
Abstract
Hydrogen sulfide (H2S) is a multifunctional second messenger involved in plant growth, development, and acquisition of stress tolerance, including heat tolerance, but the mechanism of H2S-induced heat tolerance in tobacco suspension-cultured cells is not completely clear. This study investigated the effects of pretreatment with the H2S donor sodium hydrosulfide (NaHS) and its precursors cysteine and potassium bisulfite (KHSO3) on the heat tolerance of tobacco suspension-cultured cells and the involvement of sulfhydryl compounds and antioxidant enzymes in conferring heat tolerance. Pretreatment with NaHS, cysteine, and KHSO3 significantly increased the survival percentage of tobacco suspension-cultured cells under heat stress, while treatment with the H2S scavenger hypotaurine in combination with NaHS eliminated heat tolerance induced by treatment with NaHS alone. In addition, NaHS treatment increased the levels of water-soluble sulfhydryl compounds such as H2S, total sulfhydryl compounds, total sulfhydryl proteins, cysteine, and glutathione (GSH) as well as the activities of antioxidant enzymes superoxide dismutase (SOD), catalase (CAT), guaiacol peroxidase (GPX), and glutathione reductase (GR) under normal culture conditions (26°C). Under heat stress at 43°C, the levels of water-soluble sulfhydryl compounds and the activities of antioxidant enzymes all dropped, but the cells treated with NaHS sustained significantly higher levels of water-soluble sulfhydryl compounds and activities of antioxidant enzymes than the control. These results suggest that the pretreatment with NaHS could improve the heat tolerance of tobacco suspension-cultured cells and that the acquisition of this heat tolerance is caused by the elevated levels of water-soluble sulfhydryl compounds and elevated activities of antioxidant enzymes induced by NaHS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Heat stress is often defined as a rise in temperature beyond a threshold level for a period of time sufficient to cause irreversible damage to cells, organs, and even the whole plant (Wahid et al. 2007; Saidi et al. 2011; Mittler et al. 2012). High temperatures lead not only to direct injuries including protein denaturation and aggregation, an increase in fluidity of membrane lipids, and the loss of membrane integrity, but also to indirect heat injuries such as oxidative stress, i.e., the excess production of reactive oxygen species (ROS) such as superoxide radical (O2 ·−), hydrogen peroxide (H2O2), and hydroxyl radical (OH·−). These ROS then cause membrane lipid peroxidation, protein oxidation, DNA damage, inactivation of enzymes in chloroplasts and mitochondria, inhibition of protein synthesis, and protein degradation and may eventually result in severe cellular injury and even cell death (Larkindale and Knight 2002; Wahid et al. 2007). To cope with heat injuries, plants have evolved several strategies such as maintaining ROS homeostasis through the synergistic effects of low-molecular-weight antioxidants such as glutathione (GSH) and antioxidant defense enzymes including superoxide dismutase (SOD), catalase (CAT), guaiacol peroxidase (GPX), glutathione reductase (GR), and ascorbate peroxidase (APX). In addition, other mechanisms are also involved in osmotic adjustment by substances such as proline, soluble sugars, and trehalose; repair and re-establishment of biomembrane integrity; synthesis of heat shock proteins; and so forth (Foyer and Noctor 2009, 2011; Jaleel et al. 2009; Grant et al. 2014). Many studies have demonstrated that increases in antioxidant defense systems (mainly including antioxidants and antioxidant defense enzymes) in plants are closely related to abiotic stress tolerance, including heat tolerance (Foyer and Noctor 2009, 2011; Jaleel et al. 2009; Grant et al. 2014).
Hydrogen sulfide (H2S) has recently been considered as a second messenger in plant cells, which has been found to play multiple physiological roles in plant growth, development, and acquisition of stress tolerance (Lisjak et al. 2010, 2013; Li 2013; Hancock and Whiteman 2014). Many functions of hydrogen sulfide have been uncovered, including physiological processes such as seed germination (Zhang et al. 2010a; Li et al. 2012a), root organogenesis (Zhang et al. 2009a), and stomata movement (Lisjak et al. 2010; Liu et al. 2011; Jin et al. 2013); alleviation of abiotic stresses such as osmotic stress (Zhang et al. 2009b), salt stress (Wang et al. 2012), oxidative stress (Shan et al. 2011; Zhang et al. 2011), heavy metal stress (Zhang et al. 2008, 2010b; Chen et al. 2013), and chilling stress (Fu et al. 2013); and resistance to pathogen infection (Bloem et al. 2011). Abiotic stress can trigger the accumulation of endogenous H2S in plants, and exogenously applied sodium hydrosulfide (NaHS) (an H2S donor) can improve the resistance of plants to multiple abiotic stresses, suggesting that H2S functions as a signal molecule in the acquisition of abiotic stress tolerance (Zhang et al. 2008, 2010b; Chen et al. 2013; Fu et al. 2013; Li 2013). In plants, there are at least five pathways involved in H2S biosynthesis. First, l-cysteine desulfhydrase catalyzes the degradation of l-cysteine to produce H2S, amine, and pyruvate. Second, d-cysteine desulfhydrase decomposes d-cysteine to H2S, similar to l-cysteine desulfhydrase. Third, sulfite is reduced by sulfite reductase to H2S using ferredoxin as an electron donor. Fourth, H2S can be released from cysteine via cyanoalanine synthase in the presence of hydrogen cyanide. Fifth, H2S is incorporated into O-acetyl-l-serine via cysteine synthase to form cysteine; the reverse reaction can release H2S (Li 2015a). For these reasons, potassium bisulfite (KHSO3) and cysteine have commonly been used as H2S precursors (Li 2015a).
In maize seedlings, pretreatment with NaHS improved the survival percentage of maize seedlings under heat stress (Li et al. 2013a). Similarly, in tobacco suspension-cultured cells, NaHS treatment also improved the resistance of tobacco cells to high temperature (Li et al. 2012b), but the resistance mechanism was not fully clear. This study investigated the effect of pretreatments with the H2S donor NaHS and the H2S precursors cysteine and KHSO3 (Li 2015a) on heat tolerance of tobacco suspension-cultured cells and the involvement of water-soluble sulfhydryl compounds and antioxidant enzymes in conferring heat tolerance. The objective was to further elucidate possible mechanisms of H2S-induced heat tolerance in suspension-cultured cells of tobacco.
Materials and Methods
Plant materials and treatments.
Calluses were developed from young stem pith of tobacco variety ‘Bright Yellow’ (Li et al. 2012b). Suspension cells derived from this callus were cultured in liquid MS (Murashige and Skoog 1962) basal medium containing 0.9 μM 2,4-dichlorophenoxyacetic acid and 0.4 μM kinetin (Sinopharm Chemical Reagent Co., Ltd, Shanghai, China) in a rotary shaker (Shanghai Precision Instrument Co., Ltd, Shanghai, China) at 120 rpm and 26°C in the dark. The liquid medium was refreshed at weekly intervals, and the suspension cells were cultured according to Li et al. (2012b). At 5 d after subculture, tobacco-cultured cells in the logarithmic phase were collected and used for the following experiments.
To investigate the effects of pretreatment with H2S donors, precursors, and scavengers on heat tolerance, the 5-d-old tobacco-cultured cells were immediately transferred into different liquid MS media containing the following chemicals: (1) 50 μM NaHS (H2S donor; Li et al. 2013a); (2) 5 mM cysteine (H2S precursor; Li 2015a); (3) 5 mM KHSO3 (H2S precursor; Li 2015a); (4) 50 μM NaHS + 100 μM hypotaurine (HT; H2S scavenger; Li et al. 2013a); (5) 100 μM HT alone; or (6) distilled water (control, 1 mL of distilled water replaced above chemicals) (media additives from Sinopharm Chemical Reagent Co., Ltd). The pH of each chemical stock solution was adjusted to 5.8 with KOH or HCl (Sinopharm Chemical Reagent Co., Ltd) before being added to cell cultures. All treatments were grown on a rotary shaker at 120 rpm and 26°C in the dark for 4 h and then half of the flasks were transferred to another rotary shaker at 120 rpm and 43°C for 7 h to heat stress the cultures (Li et al. 2012b). The remaining flasks were maintained on the rotary shaker at 26°C for another 7 h. After 7 h, the cultures were harvested and the survival percentage of cells was determined as described below.
Determination of survival percentage.
Survival percentage of cells was determined according to Li et al. (2012b). In brief, a 0.2-mL aliquot of cell suspension was transferred to a test tube with 0.3 mL of 3% sucrose and 0.5 mL of 0.5% trypan blue solution (w/v) (Sinopharm Chemical Reagent Co., Ltd). After 5 min, 10 μL of the trypan blue–cell suspension mixture was transferred to a microscope slide, and the viable (unstained) and nonviable (blue-stained) cells were counted. For each sample, 1000 cells were scored. Cell viability was calculated as the percentage of cells that did not stain with trypan blue.
Measurement of sulfhydryl compound contents.
To further understand the effect of NaHS treatment on the contents of sulfhydryl compound such as H2S, total sulfhydryl compounds (TSC), total sulfhydryl proteins (TSP), cysteine, and GSH, cells were collected at various times during NaHS treatment (0, 2, and 4 h) and heat stress (3 and 7 h at 43°C). The cells were filtered and washed using the abovementioned fresh liquid MS basal medium without organics and plant hormones, and sulfhydryl compound contents were determined according to the following methods.
H2S content was measured by the formation of methylene blue from dimethyl-p-phenylenediamine in H2SO4 according to Li (2015b) and Li et al. (2013a). Filtered cells (0.2 g) were ground and extracted in 2 mL phosphate-buffered saline (50 mM, pH 6.8) containing 0.1 mM EDTA and 0.2 mM ascorbic acid (Sinopharm Chemical Reagent Co., Ltd). The homogenate was mixed in a test tube containing 100 mM phosphate-buffered saline (pH 7.4), 10 mM l-cysteine, and 2 mM phosphopyridoxal (Sinopharm Chemical Reagent Co., Ltd) at 25°C, and the released H2S was absorbed in a zinc acetate trap (1 mL of 10 mM zinc acetate [Sinopharm Chemical Reagent Co., Ltd] in a small glass tube fixed to the bottom of a reaction bottle). After 30 min, the 0.3 mL of 5 mM dimethyl-p-phenylenediamine dissolved in 3.5 mM H2SO4 and 0.3 mL of 50 mM ferric ammonium sulfate in 100 mM H2SO4 (Sinopharm Chemical Reagent Co., Ltd) were added to the trap, respectively. After incubation for 15 min at 25°C, the amount of H2S in the zinc acetate trap was determined colorimetrically at 667 nm using a spectrophotometer (Unico Instrument Co., Ltd, Shanghai, China). A calibration curve was made according to the above methods, and H2S content in cells was expressed as nanomoles per gram of fresh weight (FW).
TSC content was determined based on the methods of De Kok et al. (1985) with some modifications. In brief, fresh cells (0.2 g) were ground in 3 mL of 7.5 mM sodium ascorbate solution with a mortar and pestle. The homogenate was centrifuged at 15,000×g for 15 min (4°C). The supernatant was reacted with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB; Sinopharm Chemical Reagent Co., Ltd) and used for the TSC assay at 412 nm. The TSC content was calculated using the extinction coefficient of 3.14 mM−1 cm−1 at 412 nm and expressed as micromoles per gram of FW.
TSP content was measured by using the same 15,000×g supernatant as for measurement of TSC. TSP content was estimated as described by De Kok et al. (1985). A 0.5-mL aliquot of supernatant was added to a mixture of 0.5 mL of 8% sodium dodecyl sulfate (SDS) and 1.0 mL of 200 mM Tris buffer, pH 8.0. Then, 0.1 mL DTNB (10 mM in 20 mM potassium phosphate buffer, pH 7.0) was added. After 15 min, the developed yellow color was measured at 412 nm with a spectrophotometer. The absorbance was corrected for the color of the 15,000×g supernatant extract in SDS and for the color of DTNB. TSP content was determined by subtracting the measured sulfhydryl content of a deproteinized 15,000×g supernatant extract from that of an SDS-treated 15,000×g supernatant extract and was expressed as micromoles per gram of FW.
Cysteine content was performed according to the methods of Gaitonde (1967). In brief, fresh cells (1 g) from control and treated cells were ground in a mortar with a pestle in 3 mL of 5% chilled perchloric acid (Sinopharm Chemical Reagent Co., Ltd) and the homogenates were centrifuged at 15,000×g for 10 min. A 1-mL aliquot of the supernatant was added to a mixture of 1 mL of glacial acetic acid and 1 mL of acid ninhydrin reagent (prepared by dissolving 250 mg of ninhydrin in a mixture of 6 mL of glacial acetic acid and 4 mL of concentrated HCl), and the reaction mixture was kept at 95°C for 30 min. After cooling, the absorbance of the reaction mixture was read at 560 nm, the amount of cysteine was calculated by using a cysteine (Sinopharm Chemical Reagent Co., Ltd) standard curve and expressed as nanomoles per gram of FW.
GSH in tobacco cells was extracted and measured according to Li et al. (2013b). Briefly, filtered cells (0.2 g) were ground in 3 mL of 5% (v/v) sulfosalicylic acid (Sinopharm Chemical Reagent Co., Ltd) with a mortar and pestle. The homogenates were centrifuged at 10,000×g for 15 min at 4°C. The supernatants were used for assays of reduced GSH, determined by the DTNB-GR recycling procedure. The increases in absorbance of the reaction mixtures were measured at 412 nm, and GSH content was calculated using the extinction coefficient of 3.14 mM−1 cm−1 at 412 and expressed as micromoles per gram of FW.
Antioxidant enzyme activity assay.
To clarify the effect of NaHS treatment on antioxidant enzyme activity, the antioxidant enzymes SOD, CAT, GPX, GR, and APX were extracted from tobacco suspension-cultured cells at several times during NaHS treatment (0, 2, and 4 h) and heat stress at 43°C (3 and 7 h) and measured according to Li et al. (2013b). In brief, filtered cells (0.2 g) were ground with a mortar and pestle in 2 mL of extraction buffer containing 50 mM Tris–HCl (pH 7.0), 0.1 mM EDTA, 1 mM ascorbic acid (AsA), 1 mM dithiothreitol (DTT), and 5 mM MgCl2 (Sinopharm Chemical Reagent Co., Ltd). The homogenates were centrifuged at 10,000×g for 15 min at 4°C. The supernatants were used for assays of antioxidant enzymes by the following methods.
SOD (EC1.11.1.6) activity was determined by measuring its ability to inhibit the photochemical reduction of nitroblue tetrazolium (NBT). The 3-mL reaction mixture contained 50 mM Tris–HCl (pH 7.8), 13.37 mM methionine, 0.1 mM NBT (Sinopharm Chemical Reagent Co., Ltd), 0.1 mM riboflavin, 0.1 mM EDTA, and 0.1 mL enzyme extract. One unit of enzyme activity was defined as the amount of the enzyme bringing about 50% inhibition of the photochemical reduction of NBT, and the activity of SOD was expressed as units per gram of FW.
CAT (EC1.11.1.6) activity was determined by measuring the decrease in the absorbance of H2O2 at 240 nm. The 3-mL reaction mixture consisted of 50 mM Tris–HCl (pH 7.0), 0.1 mM EDTA, and 0.1 mL enzyme extract. The reaction was initiated by adding 12.5 mM H2O2 (final concentration; Sinopharm Chemical Reagent Co., Ltd). CAT activity was computed using the extinction coefficient of 0.04 mM−1 cm−1 at 240 and expressed as micromoles per gram of FW per minute.
GPX (EC1.11.1.7) activity was estimated by measuring the increase in absorbance at 470 nm due to guaiacol oxidation. The reaction mixture contained 50 mM Tris–HCl (pH 7.0), 10 mM guaiacol (Sinopharm Chemical Reagent Co., Ltd), and 5 mM H2O2. The reaction was initiated by adding 0.1 mL enzyme extract to the reaction mixture. GPX activity was measured using the extinction coefficient of 26.6 mM−1 cm−1 at 470 nm and expressed as micromoles per gram of FW per minute.
GR (EC1.6.4.2) was assayed by monitoring the increase in absorbance at 340 nm. The reaction mixture contained 50 mM Tris–HCl (pH 7.5), 0.1 mM EDTA, 5 mM MgCl2, 0.2 mM NADPH (Sinopharm Chemical Reagent Co., Ltd), 0.1 mL enzyme extract, and distilled water to make up a final volume of 1 mL. The reaction was initiated by adding 0.5 mM GSSG (oxidized glutathione, final concentration). GR activity was calculated using the extinction coefficient of 6.2 mM−1 cm−1 at 340 and expressed as micromoles per gram of FW per minute.
APX (EC1.11.1.1) activity was measured by monitoring the rate of AsA oxidation at 290 nm. The assay mixture contained 50 mM Tris–HCl (pH 7.0), 0.1 mM H2O2, 0.1 mM EDTA, and 0.1 mL enzyme extract. The reaction was initiated by adding 0.5 mM AsA (final concentration). APX was detected according to the reduction in absorbance at 290 nm per unit time due to the oxidation of AsA, and APX activity was measured using the extinction coefficient of 2.8 mM−1 cm−1 at 290 and expressed as micromoles per gram of FW per minute.
Statistical analysis.
The experiment was set up according to a completely randomized design with at least three replications. The data were processed statistically using one-way analysis of variance (ANOVA). The figures were drawn by SigmaPlot 12.5 (Systat Software Inc., London, UK). In each figure, error bars represent standard error, and each data point represents the mean ± SE of at least three independent experiments.
Results
Effect of H 2 S donor, precursor, and scavenger treatments on survival percentage of tobacco suspension-cultured cells under normal and heat stress conditions.
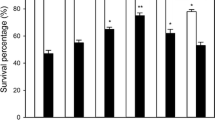
The 5-d-old-cultured cells were exposed to heat stress at 43°C for 7 h in a rotary shaker after being treated with NaHS, cysteine, KHSO3, hypotaurine (HT) alone, or NaHS in combination with HT for 4 h. As shown in Fig. 1a , under normal culture conditions at 26°C, application of NaHS, cysteine, KHSO3, HT alone, or NaHS in combination with HT had no significant effect on survival percentage of suspension cells compared with the control treatment, illustrating that these treatments had no toxic effects on tobacco cells. Under high-temperature stress conditions, NaHS, cysteine, and KHSO3 pretreatments increased the survival percentage of tobacco suspension cells (P < 0.01 for NaHS and cysteine; P < 0.05 for KHSO3; Fig. 1a ). Addition of HT in combination with NaHS eliminated NaHS-induced heat tolerance, and HT alone was not significantly different from the control (Fig. 1a ). These results demonstrate that pretreatment with H2S and its precursors could improve tolerance of tobacco suspension-cultured cells to heat stress.

Effects of pretreatment with NaHS, cysteine (Cys), KHSO3, hypotaurine (HT), or NaHS in combination with HT. (a) Survival percentage of tobacco suspension-cultured cells under normal culture conditions (26°C) or heat stress at 43°C. (b) Content of endogenous H2S under normal culture conditions. The 5-d-old suspension cells were either subjected to heat stress at 43°C or maintained at the normal temperature for 7 h after being treated with 50 μM NaHS, 5 mM cysteine, 5 mM KHSO3, and 100 μM HT or NaHS in combination with HT for 4 h. Error bars represent standard error and each data point represents the mean ± SE of three experiments. Asterisks indicate significant differences (*P < 0.05; **P < 0.01) from the control without NaHS treatment.
Effect of H 2 S treatment on contents of endogenous H 2 S, sulfhydryl compounds, sulfhydryl proteins, cysteine, and glutathione under normal and heat stress conditions.
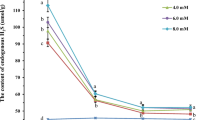
To investigate the effects of H2S donor, precursor, and scavenger pretreatments on endogenous H2S levels, suspension-cultured cells were treated with NaHS, cysteine, KHSO3, HT alone, or NaHS in combination with HT for 4 h, and then the endogenous content of H2S was determined. Pretreatment with NaHS, cysteine, or KHSO3 increased the endogenous level of H2S in tobacco suspension-cultured cells (Fig. 1b ), whereas treatment with HT in combination with NaHS eliminated the accumulation of endogenous H2S compared with the control (Fig. 1b ). These results parallel those for survival under heat stress (Fig. 1a ) and indicate that H2S donor and precursor treatments can increase the endogenous level of H2S in tobacco suspension-cultured cells. During NaHS treatment, the accumulation of endogenous H2S increased as treatment time increased (P < 0.01 for 2- and 4-h treatments vs. control; Fig. 2). Under high-temperature stress at 43°C, H2S content decreased in both treated and control cells, but H2S content in treated cells was higher than that of the untreated control at both 3 and 7 h (P < 0.01 and P < 0.05, respectively; Fig. 2), indicating that NaHS treatment could increase the content of endogenous H2S and alleviate its decrease under heat stress.

Effects of NaHS pretreatment on the content of endogenous H2S in tobacco suspension-cultured cells under normal and heat stress conditions. The 5-d-old suspension cells were treated with 50 μM NaHS and then subjected to heat stress at 43°C. Endogenous H2S content was measured during NaHS and heat stress treatments. Error bars represent standard error and each data point represents the mean ± SE of three experiments. Asterisks indicate significant differences (*P < 0.05; **P < 0.01) from the control without NaHS treatment.
In addition, during NaHS treatment, the contents of TSC, TSP, cysteine, and GSH increased as treatment time increased (P < 0.01 at 4 h; Figs. 3, 4, 5, and 6). The levels of TSC, cysteine, and GSH increased more rapidly than those of TSP, reaching very significant differences from the control (P < 0.01) at 2 h of NaHS treatment (Figs. 3, 4, 5, and 6). Under high-temperature stress conditions, the contents of TSC, TSP, cysteine, and GSH in NaHS-treated and untreated cells all decreased as time under heat stress increased, but remained significantly higher than those of the control after 7 h of heat stress treatment (P < 0.05 for TSP, TSC, and cysteine; P < 0.01 for GSH; Figs. 3, 4, 5, and 6). These patterns were similar to those observed for the change in endogenous H2S content induced by NaHS and heat stress (Fig. 2). All of these data demonstrate that pretreatment with H2S could increase the content of water-soluble sulfhydryl compounds in suspension-cultured cells of tobacco and alleviate their decrease under heat stress.

Effects of NaHS pretreatment on the content of total sulfhydryl compounds (TSC) in tobacco suspension-cultured cells under normal and heat stress conditions. The 5-d-old suspension cells were treated with 50 μM NaHS, and then subjected to heat stress at 43°C. TSC content was measured during NaHS and heat stress treatments. Error bars represent standard error and each data point represents the mean ± SE of three experiments. Asterisks indicate significant differences (*P < 0.05; **P < 0.01) from the control without NaHS treatment.

Effects of NaHS pretreatment on the content of total sulfhydryl proteins (TSP) in tobacco suspension-cultured cells under normal and heat stress conditions. The 5-d-old suspension cells were treated with 50 μM NaHS and then subjected to heat stress at 43°C. TSP content was measured during NaHS and heat stress treatments. Error bars represent standard error and each data point represents the mean ± SE of three experiments. Asterisks indicate significant differences (*P < 0.05; **P < 0.01) from the control without NaHS treatment.

Effects of NaHS pretreatment on the content of cysteine in tobacco suspension-cultured cells under normal and heat stress conditions. The 5-d-old suspension cells were treated with 50 μM NaHS and then subjected to heat stress at 43°C. Cysteine content was measured during NaHS and heat stress treatments. Error bars represent standard error and each data point represents the mean ± SE of three experiments. Asterisks indicate significant differences (*P < 0.05; **P < 0.01) from the control without NaHS treatment.

Effects of NaHS pretreatment on the content of glutathione (GSH) in tobacco suspension-cultured cells under normal and heat stress conditions. The 5-d-old suspension cells were treated with 50 μM NaHS and then subjected to heat stress at 43°C. GSH content was measured during NaHS and heat stress treatments. Error bars represent standard error and each data point represents the mean ± SE of three experiments. Asterisks indicate significant differences (**P < 0.01) from the control without NaHS treatment.
Effect of H 2 S treatment on antioxidant enzyme activity under normal and heat stress conditions.
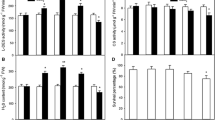
The activities of five antioxidant enzymes (SOD, CAT, GPX, GR, and APX) were measured under normal and heat stress conditions. Under normal culture conditions, the activities of SOD, CAT, GPX, and GR were increased by NaHS treatment compared with the control (P < 0.01 at 4 h; Figs. 7, 8, 9, and 10), similar to the trends for water-soluble sulfhydryl compounds (Figs. 2, 3, 4, 5, and 6). The activities of SOD and CAT increased faster than those of GPX and GR, reaching significance (P < 0.05) after 2 h (Figs. 7, 8, 9, and 10). No significant difference in the activity of APX was observed in NaHS-treated cells compared with the control (Fig. 11). Under high-temperature stress conditions, the activities of all five antioxidant enzymes declined, but NaHS-treated cells maintained significantly higher antioxidant enzyme activities after 7 h for all of the enzymes except APX (P < 0.05 for CAT and GR; P < 0.01 for SOD and GPX; Figs. 7, 8, 9, 10, and 11). Thus, the trends for all enzymes except APX were similar to those observed for water-soluble sulfhydryl compounds (Figs. 2, 3, 4, 5, and 6). The activities of CAT and GR in NaHS-treated cells under heat stress decreased faster than those of SOD and GPX (Figs. 7, 8, 9, 10, and 11). These results demonstrate that H2S pretreatment could increase the activities of antioxidant enzymes in suspension-cultured cells of tobacco under normal conditions and alleviate their decrease under heat stress.

Effects of NaHS pretreatment on the activity of superoxide dismutase (SOD) in tobacco suspension-cultured cells under normal and heat stress conditions. The 5-d-old suspension cells were treated with 50 μM NaHS and then subjected to heat stress at 43°C. SOD activity was determined during NaHS and heat stress treatments. Error bars represent standard error and each data point represents the mean ± SE of three experiments. Asterisks indicate significant differences (*P < 0.05; **P < 0.01) from the control without NaHS treatment.

Effects of NaHS pretreatment on the activity of catalase (CAT) in tobacco suspension-cultured cells under normal and heat stress conditions. The 5-d-old suspension cells were treated with 50 μM NaHS and then subjected to heat stress at 43°C. CAT activity was determined during NaHS and heat stress treatments. Error bars represent standard error and each data point represents the mean ± SE of three experiments. Asterisks indicate significant differences (*P < 0.05; **P < 0.01) from the control without NaHS treatment.

Effects of NaHS pretreatment on the activity of guaiacol peroxidase (GPX) in tobacco suspension-cultured cells under normal and heat stress conditions. The 5-d-old suspension cells were treated with 50 μM NaHS and then subjected to heat stress at 43°C. GPX activity was determined during NaHS and heat stress treatments. Error bars represent standard error and each data point represents the mean ± SE of three experiments. Asterisks indicate significant differences (**P < 0.01) from the control without NaHS treatment.

Effects of NaHS pretreatment on the activity of glutathione reductase (GR) in tobacco suspension-cultured cells under normal and heat stress conditions. The 5-d-old suspension cells were treated with 50 μM NaHS and then subjected to heat stress at 43°C. GR activity was determined during NaHS and heat stress treatments. Error bars represent standard error and each data point represents the mean ± SE of three experiments. Asterisks indicate significant differences (*P < 0.05; **P < 0.01) from the control without NaHS treatment.

Effects of NaHS pretreatment on the activity of ascorbate peroxidase (APX) in tobacco suspension-cultured cells under normal and heat stress conditions. The 5-d-old suspension cells were treated with 50 μM NaHS and then subjected to heat stress at 43°C. APX activity was determined during NaHS and heat stress treatments. Error bars represent standard error and each data point represents the mean ± SE of three experiments.
Discussion
Sulfur, an essential macronutrient in plant life cycle, is taken up as sulfate and is assimilated into cysteine, which is an amino acid at the crossroads of primary metabolism, synthesis of proteins such as sulfhydryl protein, and the formation of other small-molecule sulfhydryl compounds such as H2S and GSH. These sulfhydryl compounds are crucial for the survival of plants under abiotic and biotic stress, through a process termed sulfur-induced resistance (SIR; Rausch and Wachter 2005; Riemenschneider et al. 2005). Among sulfhydryl compounds, volatile H2S, a key intermediate in sulfur assimilation, plays an important role in SIR, which is involved in the acquisition of tolerance to stresses such as drought (Zhang et al. 2009b), salt (Wang et al. 2012), heavy metals (Zhang et al. 2008, 2010b; Chen et al. 2013), and chilling (Fu et al. 2013). Both previous and current results showed that H2S donor and precursor (Li 2015a) treatments could increase the level of endogenous H2S, which in turn improved the tolerance of tobacco suspension-cultured cells to high-temperature stress (Li et al. 2012b; Fig. 1a ). However, the mechanisms of SIR in plant cells are not yet known.
Abiotic stress factors such as high temperature lead to excess production of ROS such as superoxide radical (O2 ·−), hydrogen peroxide (H2O2), and hydroxyl radical (OH·−) and produce oxidative stress, which causes peroxidation of membrane lipids, protein oxidation, and DNA damage (Foyer and Noctor 2009; Jaleel et al. 2009; Scheibe and Dietz 2012). Therefore, ROS homeostasis controlled by disulfide/thiol exchange reactions involving the GSH pool and antioxidant enzymes such as SOD, CAT, GPX, and GR is crucial for the survival of plants under heat stress and other abiotic stresses (Foyer and Noctor 2009; Jaleel et al. 2009; Scheibe and Dietz 2012). Sulfhydryl compounds containing thiol residues with reversible oxidation–reduction potential effectively scavenge ROS in a series of biochemical reactions (Foyer and Noctor 2009). The redox buffer GSH protects the cytosol and other cellular compartments against ROS. In the present work, pretreatment with H2S increased the contents of TSC, TSP, cysteine, and GSH and alleviated their decrease under heat stress (Figs. 2, 3, 4, 5, and 6); this is one basis for H2S-induced heat tolerance of tobacco suspension-cultured cells.
H2S-increased antioxidant capacity is being uncovered in various plant species. In strawberry (Fragaria × ananassa), pretreatment of roots with NaHS resulted in expression of genes encoding glutathione biosynthesis enzymes (glutamylcysteine synthetase, l-galactose dehydrogenase, and glutathione synthetase) and key antioxidant enzymes (APX, CAT, SOD, and GR), which maintained high glutathione redox states, which in turn increased the resistance of plants to subsequent salt and non-ionic osmotic stresses (Christou et al. 2013). Similarly, pretreatment of cucumber (Cucumis sativus) seedlings with NaHS differentially activated the total and isozymatic activities and corresponding transcripts of SOD, CAT, GPX, and APX, thus resulting in the alleviation of oxidative damage induced by boron (Wang et al. 2010). In wheat (Triticum aestivum) seedlings, application of NaHS increased the contents of GSH and total glutathione and the activities of APX, GR, dehydroascorbate reductase (DHAR), and γ-glutamylcysteine synthetase (γ-GCS), a key enzyme of GSH biosynthesis under water stress, which in turn decreased the malondialdehyde (MDA) accumulation induced by water deficiency compared to the control without NaHS treatment (Shan et al. 2011). Pretreatment with exogenous NaHS increased the activities of SOD, CAT, APX, and GPX in wheat seedlings and reduced the Cr-induced increase in overproduction of MDA and H2O2, which alleviated the decrease in the germination rate of wheat seeds under Cr stress in a dose-dependent manner (Zhang et al. 2010a). In addition, NaHS treatment alleviated the ROS burst and cell damage induced by salt, osmotic, and cold stress by modulating the metabolism of several antioxidant enzymes (CAT, POD, and GR), the non-enzymatic glutathione antioxidant pool, and the redox state (Shi et al. 2013). In addition to these examples, short-term fumigation of spinach (Spinacia oleracea) seedlings with H2S resulted in a rapid accumulation of TSC, TSP, and GSH (De Kok et al. 1985). In the present study, pretreatment with NaHS increased the level of endogenous H2S, which in turn increased the contents of TSC, TSP, cysteine, and GSH (Figs. 2, 3, 4, 5, and 6) and the activities of SOD, CAT, GPX, and GR (Figs. 7, 8, 9, and 10), which was consistent with previous reports (Zhang et al. 2010a; Shan et al. 2011; Christou et al. 2013). On the other hand, no significant difference in the activity of APX in tobacco cells was observed (Fig. 11), inconsistent with previous reports (Zhang et al. 2010a; Shan et al. 2011), which may be relative to experimental system and plant species.
Under high-temperature stress, all of the antioxidant enzyme activities in treated and untreated cells declined, but SOD, CAT, GPX, and GR activities in NaHS-treated cells were maintained at higher levels than in the corresponding controls (Figs. 7, 8, 9, 10, and 11), and the contents of sulfhydryl compounds TSC, TSP, cysteine, and GSH showed similar trends (Figs. 3, 4, 5, and 6). In contrast to the activities of the other antioxidant enzymes observed, that of APX did not differ significantly from the control under either NaHS treatment or subsequent high-temperature stress (Fig. 11). All of these results showed that H2S treatment could increase the contents of sulfhydryl compounds and the activities of antioxidant enzymes and alleviate their decrease under heat stress, which in turn increased the resistance of plants to heat stress.
Based on the reports cited above, the increase in antioxidant capacity induced by H2S might be achieved through the following pathways: (1) H2S may enhance antioxidant capacity by acting as a precursor of cysteine, which is inserted into sulfhydryl compounds such as sulfhydryl proteins and GSH (Figs. 3, 4, 5, and 6; Riemenschneider et al. 2005). (2) H2S may increase antioxidant capacity by regulating transcription. H2S as a second messenger triggered increases in activities and transcript levels of antioxidant enzymes and biosynthesis enzymes of sulfhydryl compounds (Figs. 7, 8, 9, and 10; Christou et al. 2013). (3) H2S may increase antioxidant capacity through posttranslational modification. As a sulfhydryl group (−SH) donor, H2S activates antioxidant enzymes and biosynthesis enzymes of sulfhydryl compounds by sulfhydrylation (Li et al. 2011).
Conclusion
In summary, the present work demonstrated that exogenously applying NaHS (H2S donor) and its precursors cysteine and KHSO3 to tobacco suspension-cultured cells significantly increased the level of endogenous H2S, which in turn increased the survival percentage of tobacco cells under heat stress, whereas treatment with the H2S scavenger HT in combination with NaHS eliminated the accumulation of endogenous H2S and heat tolerance induced by H2S. In addition, NaHS treatment increased the levels of water-soluble sulfhydryl compounds and the activities of antioxidant enzymes under normal culture conditions and, subsequently, alleviated the decreases of these compounds and enzyme activities in tobacco suspension-cultured cells under heat stress at 43°C. These results suggest that pretreatment with H2S could increase the heat tolerance of tobacco suspension-cultured cells. Further, they suggest that the acquisition of heat tolerance was caused by the increase in contents of water-soluble sulfhydryl compounds and activity of antioxidant enzymes induced by NaHS.
References
Bloem E, Rubekin K, Haneklaus S, Banfalvi Z, Hesse H, Schnug E (2011) H2S and COS gas exchange of transgenic potato lines with modified expression levels of enzymes involved in sulphur metabolism. J Agron Crop Sci 197:311–321
Chen J, Wang WH, Wu FH, You CY, Liu WT, Dong XK, He JX, Zheng HL (2013) Hydrogen sulfide alleviates aluminum toxicity in barley seedlings. Plant Soil 362:301–318
Christou A, Manganaris GA, Papadopoulos I, Fotopoulos V (2013) Hydrogen sulfide induces systemic tolerance to salinity and non-ionic osmotic stress in strawberry plants through modification of reactive species biosynthesis and transcriptional regulation of multiple defence pathways. J Exp Bot 64:1953–1966
De Kok LJ, Bosma W, Maas FM, Kuiper PJC (1985) The effect of short-term H2S fumigation on water-soluble sulphydryl and glutathione levels in spinach. Plant Cell Environ 8:189–194
Foyer CH, Noctor G (2009) Redox regulation in photosynthetic organisms: signaling, acclimation, and practical implications. Antioxid Redox Signal 11:861–905
Foyer CH, Noctor G (2011) Ascorbate and glutathione: the heart of the redox hub. Plant Physiol 155:2–18
Fu PN, Wang WJ, Hou LX, Liu X (2013) Hydrogen sulfide is involved in the chilling stress response in Vitis vinifera L. Acta Soc Bot Pol 82:295–302
Gaitonde MK (1967) A spectrophotometric method for the direct determination of cysteine in the presence of other naturally occurring amino acids. Biochem J 104:627–633
Grant OM, Brennan DP, Salas CDM, Dix PJ (2014) Impact of enhanced capacity to scavenger reactive oxygen species on cold tolerance of tobacco. Int J Plant Sci 175:544–554
Hancock JT, Whiteman M (2014) Hydrogen sulfide and cell signaling: team player or referee? Plant Physiol Biochem 78:37–42
Jaleel CA, Riadhm K, Gopi R, Manivannan P, Ines J, Al-Juburi HJ, Zhao CX, Shao HB, Panneerselvam R (2009) Antioxidant defense responses: physiological plasticity in higher plants under abiotic constraints. Acta Physiol Plant 31:427–436
Jin ZP, Xue SW, Luo YN, Fang BH, Tian HH, Li H, Pei YX (2013) Hydrogen sulfide interacting with abscisic acid in stomatal regulation responses to drought stress in Arabidopsis. Plant Physiol Biochem 62:41–46
Larkindale J, Knight MR (2002) Protection against heat stress-induced oxidative damage in Arabidopsis involves calcium, abscisic acid, ethylene, and salicylic acid. Plant Physiol 128:628–695
Li L, Rose P, Moore PK (2011) Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol 51:169–187
Li ZG (2013) Hydrogen sulfide: a multifunctional gaseous molecule in plants. Russ J Plant Physiol 6:733–740
Li ZG (2015a) Analysis of some enzymes activities of hydrogen sulfide metabolism in plants. Methods Enzymol 555:253–269
Li ZG (2015b) Quantification of hydrogen sulfide concentration using methylene blue and 5,5′-dithiobis(2-nitrobenzoic acid) methods in plants. Methods Enzymol 554:101–110
Li ZG, Gong M, Liu P (2012a) Hydrogen sulfide is a mediator in H2O2-induced seed germination in Jatropha curcas. Acta Physiol Plant 34:2207–2213
Li ZG, Gong M, Xie H, Yang L, Li J (2012b) Hydrogen sulfide donor sodium hydrosulfide-induced heat tolerance in tobacco (Nicotiana tabacum L.) suspension cultured cells and involvement of Ca2+ and calmodulin. Plant Sci 185(186):185–189
Li ZG, Yang SZ, Long WB, Yang GX, Shen ZZ (2013a) Hydrogen sulphide may be a novel downstream signal molecule in nitric oxide-induced heat tolerance of maize (Zea mays L.) seedlings. Plant Cell Environ 36:1564–1572
Li ZG, Yuan LX, Wang QL, Ding ZL, Dong CY (2013b) Combined action of antioxidant defense system and osmolytes in chilling shock-induced chilling tolerance in Jatropha curcas seedlings. Acta Physiol Plant 35:2127–2136
Lisjak M, Srivastava N, Teklic T, Cival L, Lewandowski K, Wilson I, Wood ME, Whiteman M, Hancock JT (2010) A novel hydrogen sulfide donor causes stomatal opening and reduces nitric oxide accumulation. Plant Physiol Biochem 48:931–935
Lisjak M, Teklic T, Wilson ID, Whiteman M, Hancock JT (2013) Hydrogen sulfide: environmental factor or signalling molecule? Plant Cell Environ 36:1607–1616
Liu J, Hou L, Liu G, Liu X, Wang X (2011) Hydrogen sulfide induced by nitric oxide mediates ethylene-induced stomatal closure of Arabidopsis thaliana. Chin Sci Bull 56:3547–3553
Mittler R, Finka A, Goloubinoff P (2012) How do plants feel the heat? Trends Biochem Sci 37:118–125
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassay with tobacco tissue cultures. Physiol Plant 15:473–479
Rausch T, Wachter A (2005) Sulfur metabolism: a versatile platform for launching defence operations. Trends Plant Sci 10:503–509
Riemenschneider A, Nikiforova V, Hoefgen R, DeKok LJ, Papenbrock J (2005) Impact of elevated H2S on metabolite levels, activity of enzymes and expression of genes involved in cysteine metabolism. Plant Physiol Biochem 43:473–483
Saidi Y, Finka A, Goloubinoff P (2011) Heat perception and signalling in plants: a tortuous path to thermotolerance. New Phytol 190:556–565
Scheibe R, Dietz KJ (2012) Reduction–oxidation network for flexible adjustment of cellular metabolism in photoautotrophic cells. Plant Cell Environ 35:202–216
Shan CJ, Zhang SL, Li DF, Zhao YZ, Tian XL, Zhao XL, Wu YX, Wei XY, Liu RQ (2011) Effects of exogenous hydrogen sulfide on the ascorbate and glutathione metabolism in wheat seedlings leaves under water stress. Acta Physiol Plant 33:2533–2540
Shi H, Ye T, Chan Z (2013) Exogenous application of hydrogen sulfide donor sodium hydrosulfide enhanced multiple abiotic stress tolerance in bermudagrass (Cynodon dactylon (L). Pers.). Plant Physiol Biochem 71:226–234
Wahid A, Gelani S, Ashraf M, Foolad MR (2007) Heat tolerance in plants: an overview. Environ Exp Bot 61:199–223
Wang BL, Shi L, Li YX, Zhang WH (2010) Boron toxicity is alleviated by hydrogen sulfide in cucumber (Cucumis sativus L.) seedlings. Planta 231:1301–1309
Wang YQ, Li L, Cui WT, Xu S, Shen WB, Wang R (2012) Hydrogen sulfide enhances alfalfa (Medicago sativa) tolerance against salinity during seed germination by nitric oxide pathway. Plant Soil 351:107–119
Zhang H, Hu LY, Hu KD, He YD, Wang SH, Luo JP (2008) Hydrogen sulfide promotes wheat seed germination and alleviates oxidative damage against copper stress. J Integr Plant Biol 50:1518–1529
Zhang H, Hu LY, Li P, Hu KD, Jiang CX, Luo JP (2010a) Hydrogen sulfide alleviated chromium toxicity in wheat. Biol Plant 54:743–747
Zhang H, Hua SL, Zhang ZJ, Hua LY, Jiang CX, Wei ZJ, Liu J, Wang HL, Jiang ST (2011) Hydrogen sulfide acts as a regulator of flower senescence in plants. Postharvest Biol Technol 60:251–257
Zhang H, Tang J, Liu XP, Wang Y, Yu W, Peng WY, Fang F, Ma DF, Wei ZJ, Hu LY (2009a) Hydrogen sulfide promotes root organogenesis in Ipomoea batatas, Salix matsudana and Glycine max. J Integr Plant Biol 51:1086–1094
Zhang H, Wang MF, Hua LY, Wang SH, Hua KD, Bao LJ, Luo JP (2010b) Hydrogen sulfide promotes wheat seed germination under osmotic stress. Russ J Plant Physiol 57:532–539
Zhang H, Ye YK, Wang SH, Luo JP, Tang J, Ma DF (2009b) Hydrogen sulfide counteracts chlorophyll loss in sweetpotato seedling leaves and alleviates oxidative damage against osmotic stress. Plant Growth Regul 58:243–250
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31360057). We appreciate the editors and reviewers for their exceptionally helpful comments about the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: David Duncan
Rights and permissions
About this article

Cite this article
Li, ZG., Long, WB., Yang, SZ. et al. Involvement of sulfhydryl compounds and antioxidant enzymes in H2S-induced heat tolerance in tobacco (Nicotiana tabacum L.) suspension-cultured cells. In Vitro Cell.Dev.Biol.-Plant 51, 428–437 (2015). https://doi.org/10.1007/s11627-015-9705-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11627-015-9705-x