
Abstract
Chromium, broadly present in metallurgical systems such as stainless steel slags and vanadium-bearing slags, tends to be in a trivalent form bound in spinel phases due to their ultrahigh stability. To explore the stability of chromium bearing spinels with different divalent cations, herein ACr2O4 (A=Mg, Zn, Mn) spinels were synthesized and stability-tested under both alkaline and acidic environments. X-ray diffraction (XRD) and refinement results reveal that MnCr2O4 exhibited the largest lattice parameters. X-ray photoelectron spectroscopy (XPS) results show that MnCr2O4 had the highest oxygen vacancy concentration, potentially triggering structural instability, consistent with the Fourier-transform infrared (FTIR) and Raman analyses. To further investigate the stability of ACr2O4 spinels in alkaline and acidic environments, we introduced CaO and SiO2, respectively, for high temperature calcinations. XRD analyses reveal that ACr2O4 spinels exhibited a high instability under an alkaline condition, with an increasing degree of instability for MgCr2O4, ZnCr2O4, and MnCr2O4 successively. We further quantified the relationship between the oxygen vacancy concentrations and the divalent cations containing in spinels as well as the spinel stabilities under an alkaline environment. Under an acidic condition, all ACr2O4 spinels demonstrated a relatively strong structural stability. The understanding of formation and stability of chromium bearing spinels under different conditions could contribute to potential modifications and applications of spinels especially with regard to resource recycling in metallurgy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chromium is an important alloying element in metallurgy and currently, approximately 96 pct of chromium is utilized in the production of stainless steel.[1] A consequential by-product arising from this manufacturing process, known as stainless steel slags, constitutes approximately 25–33 pct of the total stainless steel production.[2,3] Due to limitations in treatment technology, a substantial quantity of stainless steel slags continues to be deposited in slag fields. It is crucial to highlight that hexavalent Cr6+, distinguished by pronounced toxicity and migration capability,[4,5] exhibits a tendency for dissemination into groundwater and soil,[6] consequently impeding its potential for further extensive utilization.[4] Conversely, another predominant form of chromium exists as Cr3+, where trivalent chromium is relatively stable and possesses lower toxicity, serving as an essential trace element in biological systems.[7]
Transforming the heavy metals including chromium into structure stabilized phases has been an effective strategy for their decontaminations, finding applications across various resource recycling systems.[8,9,10,11] Previous studies have affirmed spinel as an ideal phase for chromium enrichment and decontamination, where chromium exists predominantly in stable trivalent states, offering notable advantages such as high acid-base resistance and leaching resistance.[9,12] By adjusting the content of specific divalent metal oxides like MgO and MnO in stainless steel slags, the precipitation of stable chromium-containing spinels like (Mg,Mn)(Al,Cr)2O4 can be induced,[13,14,15,16] thereby inhibiting the generation of CaCrO4 and other chromium-containing phases (Cr6+), mitigating the toxicity of stainless steel slags. This method is not only applicable for the immobilization and purification of pollutants but also plays a crucial role in improving environmental sustainability and resource recycling in chromium metallurgy.[12,16] With regard to the complex spinel evolutions in stainless steel slags, MgCr2O4 and MnCr2O4 account for two basic spinels to guide the target spinel transformations.[12,14] However, their reactions with other key compositions in stainless steel slags such as CaO and SiO2 have not been investigated, which motivated our current fundamental study. Those reactions are also closely related to their further evolutions and stabilities under various environmental conditions.
In addition to stainless steel slags, chromium bearing spinels work in many other metallurgical systems such as vanadium bearing slags, steel dusts and refractory materials, etc. For the vanadium extraction from vanadium titano-magnetite, chromium bearing spinel in vanadium bearing slags is an important medium phase (especially for high-chromium vanadium titano-magnetite), formed by converting of liquid iron alloy.[17,18] This spinel phase should be further roasted with agents such as Na2CO3 and CaO to break up the spinel structure and to enhance the consequent leaching process for vanadium separation.[19,20] Treatment of steel dusts is another important resource recycling system for the steel metallurgy. The elemental composition of dusts varies across different subprocess types,[19] leading to variations in treatment technology. Notably, electric arc furnace dust exhibits abundant zinc,[20] dominantly in the form of complex spinels. The breakup of dust spinels also accounts for a key clue for extraction of valuable zinc from steel dusts.[21,22] Simultaneously, all those approaches align with the requirements of a resource-efficient and environmentally friendly future metallurgy.
This study involved the design and synthesis of three distinct chromium bearing spinels with varying divalent cations, namely ACr2O4 (A=Mg, Zn, Mn), in accordance with the prevalent chromium bearing spinels in stainless steel slags system. The principal objective was to explore the impact of diverse bivalent cations in the single-phase spinel structure on the stability of chromium bearing spinels. Extensive characterizations were conducted using advanced techniques. To evaluate the variation in stability performance of those chromium bearing spinels under alkaline and acidic environments, high-temperature calcination experiments were conducted by introducing CaO and SiO2, respectively. Those characterizations provided a comprehensive understanding of the stability behavior of the chromium bearing spinels under different environmental conditions.
Materials and Methods
Synthesis of ACr2O4 (A=Mg, Zn, Mn) Spinels
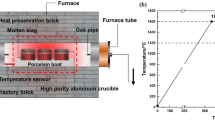
In this study, we synthesized and stability-tested three types of chromium bearing spinels with varying divalent cations, namely ACr2O4 with A=Mg, Zn, Mn. Magnesium(II) oxide (MgO, 99.9 pct, Macklin, Shanghai, China), zinc(II) oxalate dihydrate (ZnC2O4·2H2O, ≥99 pct, Macklin, Shanghai, China), manganese(II) oxide (MnO, 99 pct, Aladdin, Shanghai, China) and chromium(III) oxide (Cr2O3, ≥99 pct, Macklin, Shanghai, China) with an analytical grade were used to synthesize samples without any additional purification steps. First, appropriate masses of A-containing compound and Cr2O3 were carefully weighed, at a precise molar ratio of A : Cr=1 : 2, and mixed in an agate ball mill tank for 10 h with a rotational speed of 800 r/min. After the ball milling, finely ground materials with particle sizes below 125 μm were produced with a larger specific surface area. Subsequently, the ball milling mixture was introduced into a corundum crucible, which was then put into a tube furnace for calcinations in a high purity argon atmosphere. The samples were first heated to 1200 °C with a heating rate of 10 °C/min, and then held for a duration of 2 h to guarantee a thorough reaction. Following this, they were gradually cooled to room temperature in the furnace to obtain ACr2O4 spinel samples (Figure 1(a)).

A schematic diagram of the synthesis process and stability tests of ACr2O4 spinels: (a) synthesis of ACr2O4 spinels, and (b) stability tests of spinels
Then we used a series of advanced techniques to characterize the synthesized spinels, particularly the role of divalent cations. Those samples were first characterized using X-ray diffraction (XRD, 2Empyrean2, PANalytica, Netherlands) technique where a Cu-K-α radiation source at 40 kV and 40 mA was equipped. The samples were scanned from 2θ of 10 to 80 deg at a scanning rate of 4 deg/min. Phase identification was firstly performed using MDI Jade 6.0 software by comparing the experimental XRD patterns with those obtained from the standard database provided by the International Centre for Diffraction Data (ICDD PDF, Release 2004). The results were further refined using GSAS-II to quantify the lattice parameters.[23] The chemical states of those spinels were further characterized using X-ray photoelectron spectroscopy (XPS, ESCALAB250Xi, Thermo Fisher, U.S.) with Mg-K-α radiation (1253.6 eV) as the excitation source. To investigate the thermodynamic driving forces influencing spinel formation under synthesis conditions, we conducted thermodynamic calculations using FactSage software (FactSage 8.1, developed by Thermfact/CRCT, Canada and GTT-Technologies).[24,25] The FactPS and FToxide databases were utilized to evaluate the variation in Gibbs free energy (ΔG) during spinel formation reactions. Additionally, the Fourier transform infrared spectroscopy (FTIR, Nicolet iS50, Thermo, U.S.) was collected in the wavenumber range of 1600–400 cm−1 to probe the spinels’ structural characteristics; and similarly, the Raman spectroscopy (Raman, InVia, Renishaw, UK) measurement was conducted utilizing an excitation wavelength of 532 nm to understand the molecular vibration patterns of spinels. Furthermore, we used scanning electron microscopy (SEM, JSM-7900F, JEOL, Japan) with energy dispersive spectroscopy (EDS, JSM-7900F, JEOL, Japan) at an accelerated voltage of 15 kV and a current of 12 mA to identify the surface morphology and elemental distributions.
Stability Tests of Spinels
Next, the stability measurements of those ACr2O4 spinels were carried out under both alkaline and acidic environments aiming for varying practical industrial systems, where an air atmosphere was used considering the practical treatment and storage conditions. To clarify the spinel stabilities under an alkaline environment, the spinel sample was first mixed with 30 wt pct calcium(II) oxide (CaO, 98 pct, Macklin, Shanghai, China) and then calcined in a tube furnace in an air atmosphere. Two different holding temperatures, namely 600 and 1200 °C, were applied to clarify the spinel stabilities under both low and high temperatures. A heating rate of 10 °C/min was employed followed by 2 h heat retention (Figure 1(b)). Following the calcination process, the obtained samples were analyzed using XRD to determine the phase compositions and FTIR to probe the vibrational modes and interactions of chemical bonds. We also used SEM-EDS to measure the microstructure evolutions of spinels upon calcinations. The test conditions of XRD, FTIR and SEM-EDS for samples after calcinations were the same as those for the raw spinels in the aforementioned section. Regarding the stability tests under an acidic environment, we used 30 wt pct silicon(IV) oxide (SiO2, 99.7 pct, Aladdin, Shanghai, China) instead of CaO, with the remaining processes the same. However, due to the ultrahigh stabilities of spinels under the acidic environment, we used two relatively higher temperatures for sample calcinations, namely 1200 and 1400 °C.
Results and Discussion
Formation of Spinels and Role of Divalent Cations
The phases of the synthesized ACr2O4 (A=Mg, Zn, Mn) spinels were first characterized using XRD. By comparing the XRD results with the standard PDF cards, Figure 2(a) proved the effective synthesis of spinel phases without the formation of additional phases. As we enlarged some localized XRD peaks such as (311), (400) and (440) crystallographic planes (Figure 2(b)), we found that in comparison to MgCr2O4 and ZnCr2O4, the peak position of MnCr2O4 exhibits a noticeable shift towards lower 2θ values, suggesting enlarged lattice parameters and an expansion of crystal structure. Figure 2(c) illustrates the Rietveld refinement process of MgCr2O4. For additional Rietveld refinement details, see electronic supplementary Figure S1. MnCr2O4 shows larger lattice parameters as well as a larger unit cell volume (601.2 Å3) than MgCr2O4 and ZnCr2O4 (577.0 and 577.5 Å3) while the differences between MgCr2O4 and ZnCr2O4 were limited. The lattice structures of those ACr2O4 spinels were closely associated with the ionic radius of their divalent cations of Mg2+, Zn2+ and Mn2+,[26] as listed in Table I, and a larger ionic radius such as Mn2+ over Mg2+ and Zn2+ could result in larger lattice parameters of the spinels. This could determine their further stabilities under various environments, as discussed later.

Crystallographic phase and chemical state analyses of ACr2O4 spinels synthesized: (a) XRD patterns of ACr2O4 spinels, (b) crystallographic planes (311), (400) and (440) within the 2θ range of 34–37, 42–45 and 61–64 deg, respectively, (c) Rietveld refinement example of MgCr2O4, (d) comparison of lattice parameters and unit cell volumes for ACr2O4 spinels obtained through XRD refinements, (e) the XPS spectra of O1s peak for ACr2O4 spinels, (f) the fitting example of O1s XPS spectrum for MgCr2O4 within the binding energy range of 525–540 eV, and (g) concentration of oxygen vacancies in different ACr2O4 spinels
We then measured the chemical states of those ACr2O4 spinels using XPS. Figure 2(e) displays the O1s spectra, covering the binding energy range of 525–540 eV, which provides direct evidences of the presence of oxygen vacancies (OVs). The peak in the range of 528–532 eV centered at 530 eV is attributed to the lattice oxygen, while the peak of 530–534 eV centered at 532 eV originates from the chemisorption of OVs. Those OVs can give rise to defect sites or voids within the crystal structure, thereby influencing their stabilities. By employing semi-quantitative fitting (Figure 2(f) and electronic supplementary Figure S2), the concentrations of surficial OVs in ACr2O4 spinels have been elucidated, yielding notable distinctions. Surficial OVs concentrations for MgCr2O4, ZnCr2O4 and MnCr2O4 were 23.54, 24.90 and 37.58 pct, respectively (Figure 2(g)). Meanwhile, we calculated the Gibbs free energy change (ΔG) for the reaction of MgO or MnO with Cr2O3 to form spinel at 1200 °C. The ΔG for the reaction of MgO with Cr2O3 was −2.13×103 kJ, while for MnO with Cr2O3, it was −1.98×103 kJ. The negative values of ΔG indicate the feasibility of synthesizing ACr2O4 spinels, with a greater negative value of ΔG indicating a transition towards a more stable phase. This further elucidates the higher stability of MgCr2O4 from the perspective of thermodynamic driving forces over MnCr2O4 and ZnCr2O4. It is particularly noteworthy that MnCr2O4 exhibits the highest OVs concentration, suggesting a more pronounced scarcity of oxygen atoms in its crystal lattice and a high level of crystal instability. In contrast, ZnCr2O4 and MgCr2O4 exhibit lower OVs concentrations, indicating relatively lower degrees of oxygen deficiency and higher stabilities. Particularly, more OVs would decrease the average valences of cations including both A and Cr in the spinels, potentially accompanied with inherent instabilities of the structures. Those obtained findings provide valuable insights into the structures and stability differences observed among ACr2O4 spinels, in accordance with the XRD results.
Based on the aforementioned discussions, it was observed that the OVs concentration in spinels is closely related to the properties of divalent cations.[27,28,29] To quantify this effect, we try to employ an exponential function \(\left( {OVs = OVs\left( {X,Y} \right) = C_{1} X^{a} Y^{b} } \right)\) to depict the relationship between OVs concentration and two key intrinsic properties of those cations, namely electronegativity (X) and ionic radius (Y). Here a, b, and C1 are parameters to be determined through data fitting. Taking the natural logarithm of the aforementioned function and fitting the experimental data, we can obtain a quantitative function between OVs concentration and divalent cations: \(OVs = 8.03 \times 10^{ - 6} X^{ - 0.17} Y^{3.49}\). Accordingly, we can first note that electronegativity has a limited effect on the OVs concentration in a chromium-bearing spinel structure. In comparison, ionic radius exerts a significant influence on the OVs concentration in spinel, with a larger ionic radius corresponding to a higher OVs concentration, working as a dominant factor. Since the presence of OVs in spinels could further affect their stabilities, through this mathematic model, it is possible to predict the OVs concentration generated as well as their stabilities when different or/and multiples divalent cations are incorporated to form chromium bearing spinels.
Subsequently, to investigate the vibrational characteristics of the metal–Oxygen bonds in ACr2O4 spinels, FTIR and Raman characterizations were performed. Figure 3(a) exhibits the FTIR spectra of ACr2O4 spinels in the wavenumber range of 1200–400 cm−1. See electronic supplementary Table S1 for a summary the observed infrared characteristic peaks in ACr2O4 spinels, along with their corresponding wavenumbers. For MgCr2O4, the peak at 648 cm−1 corresponds to the vibrational mode of Cr(III)–O bonds, while the peak at approximately 513 cm−1 is associated with Mg(II)–O bonds.[30] In the FTIR spectra of ZnCr2O4, the prominent peak observed at 631 cm−1 is assigned to the stretching vibrations of Cr(III)–O bonds, signifying the displacement of oxygen anions in relation to trivalent chromium cations along the octahedral chain direction. The peak at 535 cm−1 is attributed to the vibrational mode of Zn(II)–O bonds.[31] In the case of MnCr2O4, the peak at 625 cm−1 corresponds to the Cr(III)–O vibrations, while the peak at 517 cm−1 is indicative of the stretching motion associated with Mn(II)–O bonds. Generally, shorter bond lengths result in higher infrared frequencies. This indicates an increase in the Cr(III)–O vibration had a shift towards lower wavenumbers in the sequence of MgCr2O4, ZnCr2O4 and MnCr2O4. This indicates an increase in the Cr(III)–O bond length and a corresponding decrease in bond strength in the sequence of MgCr2O4, ZnCr2O4 and MnCr2O4, consistent with the refined XRD results of lattice parameters and cell volumes.

The vibrational characteristics of metal–Oxygen bonds in ACr2O4 spinels: (a) FTIR spectra of ACr2O4 spinels in the wavenumber range of 2000–400 cm−1, and (b) Raman spectra of ACr2O4 spinels in the Raman shift range of 1001000 cm−1. There are five Raman active vibration modes in spinels (see electronic supplementary Table S2). The A1g mode corresponds to the symmetric stretching vibrations of Cr–O bonds in the CrO6 octahedron. The Eg mode represents the asymmetric stretching vibrations of Cr–O bonds in the CrO6 octahedron. The F2g mode at low Raman shift signifies the bending vibrations of A–O bonds in the AO4 tetrahedron, while the middle Raman shift F2g modes correspond to the stretching vibrations of Cr–O bonds.[28]
From the Raman spectra in Figure 3(b), five Raman active modes in ACr2O4 spinels could be observed, namely 1A1g, 1Eg, and 3F2g modes, consistent with previous studies.[33,34] For MgCr2O4, the prominent peak at 445 cm−1 corresponds to the Eg mode, associated with the motion of oxygen ions in the crystal lattice. This mode is characterized by an asymmetrical stretching of the oxygen ions relative to the surrounding metal ions. Additionally, two of the F2g modes assigned to Cr–O stretching vibrations are identified, with peaks at 540 and 611 cm−1. Another notable feature is the intense A1g band observed at 682cm−1, attributed to the symmetric Cr–O tensile vibrations from the CrO6 groups.[34,35] Regarding ZnCr2O4, the peak at 180 cm−1 corresponds to the Zn–O bending vibrations of the ZnO4 tetrahedron (F2g). The medium-intensity band at 509 cm−1 and the weak-intensity band at 602 cm−1 can both be attributed to the Cr–O stretching vibration (F2g). And a strong band at 680 cm−1 is associated with the symmetric Cr–O stretching vibrations (A1g).[32,33] As for MnCr2O4, the Raman spectra reveal distinct peaks at 192, 501 and 571 cm−1, corresponding to three of the F2g modes, and the strongest peak observed at 671 cm−1 represents its A1g mode. Shorter bond lengths typically correspond to higher Raman shift frequencies. The Raman shifts for F2g and A1g vibration modes of MgCr2O4, ZnCr2O4 and MnCr2O4 consistently shift towards lower Raman shift, indicating a sequential increase in the bond lengths of A–O and Cr–O, in agreement with the findings obtained from FTIR spectra and XRD refinement analyses.
At last, the morphologies of ACr2O4 spinels synthesized were measured by SEM and the elemental distributions by EDS, as shown in Figure 4. As noted from the EDS results, A, Cr and O elements were distributed uniformly and consistently in ACr2O4 spinels, indicating the formation of pure spinels without any segregations, consistent with the XRD results. Based on the SEM images, we could also observe that compared with MgCr2O4 and MnCr2O4, ZnCr2O4 spinel demonstrates a larger grain size, indicating a higher reactivity between ZnC2O4·2H2O and Cr2O3 over those oxides of MnO and MgO used for MgCr2O4 and MnCr2O4, and an accelerated growth rate during the calcination process. Additionally, the grain sizes of ACr2O4 spinels could further affect their structure stabilities, including their reactivity with CaO and SiO2, as discussed later.

Microscopic features of ACr2O4 spinels by SEM and elemental distributions by EDS: (a) MgCr2O4, (b) ZnCr2O4, and (c) MnCr2O4
Stability of Spinels Under an Alkaline Environment
Since the spinels could be present in various environments regarding industrial systems, their stabilities could be quite unique. In this study, we clarify the stabilities of ACr2O4 spinels under two typical environments, namely an alkaline environment established by a CaO addition and an acidic by SiO2. We first tested the stabilities under an alkaline environment. Figure 5(a) illustrates the XRD results after alkaline treatment at a low temperature of 600 °C. It first reveals the presence of Ca in two distinct phases, namely CaCrO4 and CaO, signifying the disruption of ACr2O4 spinels under alkaline conditions. However, the XRD patterns showcase the exclusive growth of the (200) crystal plane at 2θ=24.5 deg in CaCrO4 only with a weak intensity. The consistent traces of the CaO phase and comparable levels of spinel phase preservation are observed across all spinel compositions, implying the strong stabilities of ACr2O4 spinels at low temperatures. Figure 5(b) presents the XRD results for samples treated at a high temperature of 1200 °C. Comparative analysis of the XRD patterns demonstrates that the exclusive presence of Ca in the CaCrO4 phase but not in CaO, indicating the thorough conversation of CaO at the elevated temperature and denoting a more ideal degree of reaction. Significantly, the growth intensity of the (200) crystal plane at 2θ=24.5 deg in CaCrO4 exhibits a substantial increase, accompanied by a higher abundance and a more ordered arrangement of crystal planes. The pronounced growth of CaCrO4 suggests a higher level of ACr2O4 spinels transformation under the calcination temperature of 1200 °C. Owing to the relatively larger ionic radius of Mn2+ (0.83 Å) over Mg2+ (0.72 Å) and Zn2+ (0.74 Å) and the higher OVs in MnCr2O4 than ZnCr2O4 and MgCr2O4, MnCr2O4 had a stronger reactivity with CaO as well as a lower stability under the alkaline environment.

Phase evolutions and vibrations of chemical bonds of ACr2O4 spinels subjected to an alkaline treatment: (a) phase evolutions of the spinels held at a low temperature of 600 °C for 2 h with the addition of 30 wt pct CaO by XRD, (b) phase evolutions of the spinels held at a high temperature of 1200 °C for 2 h with the addition of 30 wt pct CaO by XRD, (c) FTIR spectra of the samples after calcination at 600 °C for 2 h with the addition of 30 wt pct CaO, and (d) FTIR spectra of the samples after calcination at 1200 °C for 2 h with the addition of 30 wt pct CaO
The samples after calcinations were further characterized by FTIR, with a summary of those results (see electronic supplementary Table S3). As observed from Figure 5(c), the large preservation of the spinel phase at the calcination temperature of 600 °C allows for the identification of distinctive vibrational modes attributed to the Cr(III)–O (630 cm−1) and A(II)–O (520 cm−1) bonds. Notably, a broad and sharp peak observed in the range of 1402–1546 cm−1 and a peak at approximately 880 cm−1 can be attributed to the characteristic signature of CaO, indicating a more residual CaO at low temperature.[36,37] The high residual spinel and CaO in the samples proved a high stability of spinels at low temperatures, consistent with the XRD results. Figure 5(d) depicts the FTIR spectra of the samples calcined at 1200 °C. Similarly, the vibrational modes associated with the Cr(III)–O and A(II)–O bonds can still be discerned in the shaded region, albeit with a less prominent reduction in transmittance compared to the spectra obtained at the calcination temperature of 600 °C. Additionally, a characteristic peak at 880 cm−1 was observed, corresponding to the vibration of Ca–O bonds in CaCrO4.[36] These observations align with the XRD results, where the Ca is exclusively detected in the CaCrO4 phase. In summary, those findings imply a substantial disruption of ACr2O4 spinels at high temperatures.
In addition to those molecular analyses, we also measured the morphology evolutions of the spinels upon calcination under alkaline conditions as well as the elemental distributions. As observed from Figures 6(a) through (c), the elemental distributions especially for A and Cr atom exhibit consistency across ACr2O4 spinels at a low temperature of 600 °C, indicating the retention of a significant portion of the spinel phase in the calcined samples. In contrast, Figures 6(d) through (f) shows that at a high temperature of 1200 °C, the elements in the calcined samples, especially A and Cr, were distributed more independently. Overall, the samples calcined at high temperatures got much denser with larger grain sizes. This proves that the introduction of CaO induced notable modifications in the original phases at high temperatures, particularly in the case of MnCr2O4, in good agreement with those XRD and FTIR results. The calcination of MnCr2O4 under alkaline conditions at 1200 °C induced a dramatic transformation in its morphology, with the initial cubic structures even giving way to the newly formed needle-shaped crystals.

Morphology evolutions and elemental distributions of ACr2O4 spinels subjected to an alkaline treatment. (a–c) morphologies and elemental distributions (A, Cr, O and Ca atoms) of ACr2O4 spinels after calcination at 600 °C: (a) MgCr2O4, (b) ZnCr2O4, and (c) MnCr2O4; (d–f) morphologies and elemental distributions of ACr2O4 spinels after calcination at 1200 °C: (d) MgCr2O4, (e) ZnCr2O4, and (f) MnCr2O4
In Figure 6(a), a morphology similar to that of MgCr2O4 in Figure 4(a) was also noted, indicating a substantial retention of MgCr2O4. At a higher temperature of 1200 °C, the extent of reaction between MgCr2O4 and CaO increased, accompanied by a slight increase in grain size, as shown in Figure 6(d). For ZnCr2O4, similar effect was observed (Figures 6(b) and (e)).[38,39] In contrast, MnCr2O4 exhibits inherent instability especially considering its smaller grain size (Figure 4(c)) compared with ZnCr2O4 (Figure 4(b)). The smaller grain size of spinels, with a larger surface ares, could cause a larger contacted area with CaO as well as an enhanced reaction[38,39] upon interaction with CaO. Those induced substantial morphological alterations, notably replacing the original cubic structure with even needle-shaped crystals formed for MnCr2O4 (Figure 6(f)).
Based on the aforementioned results, we could further quantify the stability of spinel at 1200 °C in an alkaline environment and their divalent cations. We use residual amounts of spinel (RSs) as an indicator of spinel stabilities. The RSs values for MgCr2O4, ZnCr2O4, and MnCr2O4 are 27.2, 25.4, and 20.2 pct, respectively. Similarly, we use an exponential function to establish the relationship between stability (RSs), electronegativity (X) and ionic radius (Y)[27,28,29]: RSs = RSs (X,Y) = C2XcYd. Here c, d, and C2 are undetermined parameters. By taking the natural logarithm of this function and fitting the experimental data, we can obtain that RSs = 11.98 X−0.056 Y−2.03. his quantitatively proves that ionic radius of divalent cations has a stronger effect on spinel stabilities with electronegativity showing a limited role. A larger ionic radius of divalent cations in spinel contribute to a decrease in spinel stability or a smaller amount of residual spinel after alkaline treatment. This agrees well with the OVs concentrations in spinels, i.e., a larger ionic radius of divalent cations causes a higher OVs concentration in spinel and a higher OVs concentration in spinel could further cause a stronger structure instability. This highlights the crucial role played by ionic radius in determining the stability of spinel structures under alkaline conditions.
Stability of Spinels Under an Acidic Environment
Next, we tested the stabilities of ACr2O4 spinels under an acidic environment where 30 wt pct SiO2 was used to create this environment. Figure 7(a) presents the XRD patterns, which reveals the coexistence of two distinct phases in each sample, namely spinel and SiO2. Notably, all peak positions and intensities exhibit a high consistency for those three samples. Furthermore, the crystallographic planes of the initial spinel phases remain unchanged after the calcination process, indicating that the growth of crystal planes is not affected by the incorporation of SiO2 at 1200 °C. Importantly, silica is exclusively observed in the SiO2 phase, without any evidence of transformation into other silicon containing phases under the given processing conditions. Most crystal planes in the SiO2 phase exhibit growth and development, with a minor fraction of crystal planes not detected. The three dominant peaks observed in the SiO2 phase at 26.6, 50.1, and 20.9 deg corresponded to the (011), (11-2), and (100) crystallographic planes, respectively. Those findings demonstrate the notable stability of ACr2O4 spinels when calcined at 1200 °C and their strong resistance to transformation under an acidic environment compared to alkaline.

Phase evolutions and vibrations of chemical bonds of ACr2O4 spinels subjected to an acidic treatment: (a) phase evolutions of the spinels held at a high temperature of 1200 °C for 2 h with the addition of 30 wt pct SiO2 by XRD, (b) phase evolutions of the spinels held at a higher temperature of 1400 °C for 2 h with the addition of 30 wt pct SiO2 by XRD, (c) FTIR spectra of the samples after calcination at 1200 °C for 2 h with the addition of 30 wt pct SiO2, and (d) FTIR spectra of the samples after calcination at 1400 °C for 2 h with the addition of 30 wt pct SiO2
At a higher calcination temperature of 1400 °C, Figure 7(b) illustrates that, similar to the results at 1200 °C, only spinel and SiO2 phases were detected in all samples. However, slight changes in the number of peaks were observed compared to the results at 1200 °C. At 1400 °C, the relative intensity of the main peak (101) of SiO2, in relation to the spinel main peak (311), served as a measure to quantify the remaining SiO2 phase content in ACr2O4 with an acid treatment (see electronic supplementary Table S4). It was observed that MnCr2O4 following acid treatment exhibited the highest SiO2 phase content. Notably, the (101) crystallographic plane of β-cristobalite was detected at 2θ=21.8 deg subsequent to the calcination of MgCr2O4 or ZnCr2O4 with SiO2. β-cristobalite, showing the same cubic structure as spinel, represents the highest temperature phase of SiO2.[40,41] The SiO2 employed in this experiment corresponds to the quartz phase (see electronic supplementary Figure S3). The appearance of (101) crystallographic plane suggests a potential transformation from quartz into β-cristobalite. Moreover, this observation indicates that during the calcination process, the interaction between spinel and SiO2 are relatively independent, with spinel having no significant influence on the phase transition of SiO2. The XRD results reveal that during calcination, the presence of Mn2+ or Cr3+ in MnCr2O4 may undergo chemical interactions with SiO2, leading to a solid dissolution of Mn2+ or Cr3+ into SiO2. This process could potentially impede the transformation of quartz into β-cristobalite.
Similar to the XRD results, considerable similarities were found in the FTIR spectra of the samples obtained after calcination at two different high temperatures. As shown in Figures 7(c) through (d), owing to the abundance of spinel phase, the vibrational modes corresponding to Cr(III)–O and A(II)–O bonds remained distinctly observable in the wavenumber range of 500–650 cm−1. The unique peak at around 1090 cm−1 could be attributed to the antisymmetric stretching vibrations of Si–O-Si,[42] although the absorption band was relatively weak in this region. Another prominent peak at around 800 cm−1 corresponded to the symmetric stretching vibrations of Si–O bonds. The vibration modes mentioned above are succinctly summarized (see electronic supplementary Table S5). Those observations indicate the presence of SiO2 in all calcined samples, and compared to alkaline conditions, the vibrations of Cr(III)–O and A(II)–O bonds were less affected by the presence of SiO2, leading to higher transmittance of Cr(III)–O and A(II)–O vibrations. Those findings suggest that spinels were more stable under an acidic environment, even when calcined at elevated temperatures.
In addition to XRD and FTIR analyses, we investigated the microstructure and elemental compositions of the spinels after calcination under an acidic environment by SEM-EDS. As depicted in Figure 8, the distribution of elements, particularly A and Cr atoms, displayed remarkable consistency among ACr2O4 spinels at high temperatures of 1200–1400 °C, which was quite different from those results under an alkaline environment. This observation suggests the substantial retention of the spinel phase in the calcined samples. Meanwhile, the noticeable separation of Si from the other three elements suggests the relatively stable existence of spinel in acidic environments. From the perspective of grain size, ACr2O4 spinels demonstrated consistency before and after the acidic treatment (Figures 4 and 8). This observation well affirmed the stable coexistence of the ACr2O4 spinels phase with the SiO2 phase under an acidic environment.

Morphology evolutions and elemental distributions of ACr2O4 spinels subjected to an acidic treatment. (a–c) morphologies and elemental distributions (A, Cr, O and Si atoms) of ACr2O4 spinels after calcination at 1200 °C: (a) MgCr2O4, (b) ZnCr2O4, and (c) MnCr2O4. (d-f) morphologies and elemental distributions of ACr2O4 spinels after calcination at 1400 °C: (d) MgCr2O4, (e) ZnCr2O4, and (f) MnCr2O4
Target Modification and Application of Spinels
Based on the aforementioned results, the stability properties of chromium bearing spinels under alkaline and acidic environments as well as the further target modifications could be summarized in Figure 9. Overall, ACr2O4 spinels would transform into CaCrO4 phase and their corresponding oxides (AxOy) when they were calcined under an alkaline condition and remain unchanged during calcination under an acidic condition (Figure 9(a)). The factors affecting the conversion degree included the internal properties and the external treatment environments (Figure 9(b)). The internal properties such as lattice parameters and OVs concentration, closely associated with the divalent A2+ cations, determined the inherent stabilities of those ACr2O4 spinels. High OVs concentrations could cause an inherent structure instability of spinels. And herein based on the calcination experiments, we proved the external environments such as calcination temperature and treatment agent (alkaline over acidic) could greatly influence their stabilities considering the particular industrial systems. An alkaline environment could cause a structure instability of chromium bearing spinels while an acidic environment could assist to the further chromium fixations.

Stability of chromium bearing spinels in alkaline and acidic environments: (a) schematic diagram of reactions between the chromium bearing spinels and 30 wt pct CaO and SiO2, and (b) internal and external factors affecting the stability of chromium bearing spinels
By adjusting the composition of stainless steel slags and controlling the external factors such as temperature, the formation and growth of chromium bearing spinels can be promoted or restrained, enhancing the selective enrichment of chromium in spinels. For instance, increasing the content of MgO in stainless steel slags can promote the preferential enrichment of chromium in the form of MgCr2O4 instead of other forms like MnCr2O4. Additionally, due to the excellent acid resistance of chromium bearing spinels, reducing the content of CaO or the binary basicity, CaO/SiO2, can further facilitate the existence of the spinel phase to a greater extent, ensuring the harmless treatment of stainless steel slags. Furthermore, for the further storage and utilization of modified stainless steel slags, the implementation of a slightly acidic environment is essential, thereby perpetuating the effective immobilization of chromium in the stainless steel slags matrix whilst mitigating against secondary contamination risks. On the contrary, the efficient recovery of valuable metal zinc from steel dusts can be achieved by introducing more CaO or Na2CO3 working as agents into the dusts coupled with raising the roasting temperature. By selectively disrupting the structure of complex spinels, the extraction of zinc can be facilitated, thereby guaranteeing the direct recycling of steelmaking byproducts in metallurgical systems. The roasting of vanadium bearing slags could share similar potential modification ideas with regard to spinels.
Moreover, our study established a foundation for the further target modification of more complex industrial systems.[3,6,9,43] Considering that generally in those complex systems, multiple divalent cations (Zn2+, Mn2+, Mg2+, Fe2+, etc.) and trivalent cations (Cr3+, Al3+, Fe3+, etc.) would coexist and interfere with each other. This increases the research difficulties to quantify the roles of those cations upon formation and evolutions, but at the same time underscores the inherent stability and adaptability of spinels as a structure capable of accommodating various types of cations. By understanding the stability characteristics of spinels, we can enhance the design and development of slags, ceramics and catalysts with heightened stability, thereby reducing emissions of pollutants and curbing resource consumption.[44,45] This target modification of spinel evolutions could contribute to a clean advancement of modern metallurgy.
Conclusions
In this study, we synthesized and investigated the intrinsic properties of chromium bearing spinels with different divalent cations, along with their stability under both alkaline and acidic environments. XRD and the refinement analyses reveal that MnCr2O4 had a larger lattice parameter and unit cell volume over MgCr2O4 and ZnCr2O4 due to the larger radius of Mn2+ over Mg2+ and Zn2+. In consistent, XPS results show that MnCr2O4 had a higher OVs concentration over MgCr2O4 and ZnCr2O4, which caused a decreased bonding strength, as evidenced by FTIR and Raman characterization. To probe the stability of ACr2O4 spinels under different environmental conditions, we introduced CaO and SiO2 to establish an alkaline environment and an acidic environment, respectively. The results elucidated that ACr2O4 spinels exhibit a diminished stability upon an alkaline calcination over an acidic treatment, with an increase of instability at high temperatures. MnCr2O4 had a stronger instability compared to MgCr2O4 and ZnCr2O4, agreeing with their microstructural characteristics. Those comprehensive results provide valuable insights into the stability and evolution behaviors of ACr2O4 spinels, which could contribute significantly to the target modification and applications of spinels.
References
F. Gu, Y. Zhang, S. Zijian, T. Yikang, S. Liu, and T. Jiang: J. Clean. Prod., 2021, vol. 296, p. 126467.
G. Kim and I. Sohn: J. Hazard. Mater., 2018, vol. 359, pp. 174–85.
H. Shen, E. Forssberg, and U. Nordström: Resour. Conserv. Recycl., 2004, vol. 40, pp. 245–71.
G.J. Albertsson, L. Teng, F. Engström, and S. Seetharaman: Metall. Mater. Trans. B, 2013, vol. 44, pp. 1586–97.
B. Dhal, H.N. Thatoi, N.N. Das, and B.D. Pandey: J. Hazard. Mater., 2013, vol. 250–251, pp. 272–91.
K. GracePavithra, V. Jaikumar, P.S. Kumar, and P. SundarRajan: J. Clean. Prod., 2019, vol. 228, pp. 580–93.
M. Nath, S. Song, A.M. Garbers-Craig, and Y. Li: Ceram. Int., 2018, vol. 44, pp. 20391–98.
L. Chen, M. Chen, Y. Sun, and X. Ma: Metall. Mater. Trans. B, 2023, vol. 54, pp. 1618–23.
Y. Guo, H. Li, J. Cheng, S. Shen, J. Diao, and B. Xie: Sep. Purif. Technol., 2021, vol. 263, p. 118396.
Y. Tang, S.S. Chui, K. Shih, and L. Zhang: Environ. Sci. Technol., 2011, vol. 45, pp. 3598–3604.
Y. Sun, L. Chen, M. Chen, and X. Ma: Metall. Mater. Trans. B, 2024, vol. 55, pp. 242–50.
Y. Li, J. Gao, X. Lan, G. Feng, Y. Zhang, and Z. Guo: Sep. Purif. Technol., 2023, vol. 310, p. 123169.
Z. Wang and I. Sohn: J. Am. Ceram. Soc., 2020, vol. 103, pp. 6012–24.
Z. Wang and I. Sohn: J. Am. Ceram. Soc., 2021, vol. 104, pp. 697–705.
Q. Zeng, J. Li, Q. Mou, H. Zhu, and Z. Xue: JOM, 2019, vol. 71, pp. 2331–37.
X. Huo, X. Zhang, Z. Ding, M. Zhang, and M. Guo: J. Hazard. Mater., 2023, vol. 446, p. 130746.
B. Liu, H. Du, S. Wang, Y. Zhang, S. Zheng, L. Li, and D. Chen: AICHE J., 2013, vol. 59, pp. 541–52.
T. Jiang, J. Wen, M. Zhou, and X. Xue: J. Alloy. Comp., 2018, vol. 742, pp. 402–12.
J. Wen, T. Jiang, X. Zheng, J.W.J. Cao, and M. Zhou: Sep. Purif. Technol., 2020, vol. 230, p. 115881.
N. Xue, Y. Zhang, J. Huang, T. Liu, and L. Wang: J. Clean. Prod., 2017, vol. 166, pp. 1265–73.
N. Ma: J. Clean. Prod., 2016, vol. 112, pp. 4497–504.
M. Omran, T. Fabritius, Y. Yu, E.P. Heikkinen, G. Chen, and Y. Kacar: J. Sustain. Metall., 2021, vol. 7, pp. 15–26.
Z. Yang, H. Li, X. Yin, Z. Yan, X. Yan, and B. Xie: Int. J. Miner. Process., 2014, vol. 133, pp. 105–11.
C.W. Bale, E. Bélisle, P. Chartrand, S.A. Decterov, G. Eriksson, A.E. Gheribi, K. Hack, I.-H. Jung, Y.-B. Kang, J. Melançon, A.D. Pelton, S. Petersen, C. Robelin, J. Sangster, P. Spencer, and M.-A. Van Ende: Calphad Calphad, 2016, vol. 54, pp. 35–53.
Y. Sun, Q. Liu, H. Wang, Z. Zhang, and X. Wang: Bioresour. Technol., 2017, vol. 223, pp. 1–9.
LibreTextsTM CHEMISTRY, 8.2: Atomic and Ionic Radius. https://chem.libretexts.org/
S. Mou, K. Huang, M. Guan, X. Ma, J. Chen, Y. Xiang, and X. Zhang: J. Power. Sources, 2021, vol. 505, p. 230067.
H. Yang, Y. Zhao, Q. Wen, R. Yang, Y. Liu, H. Li, and T. Zhai: ACS Appl. Mater. Interfaces, 2021, vol. 13, pp. 14302–11.
J. Yan, J. Zhu, D. Chen, Z. Zeng, L. Jiang, S. Liu, X. Zhang, S. Yu, and F. Du: J. Mater. Chem. A, 2022, vol. 10, pp. 9419–26.
L. Mao, H. Cui, C. Miao, H. An, J. Zhai, and Q. Li: J. Mater. Cycles Waste Manag., 2015, vol. 18, pp. 573–81.
M. Stefanescu, M. Barbu, T. Vlase, P. Barvinschi, L. Barbu-Tudoran, and M. Stoia: Thermochim. Acta, 2011, vol. 526, pp. 130–36.
H. Song, D. Laudenschleger, J.J. Carey, H. Ruland, M. Nolan, and M. Muhler: ACS Catal., 2017, vol. 7, pp. 7610–22.
Z. Wang, P. Lazor, S.K. Saxena, and G. Artioli: J. Solid State Chem., 2002, vol. 165, pp. 165–70.
Z. Wang, H.S.C. O’Neill, P. Lazor, and S.K. Saxena: J. Phys. Chem. Solids, 2002, vol. 63, pp. 2057–61.
D. Lenaz and V. Lughi: Phys. Chem. Miner., 2013, vol. 40, pp. 491–98.
M. Galván-Ruiz, J. Hernández, L. Baños, J. Noriega-Montes, and M.E. Rodríguez-García: J. Mater. Civ. Eng., 2009, vol. 21, pp. 694–98.
W. Guan, F. Ji, Y. Cheng, Z. Fang, D. Fang, P. Yan, and Q. Chen: J. Nanomater., 2013, vol. 2013, pp. 1–7.
J. Li, Q. Zeng, Q. Mou, and Y. Yu: High Temp. Mater. Proc., 2019, vol. 38, pp. 867–72.
J. Li, Q. Mou, Q. Zeng, and Y. Yu: Processes, 2019, vol. 7, p. 487.
D.M. Hatch and S. Ghose: Phys. Chem. Minerals, 1991, vol. 17, pp. 554–62.
K. Lin, H. Ding, and M.J. Demkowicz: J. Nucl. Mater., 2016, vol. 479, pp. 224–31.
I. Ramalla, R.K. Gupta, and K. Bansal: Int. J. Eng. Technol., 2015, vol. 4, pp. 509–19.
R. Wang, L. Jiang, Y. Wang, and A.P. Roskilly: J. Clean. Prod., 2020, vol. 274, p. 122997.
X. Liu, X. Cui, K. Dastafkan, H. Wang, C. Tang, C. Zhao, A. Chen, C. He, M. Han, and Q. Zhang: J. Energy Chem., 2021, vol. 53, pp. 290–302.
J. Zhang, X. Liu, L. Deng, X. Liu, F. Li, R. Wang, and C. Chen: J. Alloys Compd., 2022, vol. 925, p. 166600.
Acknowledgments
The authors thank the National Natural Science Foundation of China (52274415) and The National Science Fund for Overseas Excellent Young Scholars (21FAA01748) for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author state that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Luo, S., Ma, X., Chen, Z. et al. A Comparison Study on Formation and Stabilities of Chromium Bearing Spinels. Metall Mater Trans B 55, 3284–3297 (2024). https://doi.org/10.1007/s11663-024-03183-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-024-03183-2