
Abstract
Solid-state reaction synthesis produced VO2+ -doped Li2Ba3(P2O7)2 pyrophosphate nanopowder at ambient temperature. The produced sample was characterized by powder x-ray diffraction (XRD), scanning electron microscopy (SEM) with energy dispersive spectroscopy (EDS), high-resolution transmission electron microscopy (HR-TEM), Fourier-transform infrared spectroscopy (FT-IR), Raman spectroscopy, UV-visible spectroscopy, electron paramagnetic resonance (EPR), and photoluminescence (PL) techniques. Powder XRD confirmed the preservation of an orthorhombic crystal structure of the sample with an average crystallite size of 34.2 nm. In addition, lattice cell parameters and internal lattice strain were evaluated. SEM and HR-TEM images revealed that a tiny agglomeration of stone-like particles appeared in the sample and also evaluated particle size distributions. FT-IR and Raman spectra exhibited characteristic vibrational modes of phosphate (PO43−) ions along with other bands. Optical absorption spectrum of the VO2+ -doped sample exhibited three characteristic peaks at 831, 635, and 416 nm. Furthermore, crystal and tetragonal field parameters were determined as Dq = 1574, Ds = − 2903, and Dt = 664 cm−1. From the EPR spectrum, calculated spin Hamiltonian parameters were \(g_{\parallel }\) = 1.954, \(g_{ \bot }\) = 1.977, \(A_{\parallel }\) = 284 \(\times\) 10−4 cm−1 and \(A_{ \bot }\) = 53 \(\times\) 10−4 cm−1. Optical and EPR spectra revealed that VO2+ ions have tetragonal distortion octahedral site symmetry and have a moderate covalent nature with ligands. Using the PL spectrum, the CIE chromaticity coordinates were determined to be in the yellowish-green region appropriate for LEDs and lighting systems.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recent advances in science and technology necessitate developing and applying different materials based on phosphate compounds.1 These compounds have catalytic, magnetic, and optical properties, and are frequently employed as monocrystals or polycrystals, ceramics, and so on.2 Special scientific interests include a study of the characteristics of materials as well as their synthesis and development.3 Inorganic phosphate has recently received much attention because of phosphate-based luminescence materials.4 The great optical absorbance and superior chemical and thermal stability of pyrophosphates make them an ideal host lattice for activators.5 The crystal field characteristics and energy level structure of transition metal ions can be obtained through optical absorption experiments. Information on the electronic structure, oxidation state, site symmetry, and bonding type of impurity ions can be obtained by electron paramagnetic resonance (EPR) and optical absorption techniques. In addition, LEDS have low power consumption, solid structure, compact size, fast response, and friendliness.6 M2O-P2O5-MexOy-type systems (where M = Li, Na, K) are recognized as versatile solvents for 3d-metal oxides and serve as reaction media for synthesizing diverse diphosphates.1 One of the transition metal ions that can exist in several oxidation states based on the host structure is vanadium.7 Variable states of vanadium include V2+, V3+, V4+ , and V5+, with V4+ being the majority state of them which appears as VO2+. Vanadium dioxide (VO2+) has garnered significant interest from scientists and technologists due to its distinct metal–insulator transition (MIT) near room temperature making it a notable thermochromic material. Vanadyl ions are well known in research because of their variety of applications in the field of lasers, electrical switches, memory devices, smart radiators, sensors, and luminescence.8 Recently, VO2+-doped phosphor materials have been reported with VO2+ exhibiting different luminescence properties.9,10,11,12 Authors have reported pure and iron-doped Li2Ba3(P2O7)2 pyrophosphate nanopowders,13 with pure and doped nanopowders exhibiting different luminescent properties.
As far as we are aware, no research on the characteristics of Li2Ba3(P2O7)2-doped with transition metal ions has been published. In this work, VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder has been synthesized using a solid-state reaction technique. Additionally, we discuss various structural and optical properties, including site symmetry, bonding nature, and luminescent behavior.
Experimental
Materials
The following chemicals were employed to produce VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder: lithium carbonate (Li2CO3, 99.9%), barium carbonate (BaCO3, 99.9%), ammonium dihydrogen orthophosphate (NH4H2PO4, 99.9%), and 0.01 mol% of vanadium pentoxide (V2O5, 99.9%) were procured from HiMedia laboratories, Mumbai, India. All the chemicals were analytically pure and used without further purification.
Sample Preparation
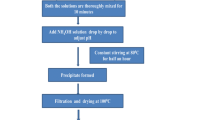
The starting chemicals, 0.739 g of Li2CO3, 5.738 g of BaCO3, and 4.601 g of NH4H2PO4 were weighed and ground into a fine powder for about 30 min using an agate mortar. Next, 0.182 g of V2O5 was added to the mixture and ground for a further 30 min. The chemical mixture obtained was placed into a silica crucible and sintered at 350°C for 1 h, at 550°C for 1 h, and finally at 750°C for 2 h in a programmable muffle furnace. Throughout this process, the sample was ground for 1 h at each temperature to eliminate volatile components such as H2O, NH3 ,and CO2. After cooling to room temperature, the mixture was further ground for 2 h to obtain the required sample. The synthesized VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder has been characterized using various spectroscopic techniques.
Characterization Techniques
The x-ray diffraction (XRD) patterns of the prepared sample were obtained using a Rigaku-Smartlab D/MAX-2500 with graphite-monochromatic CuKα (λ = 0.15406 nm) radiation in the angular range of 2θ = 10°–70°, employing a step size of 0.01°, and with a speed of 10° per min. A JEOL JSM-IT 500 scanning electron microscope (SEM) with energy-dispersive x-ray spectroscopy (EDS) was used to observe the surface morphology and chemical composition. Images from selected area electron diffraction (SAED) and high-resolution transmission electron microscopy (HR-TEM) were obtained at 200 KV using a JEOL JEM-2100. The attenuated total reflectance approach was used to measure the Fourier-transform infrared (FT-IR) spectrum in the 4000–400 cm−1 spectral band at room temperature using a JASCO FT/IR-4700 instrument. Furthermore, a RENISHAW micro-RAMAN spectrometer was used to produce Raman spectra between 0 cm−1 and 3500 cm−1 . Optical absorption spectra were captured at room temperature within 200–1400 nm wavelength range using a JASCO V-670 UV-Vis-NIR spectrophotometer. A JEOL JES-FA 100 spectrometer operating at X-band frequencies with a 100-KHz field modulation was used to obtain the EPR spectrum at room temperature. Two 450-W Xenon lamps were used as excitation sources and a HORIBA Jobin-Yvon Fluorolog-3 spectrofluorometer was used to record the photoluminescence (PL) spectra at room temperature.
Results and Discussion
XRD Analysis
The diffraction peaks are ascribed to a mixed phase including the monoclinic structure of Li2BaP2O7 (Pearson’s Crystal Database No.1003786) and the hexagonal structure of Ba2P2O7 (JCPDS: 01-083-0990). In the XRD pattern of the sample, peaks observed at 2θ = 14.49, 19.28, 22.71, 25.17, 25.85, 28.42, 36.61, 39.79, 41.89, 42.91, 46.41, 53.06, 59.95, and 66.02 are assigned to the diffraction planes (0 2 1), (1 2 0), (1 2 1), (2 0 0), (0 0 4), (0 1 5), (1 3 4), (1 0 3), (1 5 3), (2 3 4), (1 4 5), (3 4 3), (2 1 8), and (0 3 9) of monoclinic wurtzite Li2BaP2O7 (*), respectively. The peaks observed at 2θ = 16.55, 22.71, 25.17, 29.61, 31.67, 33.12, 38.73, 41.89, 48.56, 50.31, 56.23, 58.17, and 62.81 correspond to the planes (1 0 1), (1 1 1), (0 0 2), (1 1 2), (3 0 0), (2 0 2), (2 1 2), (3 0 2), (3 2 0), (3 2 1), (3 1 3), (3 3 0), and (3 0 4) of a hexagonal phase of Ba2P2O7 (#), respectively. For the undoped sample, XRD peaks observed at 2θ = 14.45, 19.20, 22.69, 25.12, 25.69, 28.39, 36.51, 39. 68, 41.85, 42.88, 46.36, 53.02, 59.90, and 65.95 correspond to the monoclinic wurtzite phase of Li2BaP2O7, while peaks observed at 2θ = 16.53, 22.69, 25.12, 29.54, 31.62, 32.89, 38.68, 41.79, 48.50, 50.27, 56.19, 58. 13, and 62.74 correspond to the hexagonal phase of Ba2P2O7. The right shift in the XRD peaks with VO2+ ion-doped Li2Ba3(P2O7)2 pyrophosphate is due to the effect of the difference in the ionic radius between the cations which causes the crystallite lattice to expand and consistent with the Bragg’s equation.14 The shift in the XRD peak position of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder is depicted in Fig. 1. The resulting diffraction pattern contains information about the spacing of atomic planes within the crystal lattice as well as any deviations or distortions caused by doping-induced stresses. The unit cell parameters have been evaluated as a = 0.517 nm, b = 1.318 nm, c = 1.611 nm, and V = 1.098 nm3. These values agreed with undoped and iron-doped pyrophosphate nanopowders,13 and reveal an orthorhombic crystalline structure with space group 2P1. This information was found using the Powd software, with the effective ionic radii of cations showing the following different coordination numbers (CN) of Li+ ion (0.076 nm, CN = 6), Ba2+ ion (0.135 nm, CN = 6), and VO2+ ion (0.079 nm, CN = 6). VO2+ ions are predicted to fill any Li+ or Ba2+ ion sites that exist in the crystal lattice. The radius percentage difference (Dr) between the dopant and substituted ions was estimated by15:
where Rm and Rd are the radii of the host cations and doped ions, respectively. The calculated Dr values between VO2+ and Li+/Ba2+ are − 3.9% and 41.5% respectively. The tolerable percentage difference in ionic radii between the activated and substituted ions should not reach above 30%.16 Obviously, the substitution of VO2+ at the Li+ sites is preferable. By using Scherrer’s equation,17 the average crystallite size has been calculated:
where λ is the x-ray wavelength (0.15406 nm), θ is the maximum intensity diffraction angle, β is the full width at half-maximum of the corresponding lattice planes, and K is the shape factor with a value of 0.9. The average crystallite size was found to be 34.2 nm which is in the nano-size range.

X-ray diffraction pattern of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder.
Dislocation density and micro-strain are important characteristics that provide insight into how a material deforms from its original structure.13,18A critical parameter for determining the difference in atom displacement from reference positions is micro-strain, and this, and dislocation density, are also calculated to understand the structural behavior of the prepared sample. The micro-strain (ε) and dislocation density (δ) for the sample were obtained using19:
A method for determining the sample's crystallite size and micro-strain was proposed by Williamson and Hall (W–H). The W–H formula is written as:
Figure 2 shows a linear relationship between 4sinθ (x-axis) and βcosθ (y-axis). The slope of this line corresponds to the micro-strain (ε) while its intercept (kλ/D) represents the crystallite size (D).20 Comparison of the dislocation density and crystallite size are obtained from Debye–Scherrer’s equation with those calculated using the W–H plot listed in Table I. The results obtained through the W–H method are consistent with those from Debye–Scherrer’s equation with slight variations to the strain component considered in the W–H equation.

Williamson-Hall plot of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder.
SEM and EDS Analyses
SEM images of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder are illustrated in Fig. 3 captured at 2 µm and 500 nm. The images depict a stone-like configurational structure. Determining the true grain size using SEM images is challenging due to the irregular shape of the grains, a high degree of agglomeration, and wide size dispersion. The aggregation could be caused by mutual interaction between the particles, van der Walls forces, and electrostatic forces.6 The average grain size of the prepared sample was determined by analyzing the SEM images at a magnification of 500 nm using image-J software, and the data were utilized to create a histogram, as shown in Fig. 4 using Origin software. The calculated average grain size is 60.8 nm. The SEM grain size is somewhat larger than the XRD crystallite size values. This observed difference is likely due to the surface structure of converging grains which leads to enhanced grain growth as well as increased porosity and surface roughness.21 The elemental chemical composition of the sample was determined using EDS analysis and corresponding recorded spectra, as shown in Fig. 5. The table of determined compositions of elements Li, O, P, Ba, and V (inset in Fig. 5) closely matches the results of experimental stoichiometric calculations. Commercial EDS detectors cannot identify elements with atomic numbers less than 5 due to instrument constraints.22 This is the reason why lithium was not found in the EDS spectrum of the current investigation.

SEM images (a) 2 µm and (b) 500 nm of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder.

Histogram of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder using SEM images of magnification 500 nm.

EDS spectrum of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder.
HR-TEM and SAED Analysis
Further analysis of the prepared sample was conducted using HR-TEM to examine the microstructures. Figure 6 shows HR-TEM images captured at different magnifications of 50 nm and 100 nm, which confirmed the presence of stone-like particles. The average particle size value of 36.3 nm was determined by utilizing Image-J software on the HR-TEM image of 100 nm and fitting the size distribution with a Gaussian model of the histogram, as shown in Fig. 7. The crystallite size was determined by Scherrer’s equation, and the W–H plot method is a good match with HR-TEM studies. Moreover, the SAED image displayed in Fig. 8a reveals the heterogeneous nature of the sample, aligning with the cross-sectional planes corresponding to planes (1 8 6), (1 5 3), (1 1 3), and (0 0 3) which are consistent with the XRD pattern. In Fig. 8b, the measured lattice d-spacings of 0.31 and 0.34 nm correspond to the (2 1 0) and (0 2 4) planes, respectively.

HR-TEM images (a) 50 nm and (b) 100 nm of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder.

Histogram of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder using HR-TEM images of magnification 100 nm.

(a) SAED pattern and (b) d-spacing of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder.
FT-IR and Raman Analysis
Figure 9a shows the room-temperature FT-IR spectrum of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder with multiple broad peaks within the range of 1500–500 cm−1. The observed bands originate from the deformation and rocking modes of PO3 as well as symmetric and asymmetric stretching vibrations of the PO3 mode and P–O–P groups.23 In the FT-IR spectrum, only a single O–H bending vibration band at 1404 cm−1 can be seen. We can distinguish the following bands within the range 1260–1000 cm−1 which are attributed to the stretching vibrations υas and υs of the PO3 group.24 The spectral range between 945 cm−1 and 720 cm−1 contains bands arising from both asymmetric and symmetric stretching vibrations of υ(P–O–P).25 The bands observed within the range of 690–630 cm−1 are associated with asymmetric deformation mode δas (PO3), while symmetric deformation δs (PO3) is documented at 547 cm−1. When compared to the pure sample, the bands at 691 and 640 cm−1 as υas of PO3 and 987 cm−1 υs of PO3 bands are not observed due to more distortion in the VO2+-doped sample.13

(a) FTIR and (b) Raman vibrational spectra of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder.
The Raman spectrum assignment in the 200–1300 cm−1 region agrees quite well with the FT-IR spectrum assignment that was previously made, as shown in Fig. 9b. According to which, the strong Raman bands in the range of 1000–1280 cm−1 can be attributed to vibrations of PO3 in both asymmetric and symmetric modes. Simultaneously, the bands in the range of 950–850 cm−1 and 800–700 cm−1 are specifically assigned to υas(P–O–P) and υs(P–O–P) vibrations, respectively.26 The bands detected within the 500–600 cm−1 range are attributed to deformations of the PO3 groups. The low-frequency band below 300 cm−1 is identified as lattice vibrations, which are mostly caused by external modes.27 A slight shift was observed when compared to the pure sample.13 The band assignments provided in Table II for the FT-IR and Raman fundamental modes of VO2+-doped sample validate the presence of diphosphate groups in the title compounds.
Optical Analysis
The UV absorption spectrum is used to investigate the production of optically mediated transitions in crystalline materials. The absorption edge in the spectrum is analyzed to determine the optical absorption coefficient, band structure type, and energy band gap. Vanadium is paired with five or six oxygen atoms in a solid or aqueous medium. One of the oxygen atoms forms a double covalent bond with vanadium to generate the vanadyl ion.28 Referring to the energy level scheme proposed by Ballhausen and Gray, the V4+ ion exhibits a d1 configuration and possesses a ground state of 2D.6 The 2D state splits into 2T2g and 2Eg in the presence of a pure octahedral crystal field. However, in the case of VO2+ , it never exhibits pure octahedral symmetry (C4v). Tetragonal symmetry results in a singlet ground state term 2T2g and splits into 2B2g and 2Eg. Two states 2B1g and 2A1g separate 2Eg and the ground state will be 2B2g among these. The UV-Vis spectrum of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder was recorded at room temperature, as shown in Fig. 10. Three bands are present in the absorption spectrum of the VO2+-doped sample at 831 (12,030 cm−1), 635 (15,744 cm−1), and 416 (24,032 cm−1), respectively. The observed bands are associated with characteristic d-d transitions29 and have been assigned to specific states as:

Optical absorbance spectrum of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder.
The following relationships are used to evaluate the crystal field parameter, Dq, and the tetragonal field parameters, Ds and Dt:
By using the above equations, the parameters have been evaluated as: Dq = 1574 cm−1, Ds = − 2903 cm−1 ,and Dt = 664 cm−1. The opposing signs found in the tetragonal field constants Ds and Dt suggest octahedral symmetry with tetragonal compression along the axis. The band head data and the corresponding transitions and calculated values are shown in Table III. The direct energy band gap (Eg) of the sample has been determined using:
where λc is the cut-off wavelength and the evaluated band gap energy is 3.75 eV at the wavelength of 331 nm. Furthermore, the band gap energy can be determined by using Tauc’s formula which demonstrates the relationship between absorption coefficients12:
where α, h, and υ represent as absorption coefficient, Plank’s constant, and the frequency of vibration, respectively. Exponent n varies depending on the types of transitions with the values ½, 2, 3/2, and 3 equivalent to authorize direct, allowed indirect, forbidden direct, and forbidden indirect respectively. A is a constant that depends on the transition probability and is known as the band tailing parameter. The band gap energy of the VO2+-doped sample has been determined graphically by plotting (αhυ)2 versus hυ, as shown in Fig. 11. The amount of band gap energy was found to be 3.78 eV. Both the experimental and calculated band gap energies are nearly equal. This type of pyrophosphate nanopowders may be used in optoelectronic devices.

Energy band gap of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder.
Additional important optical characteristics that are connected to the VO2+-doped sample are the refractive index (R.I) and metallization criteria (M). The following expression was used to determine the refractive index (n) value for prepared nanopowder based on the optical energy band gap:
The calculated refractive index (n) is 4.36 by using this equation. The metallization criteria (M) quality is measured using:
It has been stated that a substance was non-metallic if the M value was less than 1 and metallic if the M value was larger than 1.30 The calculated M value is 0.909, according to which, the prepared pyrophosphate nanopowder was non-metallic in nature; these values are in agreement with reported values.13
EPR Analysis
Figure 12 shows the EPR spectrum of the VO2+-doped sample at room temperature which consists of parallel and perpendicular components. The VO2+ ions belong to the paramagnetic family and have electron spin (S = ½), nuclear spin (I = 7/2), C4v symmetry, and just one stable natural isotope 51 V which gives them a 3d1 unpaired electronic configuration.29 Usually, VO2+-doped compounds have tetragonal distortion along the axis and octahedral symmetry. \(g_{\parallel } < g_{ \bot } < g_{e}\) is a necessary condition for tetragonally distorted octahedral site symmetry. The computed spin Hamiltonian parameters have been determined to be \(g_{\parallel }\) = 1.954, \(g_{ \bot }\) = 1.977, and they satisfy the specified criteria. Furthermore, the hyperfine coupling constant values \(A_{\parallel }\) and \(A_{ \bot }\) were found to be 172 × 10−4 cm−1 and 53 × 10−4 cm−1 , respectively, which satisfies the requirement that \(A_{\parallel } > A_{ \bot }\). Through observation, \(\frac{{\Delta_{\parallel } }}{{{\Delta }_{ \bot } }} = 1.3\) , and this value indicates tetragonal distortion of the VO2+ ions with the host lattice.

EPR spectrum of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder.
The following equations are proposed to compute the Fermi contact term, κ, the hyperfine coupling constant, P, and the molecular orbital coefficients such as \(\beta_{1}^{2}\), \(\beta_{2}^{2}\) , and γ2 based on optical absorption and EPR data.8:
where λ is the spin–orbit coupling constant (170 cm−1) and \(g_{e}\) is the free electron (2.0023). The energy values for 2B2g → 2Eg and 2B2g → 2B1g transitions are associated with \({\Delta }_{ \bot }\) and \({\Delta }_{\parallel }\). \(\beta_{1}^{2}\) denotes the degree of in-plane σ bonding, \(\beta_{2}^{2}\) denotes the degree of in-plane π bonding, and γ2 measures out-of-plane π bonding. The hyperfine coupling constant, P, is found in the radial distribution of singular electrons linked to the wave function, which is expressed as \(P \, = \, g_{e} g_{N} \beta_{e} \beta_{N} \left\langle {r^{ - 3} } \right\rangle\) where words in the expression have their usual meaning. The degree of electron orbital deformation at the vanadium nucleus is measured by the Fermi constant term, κ. Calculating the P value from the equation requires ignoring the second-order terms and taking into account negative values for \(A_{\parallel }\) and \(A_{ \bot }\)7:
The free ion P value for VO2+ is 160 × 10−4 cm−1. In this case, the computed P value is lower than the vanadyl free-ion value of 138 × 10−4 cm−1. This indicates the existence of a covalent nature in the vanadyl complexes such that the smaller the P value, the greater the covalency. Additional isotropic values for g and A can be derived from:
The evaluated values of giso and Aiso are 1.969 and 92.67 \(\times\) 10−4 cm−1 , respectively.
The Fermi constant term, κ, is determined to be 0.64. The obtained \(g_{\parallel }\), \(g_{ \bot }\), \(A_{\parallel }\), \(A_{ \bot }\), \(\beta_{1}^{2}\), \(\beta_{2}^{2}\) , and γ2 values are represented in Table IV. The bonding nature is measured by \(\beta_{1}^{2}\), which has a value of 1 for complete ionic and 0.5 for complete covalence. The presence of covalent bonding in in-plane σ and π bonds is suggested by the \(\beta_{1}^{2}\) and \(\beta_{2}^{2}\) values which range from 0.5 to 1.0. Vanadyl oxygen bonds and their ligands are covalent due to the out-of-plane π-bond, which is shown by the γ2 value. The near unity \(\beta_{2}^{2}\) value indicates both weak π and strong ionic bonding between the vanadyl ions and pyrophosphate ligands. The weak contribution of vanadium’s 4 s orbital to vanadyl bond in the produced pyrophosphate is shown by the deviating κ value.
The covalency rates are described by 1 − \(\beta_{1}^{2}\) and 1 − γ2, which provide information about in-plane σ bonding and out-of-plane π bonding of vanadyl–oxygen and vanadium ions (V4+), respectively. For this instance, (1 − \(\beta_{1}^{2}\)) = 0.44 and (1 − γ2) = 0.75 demonstrate that in-plane σ-bonding is of a strong covalent nature, while out-plane π bonding is of a moderate covalent nature.
PL Analysis
The presence of PL emission bands could be caused by a variety of defects such as vacancies, dislocations, strain, atomic bond distortions in the reaction medium, the environment, and crystallites.7 The emission and excitation spectra of a prepared sample is shown in Fig. 13. Three emission peaks are seen in the UV and visible regions of the emission spectrum under the excitation wavelength of 321 nm. The minor peak, characterized by its lower intensity, is situated at 354 nm (3.50 eV) within the UV region, whereas the blue and green visible regions exhibit two broad peaks centered at 440 nm (2.82 eV) and 530 nm (2.34 eV), respectively. The main cause of visible luminescence at 440 and 530 nm is intrinsic defects like oxygen vacancies, surface traps, and interstitials. A prominent UV emission in this investigation at 354 nm suggests that the sample has improved optical qualities and better crystal quality. The luminescence emission peaks at 440 and 530 nm were assigned to the following allowed transitions 2A1g → 2B2g and 2B1g → 2B2g , respectively.13,29

Excitation and emission spectra of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder.
To understand the luminescent properties of the prepared sample, CIE 1931 chromaticity coordinates have been calculated based on the emission spectrum. The color coordinates of the sample are situated within the yellowish-green region with respective CIE coordinates of x = 0.302 and y = 0.409. This position is displayed on the CIE chromaticity diagram by using OSRAM-IP software, as shown in Fig. 14. The correlated color temperature (CCT) is calculated using the McCamy equation31 and the CIE 1931 chromaticity coordinates (x, y):
here \(n = \frac{{\left( {x - x_{I} } \right)}}{{\left( {y - y_{I} } \right)}}\) represents inverse slope of the line with xI = 0.3320 and yI = 0.1858. The corresponding color purity can be determined using32 :

CIE 1931color chromatic image of VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder.
Among these, (x, y) represents the CIE chromaticity coordinates of the sample, (xI, yI) represents the CIE chromaticity coordinates of illuminant wavelength, while (xd, yd) corresponds to the color coordinates of the dominant wavelength and calculated color purity of 16%. The color rendering index (CRI) is an essential photometric for interpreting emission spectrum data. According to the CRI value, VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder exhibits acceptable emission properties. In the present study, the CCT, color purity, and CRI values are determined to be 6490 K, 16% ,and 41%, respectively. These results indicate that the prepared sample emits a yellowish-green color making it potentially useful in display panels and other LED display devices.
Conclusions
VO2+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowder was synthesized using a solid-state reaction technique. The XRD pattern of the produced sample revealed an orthorhombic structure with space group 2P1. The average crystallite size in the nano-range was determined and lattice cell parameters were evaluated for the prepared sample. SEM micrographs revealed irregularly agglomerated stone-like structures, and the elements Li, O, P, Ba, and V were verified to be present in the sample by the EDS spectrum. The HR-TEM images of the sample displayed stone-like agglomerated formations and the average particle size corresponded well with results obtained by the Scherrer and W–H techniques. The fundamental modes of the phosphate ions were confirmed by FT-IR and Raman analyses. The crystal and tetragonal field parameters were determined as Dq = 1574 cm−1, Ds = − 2903 cm−1 , and Dt = 664 cm−1. Optical absorption and EPR studies revealed that the VO2+ ions were partially covalent and exhibited tetragonal compression in octahedral site symmetry concerning nearby ligands in the host lattice. The refractive index (n), determined as 4.36 with the help of an optical energy band gap of 3.78 eV, suggests that the material is useful for optical detectors and photo-electrical cells. The CIE chromaticity diagram confirms a yellowish-green emission at 321 nm excitation wavelength for the synthesized sample. CCT, CRI, and color purity values were 6490 K, 41%, and 16%, and, based on these results the sample would be useful for light-emitting diodes.
Data Availability
Data will be provided upon request.
References
V.I. Maksin, R.V. Lavrik, T.I. Ushchapivska, and O.V. Petrenko, Growing of single-crystals of double sodium-manganese (II) pyrophosphate. Chem. Technol. 1, 76 (2022). https://doi.org/10.32434/0321-4095-2022-140-1-76-82.
I.V. Zatovsky, N.S. Slobodyanik, T.I. Ushchapivska, and W. Han, Formation of complex phosphates K2MIIISn(PO4)3 from solutions in melts under crystallization conditions. Funct. Mater. 24(2), 298 (2017). https://doi.org/10.15407/fm24.02.298.
S. Krivileva and V. Moiseev, Functional materials for medical and biological purposes on the system CaO–CaF2–P2O5–H2O and additives. Funct. Mater. 25(2), 358 (2018). https://doi.org/10.15407/fm25.02.358.
W. Tang, Z. Zhang, Y. Ma, and D. Qin, Color tuning in (Ca1− xSrx)8MgLu (PO4)7: Eu2+, Mn2+ phosphor via host composition design and energy transfer. Ceram. Int. 43(12), 9117 (2017). https://doi.org/10.1016/j.ceramint.2017.04.059.
F. Meng, X. Zhang, Y. Xu, J. Yang, and Z. Cheng, Photoluminescence of Eu2+, Ce3+ and Tb3+ in a new potassium barium phosphate K2Ba3(P2O7)2 host lattice. J. Alloys Compd. 729, 724 (2017). https://doi.org/10.1016/j.jallcom.2017.09.224.
T. Rajyalakshmi, Sk. Johny Basha, V. Khidhirbrahmendra, U.S. Udayachandran Thampy, and R.V.S.S.N. Ravikumar, Synthesis and investigations for white LED material: VO2+-doped calcium Cadmium phosphate hydrate nanophosphor. J. Mol. Struct. 1205, 127605 (2020). https://doi.org/10.1016/j.mostruc.2019.127605.
V. Khidhirbrahmendra, Sk. Johny Basha, M. Avinash, and R.V.S.S.N. Ravikumar, Investigations of VO2+-doped SrZn2(PO4)2 nanophosphors by solution combustion synthesis. J. Alloys Compd. 787, 276 (2019). https://doi.org/10.1016/j.jallcom.2019.02.073.
P. Krishna-Kishore-Kumar, Y. Ramesh-Babu, G.V. Zyryanov, and R.V.S.S.N. Ravikumar, Synthesis and spectral characterizations of VO2+ ions-doped CaZn2(PO4)2 nanophosphor. SN Appl. Sci. 871, 1 (2019). https://doi.org/10.1007/s42452-019-0903-8.
R. Koutavarapu, N. Sita-Maha-Lakshmi, M. Avinash, M.C. Rao, R.V.S.S.N. Ravikumar, and J. Shim, Novel yellow light emission from vanadyl ions-doped calcium-lithium hydroxyapatite nanopowders: structural, optical, and photoluminescence properties. Chem. Pap. 75, 3989 (2021).
K. Kumar, A. Datta, J. Rohilla, S. Thakur, R. Singh, and V. Kaur, Engineered organotin (IV) and vanadium (V) derivatives with distinct coordination modes and luminescent properties for the efficient detection and quantification of permanganate ions. Spectrochim. Acta A Mol. Biomol. Spectrosc. 294, 122521 (2023).
Y.V.K. Suresh, N.Ch. Anjaneyulu, A. Rajendrakumar, A.V. Chandrasekhar, and R.V.S.S.N. Ravikumar, Structural, optical, and luminescent studies of vanadyl-doped strontium tin phosphate by solid-state reaction method. Phys. Chem. Res. 12(3), 771 (2024). https://doi.org/10.22036/pcr.2024.396888.2338.
M. Nourmohammadian and F.C. Kalangestani, Wettability behavior, photoluminescence and optical properties studies of nanostructured vanadium-doped magnesium oxide thin films: an effect of V concentrations. Opt. Quantum Electron. 55(1), 20 (2023). https://doi.org/10.1007/s11082-022-04148-y.
N.Ch. Anjaneyulu, Ch. Aswini, K. Josephus-Alex Libnah, and R.V.S.S.N. Ravikumar, Crystal structures and photoluminescence properties of undoped and Fe3+-doped Li2Ba3(P2O7)2 pyrophosphate nanopowders. Nano-Structures & Nano-Objects. 38, 101179 (2024). https://doi.org/10.1016/j.nanoso.2024.101179.
J.S. Revathy, N.S. Chitra-Priya, K. Sandhya, and D.N. Rajendran, Structural and optical studies of cerium-doped gadolinium oxide phosphor. Bull. Mater. Sci. 44, 1 (2021). https://doi.org/10.1007/s12034-020-02299-w.
Y. Song, N. Guo, J. Li, R. Ouyang, Y. Miao, and B. Shao, Photoluminescence and temperature sensing of lanthanide Eu3+ and transition metal Mn4+ dual-doped antimoniate phosphor through site beneficial occupation. Ceram. Int. 46, 22164 (2020). https://doi.org/10.1016/j.ceramint.2020.05.293.
P. Phogat, S.P. Khatkar, R.K. Mail, J. Sushma, P. Dalal, and V.B.T. Hooda, Crystal chemistry and photoluminescent investigation of novel white light emanating Dy3+-doped Ca9Bi(VO4)7 nanophosphor for ultraviolet based white LEDs. Mater. Chem. Phys. 270, 124828 (2021). https://doi.org/10.1016/j.matchemphys.2021.124828.
N.M. Abd-Alghafour, G.A. Naeem, A.S. Ibraheam, N. Afzal, S.M. Mohammad, and R.F. Muslim, Fabrication and characterization of ethanol gas sensor based on hydrothermally grown V2O5 nanorods. Optik 222, 165441 (2020). https://doi.org/10.1016/j.ijleo.2020.165441.
P. Kaur, A. Kaur, S. Singh, S. Thakur, and L. Singh, Comprehensive analysis of crystal structure, optical and luminescent behavior of Fe-doped MgO nanophosphors. Optik 219, 164742 (2020). https://doi.org/10.1016/j.ijleo.2020.164742.
A. Rajendrakumar, NCh. Anjaneyulu, G. Vasu, R.V.S.S.N. Ravikumar, and N. Arundhathi, Divalent copper ions-doped strontium magnesium phosphate nanopowder: synthesis and characterization. J. Mater. Sci. Mater. Electron. 34, 1958 (2023). https://doi.org/10.1007/s10854-023-11350-5.
S. Kinra, M.P. Ghosh, S. Mohanty, R.K. Choubey, and S. Mukherjee, Correlating the microstructural and optical properties of vanadium ion-doped ZnO nanocrystals. Bull. Mater. Sci. 45(2), 65 (2022). https://doi.org/10.1007/s12034-021-02650-9.
P. Norouzzadeh, Kh. Mabhouti, M.M. Golzan, and R. Naderali, Comparative study on dielectric and structural properties of undoped, Mn-doped and Ni-doped ZnO nanoparticles by impedance spectroscopy analysis. J. Mater. Sci. Mater. Electron. 31, 7335 (2020). https://doi.org/10.1007/s10854-019-02517-0.
M. Rajesh Yadav, P. Radha Krishna, G. Venkata-Lakshmikanth, and R.V.S.S.N. Ravikumar, Influence of iron-doped on structural and optical absorption studies of calcium borophosphate (CaBP) nanophosphors. Macromol. Symp. 393, 1900214 (2020). https://doi.org/10.1002/masy.201900214.
M. Fhoula, T. Koubaa, and M. Dammak, White photoluminescence and energy transfer properties of dysprosium and europium singly and codoped Na2ZnP2O7 phosphors. Opt. Laser Technol. 130, 106352 (2020). https://doi.org/10.1016/j.optlastec.2020.106352.
I.V. Zatovsky, N.Y. Strutynska, Y.A. Hizhnyi, V.N. Baumer, I.V. Ogorodnyk, N.S. Slobodyanik, I.V. Odynets, and N.I. Klyui, New complex phosphates Cs3MIIBi(P2O7)2 (MII – Ca, Sr and Pb): synthesis, characterization, crystal and electronic structure. Dalton Trans. 47, 2274 (2018). https://doi.org/10.1039/C7DT04505K.
M. Beltaif, M. Krichen, M. Megdiche, and M. Dammak, A comparative and correlation structure-Ac conductivity analysis of doped 1% Eu3+ and undoped ceramic of Li2BaP2O7. Ionics 25, 3247 (2019). https://doi.org/10.1007/s11581-018-2826-6.
H. Lamsaf, R. Fausto, B.F.O. Costa, J. Toyir, E.H. Elghadraoui, M. Ijjaali, and A. Oulmekki, Synthesis and physicochemical characterization of a new mixed-valence IronIII-ZincII diphosphate: Zn2+ Fe3+2(P2O7)2. Mater. Chem. Phys. 216, 22 (2018). https://doi.org/10.1016/j.matchemphys.2018.05.060.
S. Ei-Arni, M. Hadouchi, A. Assani, M. Saadi, M. Ei Marssi, A. Lahmar, and L. Ei Ammari, Structural and Magnetic Investigations of the Novel Pyrophosphate Na7Ni3Fe(P2O7)4. Magneto. CHEM. 9(7), 162 (2023). https://doi.org/10.3390/magnetochemistry9070162.
I. Cirik, Y. Celik, and B. Karabulut, Theoretical and experimental EPR study of VO2+-doped Ammonium hydrogen tartrate. Z. Naturforsch. 70(8), 637 (2015). https://doi.org/10.1515/zna-2015-0141.
G. Dharmana, M.P. Srinivasa Rao, and D.M. Potukuchi, Visible light driven robust photocatalytic activity in vanadium-doped ZnO/SnS core-shell nanocomposites for decolorization of MB dye towards wastewater treatment. Inorg. Nano-Met. Chem. 52(8), 1059 (2022). https://doi.org/10.1080/24701556.2022.2075386.
K.C. Sushma, S. Kumar, G. Nagaraju, D.P. Aarti, M.B. Madhusudana Reddy, M.S. Rudresha, and R.B. Basavaraj, Color tunable SrZrO3:Sm3+ nanopowders with satisfactory photoluminescent, band engineering properties for warm white LEDs and advanced forensic applications. J. Mol. Struct. 1254, 132302 (2022). https://doi.org/10.1016/j.molstruc.2021.132302.
Y. Liu, G. Zhang, J. Huang, X. Tao, G. Li, and G. Cai, Daylight-white-emitting and abnormal thermal antiquenching phosphors based on a layered host SrIn2(P2O7)2. Inorg. Chem. 60(4), 2279 (2021). https://doi.org/10.1021/acs.inorgchem.0c03121.
X. Chen, Q. Xu, F. Hussain, C. Yang, W. Sheng, X. Luo, B. Liu, S. Sun, D. Wang, and K. Song, High thermal stability and color purity of Y2SrAl4SiO12:Eu3+ garnet-variant-structured phosphor for warm white Light LED-Lamp. Crystals 12(10), 1382 (2022). https://doi.org/10.3390/cryst12101382.
Acknowledgments
One of the authors, N.Ch. Anjaneyulu is thankful to CSIR-UGC-NET for providing financial support through the NET-JRF scheme (NTA Ref. No. 201610120900 dated 01.04.2021) to carry out the present research work.
Funding
There are no funding sources for this work.
Author information
Authors and Affiliations
Contributions
NCA: Conceptualization, methodology, software, investigation, writing-original draft. GV: Analysis, analyzing data and editing. AVC: Visualization and formal analysis. RVSSNR: Supervised the research and critical revision of the article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Anjaneyulu, N.C., Vasu, G., Chandrasekhar, A.V. et al. Influence of VO2+ Ion-Doped Li2Ba3(P2O7)2 Pyrophosphate Nanopowder: Structural, Morphological, Optical, and Photoluminescence Properties. J. Electron. Mater. 53, 6151–6163 (2024). https://doi.org/10.1007/s11664-024-11263-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11664-024-11263-x