Sigma (σ)) are described with three-sublattice models. One should be aware that the maximum dissolution of 10 wt.%Mo in FeP and 5 wt.% Fe in MoP, reported by Guerin and Sergent,[12] at 800 °C, was not considered. This simplifies the calculations but the main reason is the lack of corresponding data at other temperatures and also the lack of any phase equilibrium experimental information for the regions situated along or nearby the FeP-MoP section line.
The studies on the Fe-Mo-P system, up to 1988, have been reviewed by Raghavan.[13] The experimental information selected in the current work for the optimization of the Mo-P and the Fe-Mo-P systems is compiled in Table 2. In addition, topological data about the Fe-Mo phase equilibria,[20–28] the integral molar mixing enthalpy values of the Fe-Mo liquid solutions from Iguchi et al.[29] and the Fe-activity in liquid Fe-Mo alloys from Ichise et al.[27] are used for the partial re-optimization of the Fe-Mo system done is this work.
Results
The liquid Fe-Mo phase description by Andersson[5] was modified in this work in order to improve the agreement between the calculated and the experimental (bcc + L)-region of the system. The system Mo-P was reoptimized using the available literature experimental data (Table 2).
The adjustable parameters of the Fe-Mo-P system, obtained in the current study, are presented in Table 3. The parameters marked with a reference code were adopted from earlier assessments. Those marked with O or E were optimized or estimated in this work as follows: O—the parameter was optimized using literature experimental data (Table 2); E—the parameter was estimated taking into account the relative stabilities of the corresponding phases and keeping realistic values of their integral and partial thermodynamic quantities.
The Gibbs energy expressions for the phases Fe3P, Fe2P and FeP, given by Shim et al.,[6] have already been simplified[1,2] by changing the reference states of Fe and P. In this work we keep the modified versions. There are no significant differences between the results of the calculations done in both ways.
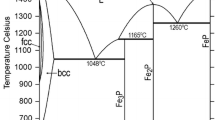
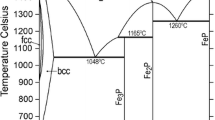
In order to verify the current optimizations, the calculated results have been compared with the original experimental data by making use of the ThermoCalc software.[32] Thus, Fig. 1 through 3 show the Fe-Mo and the Mo-P phase diagrams, whose parameters were modified in the current study, and the Fe-P phase diagram calculated by Ref 1,6. The agreements with the experimental data are good.



The new liquid phase description of the Fe-Mo system (Fig. 1) yields solid-liquid equilibria, which only slightly deviate from those of Andersson.[5] Nevertheless, in Fig. 4 one can see that (bcc + L)-region shows improved agreement with the experimental data of Gibson et al.[21] That new description also fits better for describing the phase equilibria in the ternary Fe-Cr-Mo, Fe-Mo-Ni and Fe-Mo-Si systems, allowing the usage of some lower-value ternary liquid-state parameters in these descriptions (to be published). For the integral molar mixing enthalpy and the liquid phase Fe-activity, the current results are quite similar to those of Andersson,[5] agreeing reasonably well with the experimental data of Ichise et al.[27] and Iguchi et al.[29]

Figures 5 through 14 and Table 4 contain the results of the calculations for the Fe-Mo-P system, together with the selected experimental data (Table 2). The agreement is rational and in most cases, it is slightly better than that obtained by Gustafsson.[7]

Liquidus projection of the Fe-Mo-P system calculated with the parameters obtained in this work, together with the experimental data for the invariants from Vogel and Horstmann[14] and the respective values calculated by Gustafsson[10] (see Table 4). The calculated liquidus isotherms between 1100 and 1900 °C (dotted lines) are shown







Isotherm of the Fe-Mo-P system (Fe-rich side) at 1200 °C calculated with the parameters obtained in this work


The liquidus projection of the system (Fig. 5) shows different representation of the reactions concerning codes U1 and E1 (Table 4). No suggestions based on experimental data have been given for the invariant points at high Mo contents. The current calculations and those of Gustafsson,[7] however, yield quite similar primary surfaces for the system.
In the vertical sections (Fig. 6 through 11) and the isothermal sections (Fig. 12, 13), reasonable agreement between the current calculations and the experimental data of Vogel and Horstmann,[14] Kaneko et al.[15] and Raghavan[13] is observed. Additional calculations, using the data of Gustafsson,[10] have been done for the six vertical sections. They show slightly different phase equilibria and slightly worse agreement with these measurements (for the sake of clarity only the liquidus lines of Gustafsson[10] are plotted in Fig. 6 through 11).
In Fig. 13, the phase equilibria suggested by Raghavan[13] at 800 °C (broken lines) agree well with the current calculations but disagree with those of Gustafsson.[10] The 800 °C isotherm has recently been studied also by Olinyuk et al.,[33] who found two ternary compounds, [Fe(1−x)Mo(x)]2P (x = 0.18–0.70) and [Fe(1−x)Mo(x)]3P (x = 0.85–0.90). According to the latter study the former compound overlaps the current (Fe,Mo)2P phase, but does not extend to the nearly binary Fe2P phase. Obviously, the solid state equilibria of the system should be revised as soon as more experimental measurements become available.
Figure 14 illustrates the satisfactory agreement between the calculated (this work) and the experimental (Yamada and Kato[16]) phosphorous activity coefficient (γ MoP ) in the liquid phase, at 1600 °C. The symbol γ MoP is used in order to describe the Mo-content effect on the P-activity coefficient and is defined as: γ MoP = γ (Fe-Mo-P)P /γ (Fe-P)P . In the latter expression the P content (mole fractions) is fixed to x P = 0.001, referring to a very low P-content, applied but not quantified, by Yamada and Kato.[16]
Ueno et al.[18] derived a theoretical equation using the pseudo-potential formalism coupled with the free energy of a hard sphere system. The P-activity values calculated with this equation agree excellent with the current calculations. All these results point toward negative deviations from the ideal solution behavior. The respective P-activity calculated with the parameters of Gustafsson,[10] (Fig. 14, dotted line) show opposite (i.e., positive) deviations from the Rault’s law).
Calculations has also been carried for the parameter ε MoP = ln(γ MoP )/x P, at 1400 °C, in ternary alloys containing about 20 at.% P. In this case, both assessments (this work and Gustafsson[10]), yield a value close to ε MoP = −2, whereas the measurements of Ban-ya[17] give that ε MoP = −7.26. Such a low value, however, indicates too high stability for the liquid phase so that the calculated (phosphide + liquid phase) equilibria of the present work could not be preserved.
Summary
A thermodynamic reoptimization of the binary sub-system Mo-P has been done. In addition, a slight modification was made for the liquid phase description of the adopted Fe-Mo assessment. Thereafter, a description of the ternary Fe-Mo-P system was obtained.
Thirteen phases (liquid, bcc, fcc, Mu, R, Sigma, Fe3P (dissolving Mo), Fe2P (dissolving Mo), Fe2Mo, FeP, Mo3P, MoP and (P)) are considered in the current work. Good or reasonable correlation between the calculated and the experimental thermodynamic and phase equilibrium data has been observed. Clear improvement is obtained for the liquid state thermodynamics in regard to the previous optimization of this system. The results of the current optimization have been added to the thermodynamic database on Fe-X-P systems.
References
J. Miettinen and G. Vassilev, Thermodynamic Description of Ternary Fe-X-P Systems. Part 1: Fe-Cr-P, JPED, 2014, 35(4), p 458-468
J. Miettinen and G. Vassilev, Thermodynamic Description of Ternary Fe-X-P Systems. Part 2: Fe-Cu-P, JPED, 2014, 35(4), p 469-475
J. Miettinen and G. Vassilev, Thermodynamic Description of Ternary Fe-X-P Systems. Part 3: Fe-Mn-P, JPED, 2014, 35(5), p 587-594
J. Miettinen, S. Louhenkilpi, H. Kytönen, and J. Laine, IDS: Thermodynamic-Kinetic-Empirical Tool for Modeling of Solidification, Microstructure and Material Properties, Math. Comput. Simulat., 2010, 80, p 1536-1550
J.-O. Andersson, A Thermodynamic Evaluation of the Fe-Mo-C System, CALPHAD, 1988, 12, p 9-23
J.-H. Shim, C.-S. Oh, and D.N. Lee, Thermodynamic Properties and Calculation of Phase Diagram of the Fe-P System, J. Korean Inst. Met. Mater., 1996, 34, p 385-393
J.-H. Shim, C.-S. Oh, and D.N. Lee, Thermodynamic Assessment of the Fe-C-P System, Z. Metallkd., 2000, 91, p 114-120
H. Ohtani, N. Hanaya, M. Hasebe, S. Teraoka, and M. Abe, Thermodynamic Analysis of the Fe-Ti-P Ternary System by Incorporating First-Principles Calculations into the CALPHAD Approach, CALPHAD., 2006, 30, p 147-158
C. Wu, W. Huang, X. Su, X. Peng, H. Wang, and Y. Liu, Experimental Investigation and Thermodynamic Calculation of the Al-Fe-P System at Low Phosphorus Contents, CALPHAD, 2012, 38, p 1-6
P. Gustafsson, Study of the Thermodynamic Properties of the C-Cu-Fe-P, Fe-Mo-P and Fe-Ni-P Systems. Report IM-2549, Institute of Metals Research, Stockholm, 1990
H. Lukas, S.G. Fries, and B. Sundman, Computational Thermodynamics: The Calphad Method, Cambridge University Press, UK, 2007
R. Guerin and M. Sergent, Synthesis and Crystallographic Study of the System MP-MoP and MP-WP (M = 3d Element), Compt. Rend., 1975, 281C, p 777-780, [in French]
V. Raghavan, Phase Diagrams for Ternary Iron Alloys, Part 3, Indian Institute of Metals, Calcutta, 1988
R. Vogel and D. Horstmann, The Iron-Iron Phosphide-Molybdenum Phosphide-Molybdenum Phase Diagram, Arch. Eisenhüttenwes., 1953, 24, p 369-374, [in German]
H. Kaneko, T. Nishizawa, K. Tamaki, and A. Tanifuji, Solubility of Phosphorus in α and γ Iron, Nippon Kinzoku Gakkai-Si., 1965, 29, p 166-170, [in Japanese]
K. Yamada and E. Kato, Effect of Dilute Concentrations of Si, Al, Ti, V, Cr Co, Ni, Nb and Mo on the Activity Coefficient of P in Liquid Iron, Trans. Iron Steel Inst. Jpn., 1983, 23, p 51-55
S. Ban-Ya, N. Maruyama, and Y. Kawase, Effects of Ti, V, Cr, Mn Co, Ni, Cu, Nb, Mo and W on the Activity of Phosphorus in Liquid Iron, J. Iron Steel Inst. Jpn., 1984, 70, p 65-72, [in Japanese]
S. Ueno, Y. Waseda, K.T. Jacob, and S. Tamaki, Theoretical Treatment of Interaction Parameters in Multicomponent Metallic Solutions, Steel Res., 1998, 59, p 474-483
L. Brewer, R.H. Lamoreaux, Molybdenum: Physico-Chemical Properties of its Compounds, and Alloys. Vienna: Atomic Energy Review, Special Issue No. 7, L. Brewer, Eds. IAEA, Vienna, 1980
J.L. Ham, An Introduction to Arc-Cast Molybdenum and its Alloys, Am. Soc. Mech. Eng. Trans., 1951, 73, p 723-732
W.S. Gibson, J.R. Lee, and W. Hume-Rothery, Liquidus-Solidus Relations in Iron-Rich Iron-Niobium and Iron-Molybdenum Alloys, J. Iron Steel Inst., 1961, 198, p 64-66
R.D. Rawlings and C.A. Newey, Study of the Iron-Molybdenum System by Means of Diffusion Couples, J. Iron Steel Inst., 1968, 206, p 273-276
G. Kirchner, H. Harvig, and B. Uhrenius, Experimental and Thermodynamic Study of the Equilibria between Ferrite Austenite and Intermediate Phases in the Fe-Mo, Fe-W, and Fe-Mo-W Systems, Metall. Trans., 1973, 4, p 1059-1067
C.P. Heijwegen and G.D. Rieck, Determination of the Phase Diagram of the Mo-Fe System Using Diffusion Couples, J. Less Common Met., 1974, 37, p 115-121
P.J. Alberty and C.W. Haworth, The Solubility of Mo in Gamma-Iron, Met. Sci., 1975, 9, p 140-149
Y. Ueshima, E. Ichise, T. Mori, On the Fe-Mo Phase Diagram. Tetsu-to-Hagane, 1979, 65, S-684-6 [in Japanese]
E. Ichise, T. Maruo, H. Sasho, Y. Ueshima, and T. Mori, Knudsen Cell-Mass Spectrometric Determination of Activities in Fe-Mo Alloys, Tetsu-to-Hagane., 1980, 66, p 1075-1083, [in Japanese]
T. Takayama, M.Y. Wey, and T. Nishizawa, Effect of Magnetic Transition on the Solubility of Alloying Elements in bcc Iron and fcc Cobalt, Trans. Jpn Inst. Met., 1981, 22, p 315-325
Y. Iguchi, S. Nobori, K. Saito, and T. Fuwa, A Calorimetric Study of Heats of Mixing of Liquid Alloys Fe-Cr, Fe-Mo, Fe-W, Fe-V, Fe-Nb, Fe-Ta, J. Iron Steel Inst. Jpn., 1982, 68, p 633-640, [in Japanese]
A.T. Dinsdale, SGTE Data for Pure Elements, CALPHAD., 1991, 15, p 317-425
Z.-P. Jin and C. Qiu, An Experimental Study and Thermodynamic Evaluation of the Fe-Mo-Ti System at 1000°C, Metall. Trans. A, 1993, 24A, p 2137-2142
J.-O. Andersson, T. Helander, L. Höglund, P. Shi, and B. Sundman, Thermo-Calc & DICTRA, Computational Tools for Materials Science, CALPHAD., 2002, 26, p 273-312
A.O. Olinyuk, Y.F. Lomnytska, M.V. Dzevenko, S.S. Stoyko, and A. Mar, Phase Equilibria in the Fe-Mo-P System at 800 °C and Structure of Ternary Phosphide (Mo1−xFex)3P (0.10 < x < 0.15), Inorg. Chem., 2013, 52, p 983-991
Acknowledgment
Financial support of the Finnish Funding Agency for Technology and Innovation (TEKES) is gratefully acknowledged by Dr J. Miettinen. The research was carried out as part of the Finnish Metals and Engineering Competence Cluster (FIMECC)’s SIMP program.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Miettinen, J., Pashkova, A. & Vassilev, G. Thermodynamic Description of Ternary FE-X-P Systems. Part 4: FE-MO-P. J. Phase Equilib. Diffus. 36, 60–67 (2015). https://doi.org/10.1007/s11669-014-0357-z
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11669-014-0357-z