
Abstract
With the release of the Phalaenopsis equestris (Schauer) Rchb.f. genome database, more in-depth studies of Phalaenopsis spp. will be carried out in the future. Transient gene expression in protoplasts is a useful system for gene function analysis, which is especially true for Phalaenopsis, whose stable genetic transformation is difficult and extremely time-consuming. In this study, juvenile leaves from aseptic Phalaenopsis seedlings were used as the starting material for protoplast isolation. After protocol refinement, the highest yield of viable protoplasts [5.94 × 106 protoplasts g−1 fresh weight (FW)] was achieved with 1.0% (w/v) Cellulase Onozuka R-10, 0.7% (w/v) Macerozyme R-10, and 0.4 M D-mannitol, with an enzymolysis duration of 6 h. As indicated by transient expression of green fluorescent protein (GFP), a transformation efficiency of 41.7% was achieved with 20% (w/v) polyethylene glycol (PEG-4000), 20 μg plasmid DNA, 2 × 105 mL−1 protoplasts, and a transfection duration of 30 min. The protocol established here will be valuable for functional studies of Phalaenopsis genes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Phalaenopsis spp., also known as moth orchid, is one of the most popular tropical flowers in the world. The recently released genome sequence of P. equestris (Schauer) Rchb.f., showed that moth orchids contain 29,431 predicted protein-coding genes (Cai et al. 2015). Annotation and functional characterization of these identified genes would help reveal the unique reproductive and ecological adaptations of this orchid species and would facilitate genetic improvement for orchid breeding (Su and Hsu 2003; Hsieh et al. 2013; Cai et al. 2015). However, stable genetic transformation in orchids is notoriously difficult to achieve and extremely time-consuming (Mishiba et al. 2005). For this reason, transient gene expression is an efficient alternative way to analyze gene function (Luehrsen et al. 1992; Chen et al. 2006; Julkifle et al. 2010; Huang et al. 2013). Along with the virus-induced gene silencing system developed in Phalaenopsis (Hsieh et al. 2013), protoplast-based transient gene expression is another useful experimental approach to study gene function, allowing for the elucidation of protein subcellular localization, interactions, and/or catalytic activities in vivo (Maddumage et al. 2002; Locatelli et al. 2003; Yoo et al. 2007; Zhang et al. 2011; Yao et al. 2016). Successful protoplast isolation in Orchidaceae species has been reported in Cymbidium (Pindel 2007), Dendrobium (Khentry et al. 2006), and Phalaenopsis (Kobayashi et al. 1993; Shrestha et al. 2007; Qiao et al. 2008). In Phalaenopsis, however, protoplast isolation efficiency needed further improvement before a protoplast-based transient gene expression system could be established.
The most commonly used method for protoplast isolation is by enzymatic digestion of the cell wall components (e.g., cellulose, hemicellulose, and pectin) (Rose 1980; Sun et al. 1992; Duquenne et al. 2007; Zhao et al. 2011; Ratanasanobon and Seaton 2013). During enzymatic digestion, the enzymolysis time and osmotic pressure have to be adjusted; the latter, by applying specific concentrations of osmotic regulators (e.g., sorbitol, sucrose, or D-mannitol) (Zhou et al. 2008; Rezazadeh and Niedz 2015). Polyethylene glycol (PEG)-mediated protoplast transfection and transient gene expression is the most widely used system for protoplast transformation, and variables, such as the PEG concentration and incubation time, should be optimized for different plant species (Kao and Michayluk 1974; Maas and Werr 1989).
The objectives of this present study were to develop an efficient protocol for isolating protoplasts derived from mesophyll cells of Phalaenopsis, followed by PEG-mediated transfection, and subsequent transient expression of reporter genes. Various key parameters were adjusted in this study and the presented optimum protocol for transient gene expression in Phalaenopsis protoplasts was highly reliable with repeatable results.
Materials and Methods
Plant materials and expression vectors
The Phalaenopsis hybrid cultivar ‘Ruili Beauty’ was used in this study. Aseptic seedlings were derived from flower stalk explants of ‘Ruili Beauty’ when the stalks were sterilized for 20 min with 1% (w/v) sodium hypochlorite (NaClO) solution diluted from a solution of sodium hypochlorite containing 5.2% active chlorine (Shanghai Lingfeng Chemical Reagent Co., Ltd., Shanghai, China), then cultured in the induction and proliferation medium, consisting of MS salts (Murashige and Skoog 1962), 3% (w/v) sucrose, 100 ml L−1 coconut water, 5.6 g L−1 agar (plant micropropagation grade, gelling strength 900 g cm−2), 7.0 mg L−1 N6-benzyladenine (6-BA), and 0.5 mg L−1 α-naphthaleneacetic acid (NAA), adjusted with NaOH to a pH of 5.6 prior to autoclaving at 122°C for 20 min. In vitro plantlets were cultured in an illumination incubator (GXZ, Ningbo Jiangnan Instrument Factory, Ningbo, China) at 25°C with a 12-h photoperiod of 80 μmol m−2 s−1 supplied by fluorescent lamp.
The green fluorescent protein (GFP) expression vector pGreen-GFP and the GFP fusion-protein cucumber transport inhibitor response 1 (TIR1; Accession number: JX901282.1) vector pGreen-CsTIR1-GFP were used in this study; the 35S promoter was used to drive the GFP reporter or target-GFP fusion genes (Xu et al. 2017). The plasmids were extracted using the MiniBEST Plasmid Purification Kit (TaKaRa, Dalian, China).
The isolation, purification, and viability of ‘Ruili Beauty’ mesophyll protoplasts
The improved method of protoplast isolation was based on a report by Huang et al. (2013). Only young leaves of aseptically grown plantlets were chosen for protoplast isolation, cut into 0.5–1.0-mm thin strips (Fig. 1A, B), and pretreated with 0.4–0.6 M D-mannitol (Solarbio, Beijing, China) for 1 h [1 g fresh weight (FW) leaf material was added to 10 mL of D-mannitol]. Then 1.0 g (10 mL)−1 leaf material was transferred into an Erlenmeyer flask with mixed enzymes and osmotic regulator [1.0–2.0% (w/v) Cellulase Onozuka R-10(Yakult Honsha Co., Ltd., Tokyo, Japan), 0.3–0.7% (w/v) Macerozyme R-10 (Yakult Honsha Co.), and 0.4–0.6 M D-mannitol] at pH 5.6, and incubated at 25°C in darkness with rotation of 0.1 g for 5, 6, or 7 h.

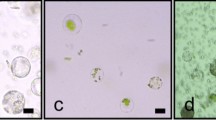
Isolation of Phalaenopsis hybrid cultivar ‘Ruili Beauty’ mesophyll protoplasts. (A) Aseptic ‘Ruili Beauty’ seedlings; red circles mark young leaves used in this study. (B) ‘Ruili Beauty’ leaves were cut into thin strips. (C) Protoplast suspension; (D) purified protoplast layer after density gradient centrifugation. (E), (F) Visualization of ‘Ruili Beauty’ protoplasts under bright-field microscopy; (G), (H) ‘Ruili Beauty’ protoplasts under fluorescence microscopy, visualizing fluorescein in live cells with excitation/emission wavelengths 470-490 and 510-530 nm. (E, G) Bars: 50 μm; (F, H) bars: 20 μm.
After enzymolysis, the solution was filtered using a 200-μm mesh sieve, centrifuged (100g, 8 min) at room temperature (23 ± 1°C), then resuspended with 5 mL of cell and protoplast washing solution, CPW-9 M [CPW salts consisted of KH2PO4 27.2 mg L−1, KNO3 101 mg L−1, CaCl2·2H2O 1480 mg L−1, MgSO4·7H2O 246 mg L−1, KI 0.16 mg L−1, CuSO4·5H2O 0.025 mg L−1; CPW-9 M was CPW salts with 9% (w/v) D-mannitol]. The centrifugation wash step was repeated twice, and the resultant protoplast pellet was resuspended in 1.0 mL of CPW-9 M, floated on the surface of a 15–25% (w/v) sucrose gradient, and centrifuged (100g, 5 min) at room temperature. The protoplast layer was collected and resuspended in 1.0 mL of CPW-9 M (Fig. 1C, D).
The collected protoplasts were diluted with CPW-9M (1:3, v/v), and yield was calculated using a hemocytometer to count a sample under the microscope. The viability of the purified protoplasts was determined using the Fluorescein diacetate (FDA) (Sigma-Aldrich® Co., St. Louis, MO) staining method: FDA solution (5.0 mg of fluorescein diacetate dissolved in 1.0 mL of acetone) was added to the purified protoplasts (1:40, v/v), incubated for 10 min at room temperature before counting the green fluorescing cells under a fluorescence microscope (OLYMPUS BX53, Tokyo, Japan) (excitation per emission wavelengths 470–490 and 510–530 nm). The viability of the protoplasts was expressed as (viable protoplasts ÷ total number of protoplasts) × 100%.
Transformation and transient gene expression in ‘Ruili Beauty’ mesophyll protoplasts
The modified PEG-mediated protoplast transfection protocol was based on a method described by Yoo et al. (2007). The ‘Ruili Beauty’ mesophyll protoplasts were resuspended in 1.0 mL of MaMg solution, consisting of 4.0 mM 2-(N-morpholino) ethanesulfonic acid (MES), 0.4 M D-mannitol, and 15 mM MgCl2, at pH 5.6. The protoplasts were incubated on ice for 30 min, then centrifuged at 70g for 4 min at room temperature. The supernatant was discarded and 0.5–1.0 mL of MaMg solution was added to adjust the protoplast density to 5 × 105–2 × 106 protoplasts mL−1.
A total of 0.2 mL of protoplast suspension was transferred to a 2.0-mL Eppendorf tube, then 5–25 μg plasmid DNA and an equal volume of 20–50% (w/v) PEG-4000 (Guangzhou Jinhuada Chemical Reagent Co., Guangzhou, China)were added in turn and incubated at room temperature in the dark for 10–60 min. To stop the transfection, a two-fold volume of W5 solution (154 mM NaCl, 125 mM CaCl2, 5 mM KCl, and 2 mM MES) was added to the tube, centrifuged (70g for 2 min) at room temperature, and the supernatant discarded; the centrifugation process was repeated once. The protoplasts were finally resuspended in 0.1 mL of W5 solution and incubated at 23 ± 1°C for 16–24 h in the dark.
The expression of the green fluorescent protein gene (GFP) was observed using a laser confocal microscope (LSM780, Zeiss, Jena, Germany) (excitation 488 nm, emission 510–530 nm). The percent transformation efficiency was calculated as (fluorescent protoplasts ÷ total protoplasts) × 100%.
Unless otherwise stated, the tissue culture chemicals including plant growth regulators, sucrose, macronutrient, and micronutrient (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) were bought from Shanghai Jiafeng Horticultural Products Co., Ltd. (Shanghai, China). Other chemicals (Xilong Chemical Reagent Co., Ltd., Shantou, China) were bought from Nanjing Shoude Biotechnology Co., Ltd. (Nanjing, China).
Statistical analysis
All variable adjustment experiments were repeated at least thrice; the range analysis of the orthogonal test was based on PASW Statistics 18 software (version 18.0, IBM Information Management, Armonk, NY).
Results and Discussion
In preliminary experiments, it was found that the growth stage of leaves and growth condition of the aseptic plants were critical for successful protoplast isolation. As indicated in Fig.1A, juvenile leaves of healthy ‘Ruili Beauty’ plants were the best starting material. The successful isolation of protoplasts depended on breaking down the cell wall and releasing the intact protoplasts. The isolated ‘Ruili Beauty’ protoplasts had a spherical or near-spherical shape with an average diameter of 39 μm (Fig. 1E, F). Maximum yield of intact protoplasts (viability confirmed by FDA staining) reached 5.94 × 106 (g FW)−1 (Fig. 1G, H) after variables were amended.
Effects of single factors on ‘Ruili Beauty’ mesophyll protoplast isolation
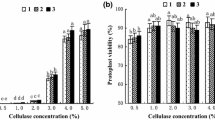
To optimize the protoplast isolation protocol, variables, such as enzyme concentration, mannitol concentration, and enzyme digestion duration, were adjusted (Fig. 2). In short, the highest yield of viable protoplasts occurred with 1.5% (w/v) Cellulase Onozuka R-10, 0.5% (w/v) Macerozyme R-10, 0.5 M D-mannitol, and 6 h of enzymolysis duration.

Effects of single factors on the yield of viable Phalaenopsis hybrid cultivar ‘Ruili Beauty’ protoplasts. The effects of Cellulase R-10 (%), Macerozyme R-10 (%), D-mannitol (M) concentrations, and enzymolysis time (h), on the yield of viable ‘Ruili Beauty’ protoplasts. Bars represent standard errors (SE).
Refinement of Phalaenopsis mesophyll protoplast isolation variables using orthogonal test
An L9 (34) orthogonal test was designed as shown in Table 1, to further optimize the abovementioned variables in protoplast isolation. A range analysis of the orthogonal test was also conducted to predicate the best combination of these variables (Table 2). From the range analysis, cellulase R-10 (R: 1.3354) and enzymolysis time (R: 1.1176) had crucial influence on the yield of viable protoplasts. By analyzing the k value via the Duncan test (Table 2), the optimal level of each factor was kA1, kB3, and kD2, respectively. As for C factors, kC1 and kC2 were significantly higher than kC3, while no significant difference existed between C1 and C2. Because C1 had a lower D-mannitol dosage compared to C2, A1B3C1D2 was deemed the best choice of isolation variables, while curtailing expenditures.
The best theoretical combination was further compared to the optimal combination of T2 (identified in Table 1) to verify the orthogonal test prediction. As shown in Table 3, the yield of 5.94 × 106 (g FW)−1of viable mesophyll protoplasts in the theoretical optimal combination had minimal cell debris and was significantly higher than that of T2 [4.83 × 106 (g FW)−1]. Therefore, the best system for the isolation of mesophyll-derived protoplasts was established as 1.0% (w/v) Cellulase Onozuka R-10, 0.7% (w/v) Macerozyme R-10, and 0.4 M D-mannitol, with an enzymolysis time of 6 h. Previously, the best yield of Phalaenopsis mesophyll protoplast was 3.8 × 106 (g FW)−1, as reported by Shrestha et al. (2007). Thus, using the optimized protocol presented here, the yield of viable protoplast was about 57.9% higher than previously reported.
Establishment of a transient gene expression system in Phalaenopsis protoplasts
To optimize the transient gene expression system in ‘Ruili Beauty’ mesophyll protoplasts, variables including concentration of PEG, density of protoplasts, amount of plasmid, and incubation time, were adjusted as shown in Fig. 3. In brief, the best transformation efficiency (~ 41.7%) was achieved by using 20% (w/v) PEG-4000 (Supplementary Table 1), 30 min of incubation duration (Supplementary Table 2), 20 μg plasmid DNA (Supplementary Table 3), and 2 × 105 mL−1 of protoplasts used for transformation (Supplementary Table 4). Using this protocol, a GFP-fusion cucumber TIR1 protein was successfully detected and localized in both the nucleus and cell membrane (Fig. 4), consistent with previously reported findings by Xu et al. (2017). This system could easily be applied to the subcellular localization of other proteins.

Effects of different factors on protoplast transformation efficiency. Effects of final polyethylene glycol (PEG)-4000 (w/v) concentration, PEG processing time (min), amount of plasmid (μg), and numbers of protoplasts (× 104) on protoplast transformation efficiency (%). Bars represent standard errors (SE).

Transient gene expression in protoplasts derived from Phalaenopsis hybrid cultivar ‘Ruili Beauty’ mesophyll cells. (A) The expression of the green fluorescent protein (GFP) gene in ‘Ruili Beauty’ mesophyll protoplasts observed under fluorescence microscopy (excitation 488 nm, emission 500–530 nm). Chlorophyll fluoresces in red (emission 650–750 nm) when excited at 488 nm. Bright-field micrograph of the same view. Merge indicates computer-generated composite of fluorescent and bright-field images. (B) Transient expression of the CsTIR1-GFP gene in ‘Ruili Beauty’ mesophyll protoplasts, observed under both fluorescence and bright-field microscopy, as described above. Bars: 50 μm.
Conclusions
The variables for protoplast isolation and PEG-mediated gene transformation were optimized for ‘Ruili Beauty’ mesophyll cells. The improved protoplast isolation protocol reached an efficiency as high as 5.94 × 106 (g FW)−1, representing a 57.9% increase over previously reported yields by Shrestha et al. (2007). Furthermore, the optimized PEG-mediated transfection protocol reached an efficiency of 41.7%, sufficient for subcellular localization studies or other investigations where a transient protoplast gene expression system would be desirable.
References
Cai J, Liu X, Vanneste K, Proost S, Tsai W-C, Liu K-W, Chen L-J, He Y, Xu Q, Bian C, Zheng Z, Sun F, Liu W, Hsiao Y-Y, Pan Z-J, Hsu C-C, Yang Y-P, Hsu Y-C, Chuang Y-C, Dievart A, Dufayard J-F, Xu X, Wang J-Y, Wang J, Xiao X-J, Zhao X-M, Du R, Zhang G-Q, Wang M, Y-Y S, Xie G-C, Liu G-H, Li L-Q, Huang L-Q, Luo Y-B, Chen H-H, Van de Peer Y, Liu Z-J (2015) The genome sequence of the orchid Phalaenopsis equestris. Nat Genet 47:65–72
Chen S, Tao L, Zeng L, Vega-Sanchez ME, Umemura K, Wang G-L (2006) A highly efficient transient protoplast system for analyzing defence gene expression and protein-protein interactions in rice. Mol Plant Pathol 7:417–427
Duquenne B, Eeckhaut T, Werbrouck S, Van Huylenbroeck J (2007) Effect of enzyme concentrations on protoplast isolation and protoplast culture of Spathiphyllum and Anthurium. Plant Cell Tissue Organ Cult 91:165–173
Hsieh M-H, Pan Z-J, Lai P-H, Lu H-C, Yeh H-H, Hsu C-C, Wu W-L, Chung M-C, Wang S-S, Chen W-H, Chen H-H (2013) Virus-induced gene silencing unravels multiple transcription factors involved in floral growth and development in Phalaenopsis orchids. J Exp Bot 64:3869–3884
Huang H, Wang Z, Cheng J, Zhao W, Li X, Wang H, Zhang Z, Sui X (2013) An efficient cucumber (Cucumis sativus L.) protoplast isolation and transient expression system. Sci Hortic 150:206–212
Julkifle AL, Rathinam X, Sinniah UR, Subramaniam S (2010) Optimisation of transient green fluorescent protein (GFP) gene expression in Phalaenopsis violacea orchid mediated by Agrobacterium tumefaciens-mediated transformation system. Aust J Basic Appl Sci 4:3424–3432
Kao KN, Michayluk MR (1974) A method for high-frequency intergeneric fusion of plant protoplasts. Planta 115:355–367
Khentry Y, Paradornuvat A, Tantiwiwat S, Phansiri S, Thaveechai N (2006) Protoplast isolation and culture of Dendrobium Sonia ‘Bom 17’. Kasetsart J –Nat Sci 40:361–369
Kobayashi T, Kameya T, Ichihashi S (1993) Plant regeneration from protoplasts derived from callus of Phalaenopsis. Plant Tissue Cult Lett 10:267–270
Locatelli F, Vannini C, Magnani E, Coraggio I, Bracale M (2003) Efficiency of transient transformation in tobacco protoplasts is independent of plasmid amount. Plant Cell Rep 21:865–871
Luehrsen KR, de Wet JR, Walbot V (1992) Transient expression analysis in plants using firefly luciferase reporter gene. Methods Enzymol 216:397–414
Maas C, Werr W (1989) Mechanism and optimized conditions for PEG mediated DNA transfection into plant protoplasts. Plant Cell Rep 8:148–151
Maddumage R, Fung RMW, Weir I, Ding H, Simons JL, Allan AC (2002) Efficient transient transformation of suspension culture-derived apple protoplasts. Plant Cell Tissue Organ Cult 70:77–82
Mishiba K-I, Chin DP, Mii M (2005) Agrobacterium-mediated transformation of Phalaenopsis by targeting protocorms at an early stage after germination. Plant Cell Rep 24:297–303
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497
Pindel A (2007) Optimization of isolation conditions of Cymbidium protoplasts. Folia Hort 19:79–88
Qiao Y-X, Zhang Y-P, Wang G-L, Chen C, Wang Y (2008) Optimization of the method for isolating protoplast of Phalaenopsis amabilisBl. Plant Physiol Comm 44:1177–1180 (in Chinese)
Ratanasanobon K, Seaton KA (2013) Protoplast isolation for species in the Chamelaucium group and the effect of antioxidant enzymes (superoxide dismutase and catalase) on protoplast viability. In Vitro Cell Dev Biol – Plant 49:593–598
Rezazadeh R, Niedz RP (2015) Protoplast isolation and plant regeneration of guava (Psidium guajava L.) using experiments in mixture-amount design. Plant Cell Tissue Organ Cult 122:585–604
Rose RJ (1980) Factors that influence the yield, stability in culture and cell wall regeneration of spinach mesophyll protoplasts. Aust J Plant Physiol 7:713–725
Shrestha BR, Tokuhara K, Mii M (2007) Plant regeneration from cell suspension-derived protoplasts of Phalaenopsis. Plant Cell Rep 26:719–725
Su V, Hsu BD (2003) Cloning and expression of a putative cytochrome P450 gene that influences the colour of Phalaenopsis flowers. Biotechnol Lett 25:1933–1939
Sun S, Furtula V, Nothnagel EA (1992) Mechanical release and lectin labeling of maize root protoplasts. Protoplasma 169:49–56
Xu J, Li J, Cui L, Zhang T, Wu Z, Zhu P-Y, Meng Y-J, Zhang K-J, Yu X-Q, Lou Q-F, Chen J-F (2017) New insights into the roles of cucumber TIR1 homologs and miR393 in regulating fruit/seed set development and leaf morphogenesis. BMC Plant Biol 17:130
Yao L, Liao X, Gan Z, Peng X, Wang P, Li S, Li T (2016) Protoplast isolation and development of a transient expression system for sweet cherry (Prunus avium L.) Sci Hortic 209:14–21
Yoo S-D, Cho Y-H, Sheen J (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2:1565–1572
Zhang Y, Su J, Duan S, Ao Y, Dai J, Liu J, Wang P, Li Y, Liu B, Feng D, Wang J, Wang H (2011) A highly efficient rice green tissue protoplast system for transient gene expression and studying light/chloroplast-related processes. Plant Methods 7:30
Zhao W, Yang W, Wei C, Sun G (2011) A simple and efficient method for isolation of pineapple protoplasts. Biotechnol Biotechnol Equip 25:2464–2467
Zhou B, Nie Y-Z, Zhang X-L, Li Y-H (2008) Protoplast isolation of Brassica rapa ‘Tsuda’ turnip and transient expression of GFP. Lett Biotechnol 19:542–544 (in Chinese)
Acknowledgments
The authors would like to thank Drs. Ji Li and Jian Xu of Nanjing Agricultural University for providing vectors and help with transient expression experiments.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 31372101).
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: Mark Jordan
Rights and permissions
About this article

Cite this article
Li, J., Liao, X., Zhou, S. et al. Efficient protoplast isolation and transient gene expression system for Phalaenopsis hybrid cultivar ‘Ruili Beauty’. In Vitro Cell.Dev.Biol.-Plant 54, 87–93 (2018). https://doi.org/10.1007/s11627-017-9872-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11627-017-9872-z