
Abstract
Long-term storage of cell stocks insures that cells are available for use whenever needed. Cryopreservation of cells is the method of choice for preservation of important or rare cell stocks. There are several factors to consider when establishing a protocol for freezing, thawing, and recovery of cells after storage. These parameters may include cell concentration, cryoprotectant choice and concentration, and thawing rate among others. Further, the assessment of cell viability and/or function prior to and following cryopreservation is imperative in order to accurately determine downstream utility as well for optimizing the cryopreservation process. This chapter is designed to provide guidance and insight into developing robust and successful protocols for preserving cells that will preserve cell stocks and provide optimal cell yield and viability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the past half-century, the fundamentals of the science of cryobiology have evolved to the point where low temperatures are now used extensively as a means to protect and preserve biological systems during enforced periods of ischemia and hypoxia. In practice, preservation is achieved using either hypothermia without freezing or cryopreservation in which the aqueous system sustains a physical phase change with the formation of ice. Survival of cells from the rigors of freezing and thawing in cryopreservation procedures is only attained by using appropriate cryoprotective agents, and in general, these techniques are applicable to isolated cells in suspension or small aggregates of cells in simple tissues. More complex tissues and organs having a defined architecture are not easily preserved using conventional cryopreservation techniques; this is principally due to the deleterious effects of ice formation in an organized multicellular tissue (Pegg et al. 1979; Pegg et al. 1987; Karlsson and Toner 1996).
Temperatures below the normal physiologic range (37°C ± 1), but above freezing, are often employed in medicine to minimize the deterioration of biological materials that occurs in vitro by lowering metabolic rates and reducing the demand for oxygen and other substrates. Hypothermic storage has proved to be very useful for short-term preservation such that cells, tissues, and organs used for transplantation and tissue engineering are often subjected to hypothermia during shipping, processing, storage, and product distribution. Unfortunately, hypothermic techniques are generally limited at present to 1 or 2 d at most, which is not adequate for effective banking and limits distribution.
Longer-term stabilization of biological materials requires the utilization of temperatures below the freezing point of water preferably below glass transition temperature (Tg) of water (− 140°C, nominally). If tissues are to be stored at such low temperatures, the exposure to hypothermia must be minimized and the conditions of exposure optimized (Brockbank 1995).
All methods currently known to be of use for storage and stabilization of cells and tissues involve the application of cryobiology (Karow 1981; Mazur 1984; Brockbank 1995). Cryobiology may be defined as the study of the effects of temperatures lower than normal physiologic ranges upon biologic systems. Simply freezing cells or tissues results in dead, non-functional materials. Little advance was made in the field of cryopreservation until Polge et al. (Polge et al. 1949) discovered the cryoprotective properties of glycerol for biological materials during freezing. Subsequently, Lovelock and Bishop (Lovelock and Bishop 1959) discovered that dimethyl sulfoxide (DMSO) was also a cryoprotectant and it is the most widely used cryoprotectant compound to date. Nevertheless, successful cryopreservation is dependent on much more than simply adding a cryoprotective agent to the cells prior to freezing, and there is now a considerable understanding of the range of variables that must be controlled and optimized to derive an effective method of cryopreservation.
Not only physical changes, water forming ice, but also chemical changes take place as the temperature is reduced and freezing occurs that subsequently affect the viability and survival of cells and tissues upon thawing. As the temperature is reduced, heat is removed and molecular processes are slowed which leads to a variety of structural and functional changes within the cells even before freezing. As a consequence, the cell experiences a cascade of biochemical and biophysical changes that sensitize the cell to further injury and can lead to irreversible damage (Baust, 2007a, b; Taylor 2007).
For example, the rate at which cells are cooled and re-warmed is known to be a major determinant of cell survival. As the temperature of the system is reduced, ice forms initially in the extracellular space. Pure water separates as ice crystals so that remaining solutes are concentrated in the remaining liquid phase. As a consequence, water moves across the plasma membrane and out of the cell in an effort to reestablish osmotic equilibrium within the extracellular space. If the cells are cooled too rapidly, less time is allowed for water to move out of the cells and intracellular ice can to form which causes irreparable damage to the cell. If cells are cooled too slowly, more water is allowed to leave the cells increasing the solute concentration within the cell (Fig. 1). This increase in solute concentration both inside and outside the cell has been termed “solution effects” injury because it encompasses a number of changes that include increase in salt concentrations which can denature proteins and membranes, precipitation of buffers, pH changes, and increased concentration of proteins allowing for the possibility of cross-linking or simple removal of structurally important water. During re-warming, the process is reversed, ice is replaced with water, and cryoprotective agents (CPA) are removed from the system. However, physical and chemical changes to bring the cells back to physiologic temperature can still cause damage. As the sample is warmed, recrystallization can occur. Recrystallization is when metastable ice crystals formed during freezing are given an opportunity to reform larger crystals during re-warming. These ice crystals can cause damage to the cells in a similar manner as those crystals that were formed during freezing. Another concern during re-warming is the removal of the cryoprotectants. The CPAs were added to the samples prior to freezing, and for compounds like DMSO, they replace the water that has been removed from the cells. As DMSO does not move across the cell membrane as readily as water, an imbalance can develop so that the cells will tend to take up water faster than the DMSO is removed causing swelling. Too much swelling can cause irreversible damage to the cell so even if the freezing protocol worked, if the re-warming is not controlled appropriately, cell survival will still not be very good.

Freezing strategies for cells in suspension. If the cells are cryopreserved by freezing, ice forms initially in the extracellular environment and the cells undergo cooling rate-dependent shrinkage due to osmotic dehydration. The slower the cooling rate, the longer intracellular water has the opportunity to move out of the cell by osmosis due to the increasing osmolality of the extracellular environment as water is incorporated into ice crystals. The cells also become concentrated at slower cooling rates as they are pushed together by the forming ice (a, b). Maximum cell viability is usually achieved at an intermediate cooling rate (b) that balances osmotic dehydration and the risk of intracellular ice formation. Rapid cooling (c) permits intracellular ice formation and usually leads to cell death upon re-warming. Very slow cooling (a) may lead to excessive cell dehydration and cell death (Coger et al. 1995).
In addition to the physical impacts of freezing (ice formation, solute concentration, and freeze-induced dehydration) on samples, there is also a biochemical/molecular-based impact. The biochemical and molecular impact on sample quality is a result of several factors including (1) a decrease in available energy and metabolic activity, (2) biochemical pathway uncoupling, and (3) activation of stress response pathways (Baust et al., 2016a, b). During the storage process, the reduction in temperature results in a slowing of metabolism and biochemical reaction/pathways as a result of decreases in kinetic energy levels (a decrease of 10°C equals a 3% decrease in available thermal energy) (Baust, 2007a, b). For mammalian cells, a simple measure which represents an average of the total change in metabolism is Q10 equating to a ~ 50% decrease in metabolism (O2 consumption) for every 10°C drop in temperature (Q10 of 2) (Fuhram and Fuhram 1959; Raison 1973). In essence, low temperature storage slows, but does not stop, all cellular biochemical reactions until the temperature drops below the glass transition temperature (Tg) of water (− 140°C, nominally). As biochemical processes are temperature dependent, even at − 20 and − 80°C, biochemical reactions still occur, to a degree, resulting in continued damage to samples. These reactions are often unregulated and incomplete resulting in the formation and accumulation of toxic intermediate compounds, such as free radicals, anaerobic metabolism byproducts, and waste products which cannot be cleared due to temperature suppression of the necessary salvage pathways or reactions. These byproducts can result in either direct damage or the activation of molecular-stress responses following thawing. Further complicating the equation is that inherent in cryopreservation is that many of the stressors experienced by a sample, if not immediately lethal, can initiate molecular death or biochemical degradation cascades upon thawing. These factors include metabolic uncoupling, free radical production, alterations in cell membrane structure and fluidity, dysregulation of cellular ion balance, release of calcium from intracellular stores, osmotic fluxes, and cryoprotective agent exposure. This list is not comprehensive but serves as an overview of the complexity of biochemical/molecular mechanisms activated during sample storage. For example, free radical production and accumulation during storage can directly damage DNA, protein, and mitochondria integrity in cells and isolated samples (O'Flaherty et al. 1997; Fujita et al. 2005; Baust et al., 2016a, b). In many cases, the accumulation of these sub-lethal stressors during cryopreservation results in the activation of apoptosis followed by a shift to secondary necrosis due to a lack of energy and continual “build up” of stressors upon warming. The manifestation of this sub-lethal damage may not be evident immediately after thawing but may take several hours to days to be detected (Baust et al. 2001). A phenomenon referred to as cryopreservation-induced delayed-onset cell death. This becomes an important factor when examining post-thaw viability as multiple assays and time points may be necessary in order to obtain an accurate determination of sample survival and function.
All these factors affect the overall survival of cells during cryopreservation. Some cells can be cryopreserved readily (i.e., hematopoietic stem cells) while other cell types are much more sensitive, such as hepatocytes. Therefore, optimization for a given cell type may be required.
Cryopreservation is a routine method for long-term storage of cell cultures. It provides a backup for active cultures and also helps prevent drift that can occur when cells stay in culture too long. Cryopreservation of many cell types is straightforward. However, other cell types can be harder to cryopreserve and may require special handling and alternate cryopreservation conditions. A discussion of the process of cryopreserving cells is presented, including factors that can affect the success of a cryopreservation protocol. Some factors to be considered are cell concentration, and cryoprotectant choice and concentration, among others. Once cryopreserved, the cells must be thawed appropriately or viability and yield will be low. Along with a discussion of the parameters that need to be considered for successfully cryopreserving the cells, further discussion is included that outlines factors to be considered when thawing cells and placing them back in culture for recovery and may include thawing rate and appropriate dilution of cryoprotectants. Further, the accurate assessment of cell viability and/or function prior to and following cryopreservation is imperative in order to properly determine downstream utility as well for optimizing the cryopreservation process. Taken together, consideration of these factors for cryopreserving, thawing, and recovery of cells will insure that an optimal protocol is developed.
Cryopreservation of Cells
Cell concentration
The concentration of cells most commonly used is 1 × 106 cell/mL. However, cell concentration can vary with cell concentrations ranging from between 5 × 105 to 1 × 107/mL. Cell concentration can be determined by the viability of the cells after thawing, the concentration of the cells to be cryopreserved, and even how much storage space is available. If viability is not an issue, a lower concentration may be preferable to provide a greater number of vials to help maintain the cell stock for long-term use. Alternately, if viability or cell density is an issue, having enough viable cells after thawing, then cryopreserving at a larger cell concentration may be required. However, a large concentration of cells in a limited volume can also affect cell viability. Called the packing effect, it is hypothesized that too tightly packed cells can experience mechanical stresses that otherwise do not occur, when the cells are at lower concentrations (Pegg et al. 1984; DeLoecker et al. 1998). So, a balance must be reached that accommodates the cell concentration required with a volume that can be cryopreserved.
Sample volume
As mentioned previously, the volume is dictated by the desired cell concentration for cryopreservation. One should not assume that if a greater cell concentration is being used, a greater concentration of cryoprotectant is required. The cryoprotectant concentration is based on what is optimal for 1.0 mL to generally work well and is easily cryopreserved in standard cryovials. Large volumes, around 80–100 mL used when cryopreserving cells in bags, will allow for more cells to be cryopreserved, but larger volumes also mean there can be more issues that can affect its cooling as well as warming. A larger volume is harder to cool at faster cooling rates. If multiple samples, particularly a large number of individual samples, are involved, it is harder for all the samples to cool at the same rate, and it is harder to coordinate ice nucleation in multiple samples. It may be better to cryopreserve a large number of samples in multiple runs with a smaller number of individual samples per run to insure that each sample will experience a more uniform and consistent cooling rate across samples. Whatever the volume that is used, be aware that some optimization may be necessary.
Cryoprotectant choice and concentration
Cryoprotectant selection for cryopreservation is usually restricted to those that have conferred cryoprotection in a variety of biological systems (dextrans, DMSO, ethylene glycol, glycerol, hydroxyethyl starch, polyvinylpyrrolidone, sucrose, and trehalose). On occasion, combinations of two cryoprotectants may result in additive or synergistic enhancement of cell survival (Karow 1981). A comparison of chemicals with cryoprotectant properties reveals no common structural features. These chemicals are usually divided into two classes. Intracellular cryoprotectants with low molecular weights penetrate and permeate cells. Cryoprotectants, such as glycerol and DMSO at concentrations from 0.5 to 3.0 M, are effective in minimizing cell damage in slowly frozen biological systems (Fig. 2). Extracellular cryoprotectants have relatively high molecular weights (greater than or equal to sucrose [342 Da]) that neither penetrate nor permeate cells. These agents are more effective at protecting biological systems cooled at rapid rates. They are often large macromolecules which affect the properties of the solution to a greater extent than would be expected from their osmotic pressure. They have a direct protective effect on the cell membrane which may be due to the oncotic forces (colloidal osmotic pressure) exerted by large molecules and alterations in the activity of the unfrozen water caused by hydrogen bonding to water molecules. Cryoprotectants and their mechanisms of action have been the subject of a number of excellent reviews (Polge et al. 1949; Brockbank 1995; Karlsson and Toner 1996).

Cell viability after cryopreservation with and without 10% serum. (Adapted from Campbell and Brockbank, 2007).
The most commonly used cryoprotectant for preserving cells is dimethyl sulfoxide or DMSO. Glycerol is another commonly used cryoprotectant and is generally used when cells do not preserve well in DMSO. For most cell types, DMSO is a great cryoprotectant and demonstrates good viability when used to cryopreserve cells. There seems to be a great debate about the quality of DMSO to be used for cryopreservation and depends largely on what a researcher has been taught. A review of the literature does not provide any studies to corroborate any one opinion concerning DMSO quality. DMSO is a great cryoprotectant. It has the ability to take up water over time which can have consequences depending on how it is being used. For the purposes of cryopreservation, the general opinion seems to be that this is not a concern and does not have an effect on freezing cells. Most cryopreservation solutions only use a percentage of DMSO that is mixed with an aqueous solution. Any water that has been taken up by the DMSO does not cause an appreciable change in the DMSO concentration of the solution nor does it seem to affect cell viability during cryopreservation. Some researchers use only high-grade DMSO provided in glass vials for single use, while others may aliquot their DMSO and freeze it for future use. Still, others keep DMSO on the shelf at room temperature and make their cryoprotectant solution fresh or even store a ready-to-use solution in the refrigerator. Recommended practice for use of DMSO would be to keep smaller volumes available and store it in a desiccant chamber at room temperature to reduce the uptake of water by DMSO over time. Then, make your cryoprotectant solution fresh and use it right away.
One of the more interesting alternative cryoprotectants being used besides DMSO is sugars, specifically disaccharide sugars such as trehalose or sucrose. The cryoprotective capabilities of some sugars, disaccharide sugars in particular, have been known for years, and early work with them demonstrated their ability to protect proteins and membrane vesicles during freezing (Rudolph and Crowe 1985; Crowe et al. 1990). Coupled with these early studies are observations made in nature regarding organisms that can survive extremes in temperature and desiccation due to their ability to accumulate large amounts of disaccharide sugars, specifically trehalose and sucrose, until more favorable conditions are available. The protective effects of trehalose and sucrose have been determined and may be classified under two general mechanisms: (1) “the water replacement hypothesis” or stabilization of biological membranes and proteins by direct interaction of sugars with polar residues through hydrogen bonding and (2) stable glass formation (vitrification) by sugars in the dry state (Crowe et al. 1987; Crowe et al. 1988; Slade and Levine 1991; Crowe et al. 1998; Crowe et al. 2001).
For cryopreservation, as cells are cooled, water is removed from the system as ice. This can include water associated with the cell membrane. The loss of structural water from the membrane can cause it to undergo a phase transition from its normal liquid crystalline state to a gel phase which usually occurs at a specific temperature (Tm) depending on the solution in which the cells are suspended. During this transition to the gel phase, the membrane becomes leaky so that intracellular contents can leak out. Addition of disaccharide sugars, in particular trehalose, depresses Tm so that the membrane can remain in the liquid crystalline state even when dried, so that upon rehydration, no phase transition takes place and no transient leaking. During cryopreservation, water is not necessarily lost, but it undergoes a phase change forming ice as the temperature drops and depending upon the rate of cooling, the cells become more or less dehydrated, rendering the cells vulnerable to damage by mechanisms similar to those proposed for desiccated cells.
The stabilizing effect of these sugars has been shown in a number of model systems, including liposomes, membranes, viral particles, and proteins (Crowe et al. 1988, Crowe et al. 1989a, Crowe et al. 1989b). Although not as well understood, a similar mechanism of action stabilizes proteins during drying (Carpenter et al. 1986, Carpenter et al. 1987a, Carpenter et al. 1987b, Carpenter and Crowe 1989). Despite their protective qualities, the use of these sugars in mammalian cells has been somewhat limited mainly because mammalian cell membranes are impermeable to disaccharides or larger sugars and there is strong evidence that sugars need to be present on both sides of the cell membrane in order to be effective (Eroglu et al. 2000; Crowe et al. 2001; Beattie et al. 1997). However, disaccharide sugars do have benefits when used as strictly extracellular cryoprotectants including the ability to form a glass when cooled at fast cooling rates.
The cryoprotectant concentration is dictated by the cell being cryopreserved. As mentioned above, most cells can be cryopreserved in DMSO, but if not, then there are many alternative cryoprotectants that can be used. For most cell types, using DMSO as the cryoprotectant of choice at a concentration of 5–10% works well. Concentrations of DMSO up to 20% have been used successfully. However, as the concentration of cryoprotectant increases, the degree of osmotic changes that can occur during the loading and unloading of the cryoprotectant must be considered as these osmotic changes can also cause damage to the cell. A compound like DMSO is able the pass through the membrane, but it does not pass as quickly as water. A temporary osmotic imbalance can occur because water will move out of the cell more quickly than the DMSO can move in. Equilibrium is eventually reached, but the osmotic imbalance must be kept to a minimum to prevent cellular damage. Addition and removal of the cryoprotectant in sequential stages of increasing cryoprotectant concentration for loading and decreasing cryoprotectant concentration for unloading will minimize any osmotic imbalance and keep cellular damage to a minimum. Another important point is to keep the cells and solutions on ice during the loading/unloading to avoid unwanted cytotoxicity from the cryoprotectants being used. Generally, solutions less than 2 M in concentration do not require a loading/unloading regimen. A 10% DMSO solution, which is 1.4 M concentration, does not necessarily require sequential loading.
It is usually a good idea to cryopreserve your cells of interest at several concentrations of your cryoprotectant choice to be confident that the best formulation is used to produce the best cell viability and yield after cryopreservation. There should be a high enough cryoprotectant concentration to provide adequate protection to the cells; however, a cryoprotectant concentration that is too high can be cytotoxic to the cells and will reduce your cell viability and yield. Also, be aware that the number of vials to be processed needs to be considered. Even when cells are kept on ice in cryoprotectant solution, cytotoxicity caused by exposure to the cryoprotectants can occur and affect cell viability after thawing. It is best to keep the exposure time minimal.
Use of serum during cryopreservation
Many researchers routinely use serum in their culture medium for growing cells. Frequently, the cryopreservation solution that is used to cryopreserve the cells is cryoprotectant added to culture medium which in the past usually contained serum. The presence of serum in culture medium promotes cell growth. The inclusion of serum during cryopreservation is helpful to cell recovery upon thawing. Some researchers will even use a cryoprotectant with only serum for cryopreservation. In recent years, serum-free media have been developed and are being used widely for a variety of cell types. With this development, there is a need to be able to cryopreserve cells in the absence of serum. Some cells do not do well with serum when cryopreserved, and some cells are required for cellular therapy and so cannot be exposed to animal-based serum. There is literature demonstrating the successful cryopreservation of cells in the absence of serum although a little more troubleshooting may be required depending on the cell type (Baust et al. 2000; Stylianou et al. 2005; Campbell and Brockbank 2007; Campbell and Brockbank 2010). The decision to use serum in your cryoprotectant formulation is largely based on how the cells have been preserved in the past and what future applications may be.
Vehicle solution
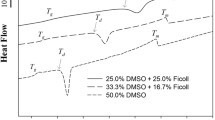
Instead of using serum in cryoprotectant solutions, solutions developed specifically for maintenance of cells at cold temperatures are now being used. Cell culture medium is designed to meet the needs of cells under physiological conditions so that they can grow and divide. As cells are cooled, their needs change and the metabolic activity of the cells also changes (Baust, 2007a, b; Taylor 2007). Initially, solutions were designed for the cold storage of whole organs. Hypothermic organ storage solutions such as Viaspan, HypoThermosol®, Euro-Collins (EC), and Unisol™ (UHK) were designed to compensate for the changing environment cells experience when temperatures drop below 37°C (Taylor 2007). Cells undergoing cryopreservation pass through a period of hypothermia before, during, and after freezing (hypothermic continuum, Baust, 2007a, b), so the role of the vehicle solution for CPAs as an interactive factor that influences the outcome of cryopreservation needs to be considered when setting up a cryopreservation scheme for your cells of interest. As can be seen from the graph, a comparison of two vehicle solutions demonstrated the impact the choice of vehicle solution can have on the viability of cryopreserved cells (Fig. 3) (Baust et al. 2000; Taylor et al. 2001). While many cell types are amenable to being cryopreserved in culture medium plus cryoprotectant, a cryoprotectant solution that does not use culture medium or serum is a good alternative choice. Some trial and error may be required to determine the best cryopreservation solution to use.

Cell viability as a function of vehicle solution and cryoprotectant concentration. The data illustrates the effect of carrier solution on cell survival. (Adapted from Taylor et al., 2001).
Cooling profile
Cooling rate is one of several variables that can impact cell survival and is usually cell type dependent. During the cooling process, freezing may be induced at any point below the equilibrium freezing point. Samples can cool beyond their freezing point before ice forms. This is often referred to as supercooling. Nucleation represents the change of liquid water to ice. Water in the system separates out from the remaining unfrozen solution and forms ice, which increases the concentration of solutes. Initiating freezing is also associated with an energy change. A localized rise in temperature, known as the latent heat of fusion, evolves as the system returns to the equilibrium freezing point of the solution (Fig. 4).

Schematic cooling curve showing nucleation and latent heat of fusion (Brockbank et al. 2007).
Samples that supercool to a significant extent undergo a more rapid cooling rate following latent heat evolution (as the sample temperature attempts to catch up with the lower temperature of its environment) than samples that nucleated near their freezing point which can affect cell viability upon thawing. Slower cooling rates, while discouraging under-cooling of the samples and the formation of ice inside the cell, allow for longer exposure times to cryoprotectants and time for osmotic changes to occur that promote cell shrinkage which conceivably have a negative impact on the stability of the cell-substrate interactions. Faster cooling rates reduce exposure to the cryoprotectant and osmotic changes to the cell but do allow supercooling to occur and can result in increased intracellular ice formation, which can be highly lethal. A balance between these two extremes has to be optimized in order for maximum cell survival to occur. A system such as 96-well plates presents unique challenges in terms of uniform cooling across the plate as well as providing an optimum cooling rate for a particular cell type. The freezing profile or freezing rate is determined by the cell type. For most cells, a cooling rate of − 1.0°C/min is adequate to produce viable cells (Fig. 5). This can be achieved using a controlled-rate freezer (CRF), which can be programmed to deliver a desired rate for freezing your cells. It can also be programmed to induce ice nucleation in the sample, which prevents super-cooIing and also initiate ice nucleation in multiple samples simultaneously for better uniformity across samples. An alternative method to controlled rate cooling is the use of an alcohol-based freezing container, such as Mr. Frosty. Mr. Frosty is an insulated container that is partially filled with isopropanol. Samples are placed into a tube holder inside the container and then the entire container is placed at − 80°C. The container cools at a controlled rate which is approximately − 1.0°C/min. Samples can then be removed from Mr. Frosty and stored long term in vapor phase liquid nitrogen. A less consistent alternative that can be employed involves using any kind of insulated container, for example, a Styrofoam box. Samples are put in the box which is then placed in a − 80°C freezer. This will produce a more controlled, slower rate of cooling, but it is not as consistent or reliable as using a controlled-rate freezer or even the Mr. Frosty. Some cell types do prefer a faster or slower cooling rate. This information can sometimes be found in the literature, or you will have to determine what works best empirically by trying several cooling rates.

Cooling rates for various cell types. Adapted from the original figure (Mazur 1984).
Storage conditions
Once cryopreserved, cells should be stored in vapor phase liquid nitrogen. Storing cells at warmer conditions, such as − 80°C, may allow for changes to occur over time that reduce the viability and functionality.
Best practices
Most cell types can be cryopreserved without difficulty using a standard protocol with 5–10% DMSO in culture medium. If this standard protocol does not work, then begin optimizing conditions until the desired results are achieved. More sensitive cell types may require more optimization to develop an adequate cryopreservation protocol. Things to remember when cryopreserving cells:
-
Use a cryopreservation solution made fresh either the same d or the d before
-
Specialized cryopreservation carrier medium may be considered in situations where increase post-thaw cell yield and viability is desirable.
-
Add the cryoprotectant solution to cells and place on ice. Leave the samples on ice until you are ready to cool them down to storage temperature in a controlled rate freezer or Mr. Frosty. This reduces potential cytotoxicity associated with the cryoprotectant solution.
-
Start by using DMSO and optimize the concentration first, then look to alternative cryoprotectants. If further optimization is still needed, then start optimizing other cryopreservation variables as mentioned in the text.
-
A higher cell density will not usually improve your cell yield after thawing. An optimized cryoprotectant solution and cooling strategy will do more to improve cell viability and yield.
-
Cooling samples using a controlled rate (typically 1°C/min) increases sample-to-sample and lot-to-lot consistency.
Thawing and Recovery of Cells
As with freezing, the thawing process has critical impact on sample quality and downstream utility (Baust et al., 2016a, b; Miyazaki and Suemori 2016; Yong et al. 2016). While relatively straightforward, there are a number of aspects of the thawing process which critically impact overall sample quality. These include air time, thaw temperature/rate, final sample temperature, thaw time, mixing consistency, and dilution process. Other issues to be cognoscente of include sterility, consistency, controllability, documentation, and cleanliness. Generally, the thaw process should be conducted as quickly as possible with minimal air time, rapid warming of a sample until the point ice has dispersed (nominally 0°C) with continuous mixing, followed by immediate dilution to reduce sample cytotoxic damage from the cryoprotective agents contained within the freeze medium.
Thawing strategies
Today, the gold standard within the cell therapy and research community for thawing samples (whether in vials, bags, straws, or other formats) is the use of a warm (37°C) water bath. While a water bath is the most common device/strategy used for thawing, there are a number of devices which can be utilized depending on the type, size, and end use of the sample. It is noteworthy that as a general rule for all frozen eukaryotic cell systems, passive sample thawing in air is not recommended as this will most likely result in complete sample loss. An overview of several thawing technologies is provided below.
Water bath thawing
Warm water baths represent the standard approach to frozen product thawing. In the research lab, simple procedures such as removing a vial from the storage freezer or dewar, immediately placing into a warm water bath, and gently agitating (mixing) for 3–5 min until thawed is standard protocol. This process requires active and continual manipulation of the sample during the thaw process to attain an optimal outcome. It is important to recognize that the hands-on requirement of this process has several risks including exposure extreme temperatures, potential cryogen outgassing, and container leakage. Further, during the water bath thawing process, a researcher must also be cognizant of the potential contamination, miss-handling (dropping or submerging), overheating, non-uniform thawing, etc. In clinical or clean room settings, the water bath is often replaced with a sterile basin filled with warm sterile water or saline solution in an effort to reduce the risk of contamination of the product, the sterile field, and potentially the patient.
Dry thawing systems
The second, less common approach to sample thawing is the use of dry thawing devices. These devices are designed to replace a water bath and provide for a more uniform, clean/sterile and in some cases documented thawing process for frozen samples. Several dry thawing systems integrate in an agitation or movement feature during the thawing to provide for a continuous and uniform mixing of the sample during thawing. Agitation during the thawing process is critical as it reduces the thermal gradients during the thaw which can be detrimental to sample quality post-thaw. Dry thawing systems currently available include DTD device (Core Dynamics), Plasmatherm (Genesis BPS), Plasmatherm (Barkley GmBH), SaharaIII (Sarstedt), ThawStar (Biocision), SmartThaw (CPSI Biotech), and Cytotherm -D Series (Cytotherm). Today, the most common area of use for dry thawing systems is the clinical blood banking arena for the thaw processing of frozen blood component and plasma products. However, the use of dry thawing systems in other areas, including cell therapy, bioprocessing, and even in the research laboratory, is growing at an exponential rate as the benefits of improved sterility, uniformity, and traceability and the elimination of end user (operator) error are being realized in process and sample quality improvement.
Thaw process stages and considerations
ᅟ
Air time
One phase of the thaw process which has tremendous impact on sample quality is that of air time. Air time is defined as the time between removal from storage (typically LN2 or − 80°C) and placement into a thawing apparatus. During this interval, a sample is exposed to passive warming resulting in a slow and prolonged thaw rate. Extended intervals of passive thawing (nominally greater than 30 s) can result in significant sample compromise. Given this, it is important that air time be kept to a minimum. There are several strategies to minimize air time including (1) positioning of the storage container and thaw apparatus next to each other to allow for direct removal of a sample from a dewar and placement into the thaw device, (2) utilization of a small ice chest of other insulated container containing either dry ice (− 80°C) or LN2 (− 196°C) to serve as a temporary holding container in which samples are placed upon removal from a dewar which then can be transported to the thaw apparatus, or (3) the use of specialized sample cold transport devices designed to maintain samples at an ultra-low storage temperature for a given interval. Such devices include LN2-based dry shippers (Cryo Associates, MiTeGen, MVE, Thermo Fisher Scientific, VWR) and ultra-low temperature transport devices (BioCision, Thermo Fisher Scientific, VWR). It is important to note that sample storage on wet ice (− 20°C) is not effective and should be avoided. Regardless of the approach utilized, control of sample temperature in the interval between removal from storage and placement into a thawing device is of critical importance.
Thaw rate
While freezing cells for cryopreservation requires very slow cooling (~ 1°C/min), thawing cells requires rapid warming rates (~ 50 to 100°C/min) to attain the most viable sample. Thawing of a typical frozen sample (standard 2 mL cryovial, 1.0 mL sample volume, stored in LN2) using a standard 37°C water bath yields an average thaw rate of ~ 65°C/min (− 196 to ~ 0°C in 3 min) (Fig. 6). It is important to recognize that the sample thaw rate is not linear but is characterized by a rapid warming rate within the first 1.5 min (~ 125°C/min) followed by a slower extended sample warming plateau (between ~ − 15 and 0°C) where samples require large input of thermal energy to go through the phase transition from solid to a liquid state (latent heat of fusion). For a typical 1.0 mL sample, this plateau lasts ~ 1.5 min resulting in a thaw rate in this stage of ~ 10°C/min. This plateau can be shortened by using elevated thawing apparatus temperatures; however, this must be balanced with the elimination of potential overheating of the sample. More recently, studies have begun to look more closely at the impact of the thaw rate on sample quality. These studies are suggesting that cell/sample-specific sample thawing rates may provide improved outcomes. To this end, several dry thawing devices allow for customization of the thaw process through the changing of the thaw environment temperature settings providing for warming rate control, similar to how programmable controlled rate coolers allow for freeze profile customization.

Thermal profile for thawing using a 37°C water bath. The warming profile of a sample upon removal from storage and immediate placement into a 37°C water bath is characterized by rapid warming for the initial ~ 1.5 min followed by a slower plateau as the sample moves through a phase change from a solid to liquid state. Once completely melted, if not removed from the bath, the sample will continue to warm which can be lethal to the cells. (adapted from Baust et al., 2016a, b).
Sample agitation
Continuous gentile agitation or mixing of samples during the entire thawing process is important to obtain high-quality samples post-thaw. Mixing of samples during the thaw process reduces the thermal gradient which forms within a sample during warming. This thermal gradient can be very steep with the warmest temperature being on the outside edges of the container and coldest in the center of the samples. If sample mixing is not practiced, during the thaw process, the thermal gradient within a sample can range, for instance, from ~ + 20 to 30°C at the outer edge of the vial to − 20°C in the center around the 2-min interval of a 3-min thaw. This thermal gradient can result in sub-optimal cell recovery for several reasons including overheating of portions of the sample (see next section) as well as due to a non-uniform sub-optimal thawing rate within the sample.
Thaw completion/sample final temperature
Regardless of the technique utilized, sample temperature at the end of the thaw interval is critical. Specifically, following thawing, samples should be cool, not warm (i.e., ~ 0 to 4°C; refrigerated temperatures). This is important as warming to room temperature or warmer exposes samples to a series of stresses, including cytotoxicity from cryoprotective agents, which can be highly lethal. The level of cytotoxicity varies depending on cell type, CPA type, and concentration and with what carrier medium is utilized. For instance, most primary and stem cells are much more sensitive to the final temperature compared to common immortalized cell lines (PC-3 or 3T3 cells). Samples cryopreserved in 10 or 20% DMSO will be more negatively affected by warmer temperatures than samples in 5% DMSO. Finally, samples frozen in specialized cryopreservation media (Unisol, CryoStor, Adesta, etc.) are much more sensitive to over-warming than samples frozen in media or serum + DMSO. This increased temperature sensitivity in the specialty cryopreservation media is due to their formulations being optimized for cellular biochemical and physiological requirements at low temperatures, which are substantially different from those under normothermic culture conditions. Given the sensitivity issues associated with sample over-warming, it is recommended that thawing be stopped as soon as all visible ice dissipates and while samples are still cool (~ 0°C). A common practice is to remove samples when only a sliver of ice remains, which serves as a visual indicator to an end user that the sample is thawed but still cold.
Dilution
The thawing process does not end with the removal of the sample from the thawing apparatus as sample handling until end use, such as placement into culture, will significantly impact outcome. Important aspects of the post-thaw handling process include (1) maintenance of sample temperature and (2) sample dilution. As with final warming temperature, it is important to maintain samples at a cool temperature (~ 4°C) until diluted for final use to minimize CPA and solution cytotoxicity. This is typically attained using a pre-cooled cold block, wet ice, or other means to keep samples at refrigerated temperatures. As with all the other stages of the thaw process, this sample holding time should be kept to a minimum as samples should be diluted and placed into culture, or other end use, as soon as possible upon thawing.
Once thawed, sample dilution should be performed as soon as possible. Several strategies for sample dilution can be utilized depending on sample type, sensitivity, and CPA concentration. These include single-step, stepwise, or gradient dilution. Generally, the dilution process should be designed to avoid or reduce subjecting the cells to severe osmotic shock as the cryoprotective medium is exchanged for growth medium. For samples cryopreserved in solutions with 5% total CPA concentration, the use of a single-step dilution of greater than 1:10 with complete culture media can be utilized. Depending on the cell type, CPA concentrations of 10% can be diluted with either a single or a stepwise (multi-step) dilution. The stepwise dilution procedure helps to reduce the osmotic shock stress which may be experienced. For solutions which contain CPA concentrations greater than 10%, the use of stepwise or gradient dilution procedures are recommended. Generally, a user should target CPA dilution to below 0.5% or less prior to placement into culture. This can be attained either through simple dilution or through the use of centrifugation and media replacement following dilution. Once dilution is complete, samples can be plated and placed into culture.
Other considerations
In addition to the above, sample retrieval and thawing procedures should also be designed to protect (1) the sterility of the culture, (2) the operator from potential explosion of a frozen vial containing LN2, (3) the operator from ultra-cold temperature exposure (frostbite), and (4) reduce the risk of operator exposure to potentially harmful CPA solutions and samples. Finally, protocols should be designed such that the entire process from sample retrieval to placement into culture or other end use is as controlled, documented, and streamlined as possible as any variations and/or delays during the thawing process can greatly affect sample quality.
Best practices
Most cell types can be rapidly thawed using a 37°C water bath without difficulty. While effective, this method is non-traceable and exposes samples to contaminants within the bath. Further, this method is highly susceptible to user variability during the thawing process. If standardization, sterility, and traceability are desired, the use of specialized thawing devices is recommended. Things to remember when thawing samples:
-
Minimize the time samples passively warm between removal from storage and placement in thawing device.
-
If using a water bath, do not completely submerge the vial. Keeping the cap and O-ring region above the water line will reduce the risk of contamination.
-
Provide gentile mixing of the samples during the warming process to minimize thermal gradients within the sample.
-
Remove sample once all ice has melted. Do not allow sample to warm to bath temperature.
-
Keep sample cool (~ 4°C) once thawed prior to dilution.
-
Dilute samples with media in a single or multi-step process (method depending on the cryoprotective agent concentration and cell type).
Cell Viability Assessment—Understanding Cryopreservation Outcome
An accurate assessment, a cell viability following cryopreservation ultimately resides on the quality and reliability of the viability and functional assays used to assess the preservation process. In the field of cryopreservation, viability assessment is continuing to evolve. Beyond viability, questions of sample uniformity, genetic integrity/alterations, and the potential for cell subpopulation selection within a sample also often arise. This section reviews the importance of assessment timing as well as current slate of viability and functional assays which can be applied to determine cell survival and function prior to and following cryopreservation. Today, it is recommended that no single assay should be used to assess viability or function of cells undergoing a preservation process. Further, the unique behavior a particular viability probe must be considered in the context of each cell type and an understanding of the impact of cryopreservation on cell function as well as proteomic and genomic profile may be desirable. As a result, often times, the recommendation is posited that two or three assays (one assay from tiers 1, 2, and 3, defined herein) be employed to best assess outcome. Further, the potential impact of the cryopreservation process on cell function (temporary or long term), molecular alternations (genomic, proteomic, epigenetic, etc.), and/or selection of sub-populations in heterogeneous samples (e.g., PBMC samples) should be appreciated. This information, in turn, will yield an improved understanding of outcome and aid in the optimization of the preservation yielding improved post-thaw cell quality and function. This is of particular importance in areas such as primary human cells, hybridomas, stem cells, cell therapy, gamete preservation, and tissue and organ preservation arenas.
Pre-freeze viability assessment
Sample quality prior to freezing greatly impacts post-thaw outcome (Snyder et al. 2004; Baust et al. 2009). Cell samples which have been stressed in culture (nutrient starved, over confluent, high passage, etc.) prior to freezing respond differently to the cryopreservation process compared to non-stressed (healthy) cells and often result in decreased sample quality post-thaw (yield, viability, and function) (Fuller 2003; Baust et al. 2009). For samples harvested freshly from a patient (not cultures) such as blood cells, tissue biopsies, liquid tumors, etc., it is often challenging to control sample quality; however, it is important to recognize that optimization of each step of the handling process can improve pre-freeze sample quality and, therefore, post-thaw outcome. Remember: garbage in = garbage out.
In order to assess sample quality prior to freezing, a common practice is to determine cell number and viability using the trypan blue or other tier 1 assay (see “Viability assessment tiers” section). During pre-freeze assessment, it is important to determine the overall cell number and sample viability so that proper calculation of cell density can be made. For cryopreservation of a typical cultured cell system (fibroblasts, endothelial cells, CHO, LNCap, etc.), a pre-freeze sample viability greater than 90% should be targeted. Once the total viable cell number is determined, samples can then be suspended in the appropriate cryopreservation media to a final concentration of 1 × 106 cells/mL. Cell concentrations ranging between 0.5 × 106 and 1 × 107 cell/mL are standard utilized in cryopreservation.
In addition to pre-freeze viability and cell number, sample quality assessment can also include baseline determination of cellular metabolic activity level, cell function, or even genomic or proteomic fingerprinting as advanced sample quality metrics.The use of these types of assays is described in more detail in the “Viability assessment tiers” section. These assessments are more commonly used to evaluate sample quality following thawing; however, incorporation of this level of analysis into pre-freeze assessment provides a larger dataset for comparison with post-thaw samples.
Assessment of post-thaw viability
Assessment of cell yield and viability immediately following re-warming of samples using probes such as trypan blue remains today’s “gold standard.” Trypan blue is a simple vital dye-based assay which allows for a quick assessment or “snap shot” of sample status. When using trypan blue as a viability assay for assessment of sample quality following cryopreservation (post-thaw), however, there are several items one must understand to properly frame the information reported by the assay, without which, inaccurate conclusions may be inadvertently reached. Trypan blue measures viability based on membrane integrity. Immediately after cryopreservation, membrane fluidity may lead to false positives and negatives depending on cell type and sensitivity. Undoubtedly, you or someone you know has experienced the scenario of thawing a vial of cells, assessing viability immediately post-thaw, placing a known cell number/seeding density into culture and then the next day, the culture growth does not reflect what was expected. This is due to the fact that a substantial level of continued cell death, which is not apparent immediately post-thaw, manifests over a 24- to 48-h period post-thaw as a result of delayed apoptosis and necrosis (Fu et al. 2001; Abrahamsen et al. 2002; Baust et al. 2002; Heng et al. 2006). This phenomenon has been widely published on and has been termed cryopreservation-induced delayed-onset cell death (CIDOCD) (Baust et al. 2001). Assessment of sample viability within a few hours post-thaw does not account for CIDOCD. The extent and timing of CIDOCD varies and is dependent on a number of factors including cell type and sensitivity, cell cycle time, preservation process utilized, cryopreservation media, concentration of cryoprotective agents, sample status/quality prior to freezing, etc. (Baust et al. 2006; Heng et al. 2009). Even under optimal cryopreservation process and protocols, CIDOCD significantly impacts outcome. As such, it is important to appreciate the impact of cryopreservation processes on both immediate and downstream (next d) viability (Baust et al. 2017).
Another important area is the recognition that cryopreservation can impact cell function (de Boer et al. 2002; Matsushita et al. 2003; ; Sugimachi et al. 2004; Sosef et al. 2005; Stylianou et al. 2005; Greco et al. 2006). Studies have shown that cryopreservation protocols can influence baseline cell function and responsiveness days to weeks following thawing (Sugimachi et al. 2004; Sosef et al. 2005). Further, studies have shown that when cryopreserving heterogeneous cell samples, such as PBMCs, leukapheresis products, whole blood, stem cells, etc., the cryopreservation process can differentially select for cellular sub-populations which may be more tolerant to the molecular and physical stresses experienced during the freeze thaw (Berens et al. 2016; Karimi-Busheri et al. 2016; Yang et al., 2016a, b). This phenomena has also been widely reported on in the area of spermatozoa cryopreservation wherein more robust (motile) sub-population selectively survive the cryopreservation process better than weaker (less motile) sub-population’s (Beirao et al. 2011; Estrada et al. 2017). Recently, the Biomedical Excellence for Safer Transfusion (BEST) Collaborative Group studied different solutions for the cryopreservation of lymphocyte infusion products and demonstrated that the cryopreservation solution utilized impacted the differential survival T-cell subpopulations within a given sample (Worsham et al. 2017). Given this, the BEST panel has recommended optimization of cryopreservation protocols for cell therapy products. While an issue in heterogeneous samples, studies investigating sub-population selection within “homogenous” samples (e.g., culture of given cell population) remain limited. Neyzen et al. (2006) has suggested that a single freeze thaw event under optimal conditions results in minimal differences in biochemical and cellular properties between cryopreserved and fresh HSCs. More recently, several studies have reported that cryopreservation can result in alterations in gene expression and epigenetic changes in samples post-thaw (Kaity et al. 2008; Yang et al., 2016a, b). As such, this is another aspect which should be considered when developing pre-freeze and post-thaw viability assessment protocols. While it is important to recognize that selection and alterations can occur, it is also important to note that this is most concerning when sub-optimal cryopreservation protocols are utilized. For instance, for a given cell type, a protocol which yields < 50% 24 h post-thaw survival is more likely to result in greater sub-population selection and/or molecular alterations than a protocol that yields 80% survival. As such, it is recommended that optimized protocols, yielding the greatest 24 h post-thaw viability possible, even for simple homogeneous cell populations, be employed to reduce the chance of selection and population genetic drift.
Viability assessment tiers
Given the plethora of downstream effects that cryopreservation can have on a cell, coupled with the complex array of cell systems being preserved, the establishment of a set of viability and function assays for sample quality assessment may be warranted (Baust et al. 2006; Van Buskirk et al. 2006). To this end, a four-tier strategy encompassing assessment of membrane integrity, molecular mechanisms, cell function, and biochemical alterations has been introduced and is outlined below (Van Buskirk 2007). This tiered strategy serves as a guide in developing a set of viability and functionality assays for sample assessment. While not all tiers are necessary, an assessment strategy which includes one assay each from tiers 1, 2, and 3 would facilitate comprehensive assessments of viability and function. Depending on the complexity of the cell or tissue system along with end-use application, consideration may be given to developing tier 4 assays as predictors of longer-term post-preservation impact.
Tier 1 assays—membrane integrity assays
Tier 1 assays are often referred to as live/dead assays. These are single endpoint tests that are limited primarily to measuring cell membrane integrity or the loss thereof. The most common tier 1 assay is trypan blue. An overview of the trypan blue assay is provided above. Another common tier 1 assay is the fluorescent probe calcein-AM. Calcein-AM is a membrane-soluble probe which once inside a cell, the AM group (acetoxymethyl (AM) ester group) is cleaved by cellular esterases allowing the molecule to fluoresce as well as trapping the Calcein within the cell. A compromise in cell membrane integrity allows the cleaved Calcein to leak from the cell into the surrounding medium leaving only the intact “viable” cells to fluoresce green. Cell fluorescence can be both qualitatively (fluorescence microscopy) and quantitatively (spectrofluorometer) assessed to determine sample viability. It should be noted that there are a variety of membrane integrity fluorescent probes including propidium iodide, BCECF-AM, and others. Calcein-AM, however, one of the most straightforward fluorescence-based tier 1 assays, demonstrates the highest signal to noise ratio, is least affected by differences in intracellular pH, and in the case of hepatocytes is not chemically altered due to the cytochrome p450 enzymes in cells. The use of tier 1 assays to determine sample viability immediately post-thaw can lead to elevated estimates of yield and survival as sub-populations of cells undergoing CIDOCD will appear viable with tier 1 assays within the first 12–24 h post-thaw.
Tier 2 assays—enzymatic and molecular mechanism assays
Tier 2 assays consist of two categories and include assessment of (1) specific molecular cell death indicators and (2) metabolic activity of samples following thawing. Assessment of molecular death indicators allows for evaluation of the mechanisms underlying the loss of cell viability and function following cryopreservation. These events are often associated with CIDOCD and as such can be utilized to provide a more detailed and accurate assessment of post-thaw survival than tier 1 assays in the initial h following thawing (Baust et al. 2001). This is due to the fact that the indicator events being assessed with tier 2 molecular assays precede the membrane lysis events measured by tier 1 assays. The assay choices for tier 2 assessment is much broader than tier 1 and somewhat dependent on the nature of the analytical question being posed. In assessing post-thaw viability, molecular tier 2 assays allow for the quantitation of cell sub-populations which remain alive versus those undergoing early stages of apoptotic or necrotic cell death. A highly effective assessment strategy to assess the live population versus apoptotic and necrotic populations is using the YoPro or Annexin V/PI assays several hours to 1 day post-thaw. These assays allow for the identification of sub-populations of cells in early stages of cell death which appear “viable cells” when tier 1 assays are applied. Other tier 2 assessment strategies include fluorescence caspase activity assays or mitochondrial status assessment using JC-1 (a fluorescent dye that qualitatively and quantitatively monitors mitochondrial activity).
While molecular assessment provides a detailed understanding and quantitation of the various sub-populations (live, apoptotic, necrotic) within a sample post-thaw, there is a series of tier 2 assays that focus primarily on assessing the living sub-population of cells based on population metabolic activity. A candidate for this type of assessment is a non-invasive metabolic indicator such as alamarBlue. The use of the alamarBlue assay provides for quantitative assessment of cell survival following thawing. This assay can be used repeatedly on the same cultures over the course of days to weeks to monitor the cell viability and repopulation. Post-thaw survival assessment using alamarBlue provides a stringent assessment of cell survival when performed 24 h following thawing. The strategy of using alamarBlue assessment 24 h post-thaw in combination with a tier 1 assay performed immediately post-thaw allows for a highly accurate characterization of sample survival and overall quality.
Tier 3 assays—functional assays
Tier 3 assays focus on cell-specific function. Numerous studies have demonstrated that a loss of function is not coincident with a loss of viability (de Boer et al. 2002; Matsushita et al. 2003; Sugimachi et al. 2004; Sosef et al. 2005; Stylianou et al. 2005; Greco et al. 2006). For instance, hepatocytes can appear perfectly viable as seen throughout long-term, post-preservation follow-up using both tiers 1 and 2 assays, yet the cell function (cytochrome p450, urea production, etc.) can be compromised (Sugimachi et al. 2004; Sosef et al. 2005). Other examples include the cryopreservation of stem cells where studies have shown a significant alteration in CD34+ and clonogenic capacity of umbilical cord blood stem cells (Stylianou et al. 2005; Greco et al. 2006). To assess the impact on cell function, tier 3 assays can be utilized. In some cases, immunological staining for cell-specific markers can be utilized as a functional assay. In other cases, differentiation potential (stem cells), cellular contraction (cardiac myocytes and myoblasts), motility, protein receptor expression (immune cells), detoxification capacity (hepatocytes), or protein secretion (pancreatic beta cells) can be incorporated to assess the downstream impact of cryopreservation on sample survival and function. Thus, in contrast to tier 1 and tier 2 where the trypan blue or calcein-AM and alamarBlue® assays may be applied as standard assays; tier 3 assays must be cell type specific.
Tier 4 assays—proteomics and genomics
The tier 4 Assays represent the newest and most complicated set that may be considered. This tier encompasses proteomics and genomics as an integrated set of assays to monitor molecular alterations, functional changes, and/or sub-population selection/enrichment which may occur within a cell population following thawing. This assessment tier remains under development; however, it offers the potential of establishing sample “viability/function fingerprints,” similar to those utilized in disease diagnostic. These advanced analyses offer the potential of enabling detailed sample-to-sample comparison over time. If such a “viability/functional fingerprint” can be identified, this analysis may improve our ability to assess stem cells and engineered tissues utilized in drug development, bioprocessing, stem cell therapy, and other research and medical applications. Thus, the ultimate value and utility of a tier 4 assay for broad-based analysis remains to be seen.
Assessment timing
One critical component of the utility of data obtained from any assay is the timing of the assessment (Van Buskirk et al. 2006; Baust, 2007a, b). For example, in a cell population following preservation, assay timing can mean the difference between apparent viability or complete cell death. In the assessment of preservation efficacy, measures of viability are typically taken immediately post-storage using tier 1 assays, such as trypan blue or calcein-AM. At this time point, tier 1 assays typically yield high degrees of viability; yet, when the same population is probed after 24 h of culture using the same assay, sample viability is highly reduced (Baust et al. 2000) (Fig. 7). As with tier 1 assays, the time in which tier 2 assays are applied can significantly affect the interpretation of the data. With the tier 2, it is important for an initial analytical time course (i.e., 1, 6, 12, and 24 h or other) to be performed to determine the optimal post-thaw assessment time point for a given system. As a rule of thumb, 1 h post-thaw assessment using tier 1 trypan blue or calcein-AM allows for determination of initial yield and viability for cell seeding purposes, the 4–8-h interval is generally most appropriate for tier 2 molecular assessment of apoptosis and necrosis (YoPro or Annexin/PI) whereas 24-h post-thaw assessment using tier 2 alamarBlue allows for determination of ultimate cell survival.

Illustration of the impact of cryopreservation-induced delayed-onset cell death on sample viability assessment at various time points following thawing. Assessment within a few hours post-thaw yields elevated survival levels, and it is not until 24–48 h (cell cycle dependent) that the true level of survival is seen. (Adapted from Baust et al. 2009).
Conclusions
Proper selection of pre-freeze and post-thaw assays is not merely of academic importance given the unique advantages and disadvantages of each assay and tier. Given the diversity, timing, and specificity of information which can be obtained, it is important to recognize their impact and integrate the use of assays from multiple tiers to determine sample quality prior to and following freezing. In this regard, an in-depth understanding of assay type, timing, and the interpretative value of resulting data will provide for the most accurate overview of sample quality. Generally, at a minimum, when considering sample quality, one should target a pre-freeze viability > 90% (tier 1 assay) coupled with secondary analysis with a tier 2, 3, or 4 assay. Assessment of post-thaw viability should include tier 1 assessment immediately post-thaw to determine sample yield and initial viability for use in seeding calculations followed by sample assessment using a tier 2 assay 24 h post-thaw to determine true cell survival. Integration of tier 3 and 4 assays conducted at 24 h and several days post-thaw (3–7 days for instance) may also be beneficial in determining any long-term effects on cell function and repopulation capacity.
Best practices
Proper assessment of cell viability and function prior to and following cryopreservation is important to understanding sample quality. This impacts not only downstream utility but also the optimization of a given cryopreservation process which is often necessary in order to obtain the highest quality samples following thawing. To this end, it is recommended that one utilize multiple assays and time points to gain an accurate picture of sample quality post-thaw. Things to remember when thawing samples are as follows:
-
Minimize sample handling prior to freezing and following thawing as stresses associated with these manipulations will impact outcome. Garbage in = garbage out!
-
Assess sample viability at multiple time points such as 1 and 24 h post-thaw.
-
Timing of CIDOCD (delayed apoptosis and necrosis) is cell type specific; however, 24-h post-thaw serves as a reference point for the survival nadir for most mammalian cells.
-
Use assays which target different measures of viability such as membrane integrity and metabolic activity or other.
-
Compare post-thaw viability measures to pre-freeze measures. Do not normalize post-thaw viability measure to post-thaw cell yields as this will result in over-estimates of survival.
-
If cell function is important, perform a direct assessment of functionality post thaw. Never assume a viable cell is a 100% functioning cell!
-
Optimize cryopreservation protocols to yield elevated post-thaw survival to minimize post-thaw subpopulation selection, molecular alterations (transient and long term), and genetic drift.
References
Abrahamsen JF, Bakken AM, Bruserud O, Gjertsen BT (2002) Flow cytometric measurement of apoptosis and necrosis in cryopreserved PBPC concentrates from patients with malignant diseases. Bone Marrow Transplant 29(2):165–171
Baust JG (2007a) Concepts in biopreservation. In: Baust JG, Baust JM (eds) Advances in biopreservation. Taylor & Francis, New York, pp 1–14
Baust JG, Snyder KK, VanBuskirk RG, Baust JM (2017) Integrating molecular control to improve cryopreservation outcome. Biopreserv Biobanking 5(2):134–141
Baust JM (2007b) Properties of cells and tissues influencing preservation outcome: molecular basis of preservation-induced cell death. In: Baust JG, Baust JM (eds) Advances in Biopreservation. Taylor & Francis, New York, pp 63–87
Baust JG, Gao D, Baust JM (2009) Changing Paradigms in Cryopreservation. Organogenesis 5(3):90–96
Baust JM, Corwin W, Snyder K, Baust JG, Van Buskirk R (2016a) Development of a novel device for the controlled dry thawing of cryopreserved cell products. Bioprocess J 5(1):30–41
Baust JM, Corwin W, Snyder K, Van Buskirk RG, Baust JG (2016b) Biobanking: the future of cell preservation strategies. In: Biobanking and Cryopreservation of Stem Cells Karimi-Busheri and Weinfeld (eds). Adv Exp Med Biol 951:13–30
Baust JM, Snyder KK, Mathew AJ, Van Buskirk RG, Baust JG (2006) Steps to improving preservation outcome: understanding key factors influencing efficacy. Bio/Technology 31(11):18–20
Baust JM, Van Buskirk RG, Baust JG (2000) Cell viability improves following inhibition of cryopreservation-induced apoptosis. In Vitro Cell Dev Biol 36(4):262–270
Baust JM, Van Buskirk RG, Baust JG (2002) Gene activation of the apoptotic caspase cascade following cryogenic storage. Cell Preserv Technol 1(1):63–80
Baust JM, Vogel MJ, Van Buskirk R, Baust JG (2001) A molecular basis of cryopreservation failure and its modulation to improve cell survival. Cell Transplant 10(7):561–571
Beattie GM, Crowe JH, Lopez AD, Cirulle V, Ricordi C, Hayek A (1997) Trehalose: a cryoprotectant that enhances recovery and preserves function of human pancreatic islets after long-term storage. Diabetes 46:519–523
Beirao J, Cabrita E, Perez-Cerezales S, Martinez-Paramo S, Herraez MP (2011) Effect of cryopreservation on fish sperm subpopulations. Cryobiology 62(1):22–31
Berens C, Heine A, Muller J, Held SA, Mayer K, Brossart P, Oldenburg J, Potzsch B, Wolf D, Ruhl H (2016) Variable resistance to freezing and thawing of CD34-positive stem cells and lymphocyte subpopulations in leukapheresis products. Cytotherapy 18(10):1325–1331
Brockbank KGM (1995) Essentials of cryobiology. In: KGM B (ed) Principles of autologous, allogeneic, and cryopreserved venous transplantation. RG Landes Company, Austin (Medical Intelligence Unit Series) and Springer-Verlag, pp 1–151
Brockbank KGM, Covault JC, Taylor MJ. (2007) ThermoFisher: cryopreservation guide. p 6
Campbell LH, Brockbank KGM (2007) Serum free solutions for the cryopreservation of cells. In Vitro Cell Dev Biol 43:269–275
Campbell LH, Brockbank KGM (2010) Cryopreservation of porcine aortic heart valve leaflet-derived myofibroblasts. Biopreserv Biobanking 8(4):211–217
Carpenter JF, Crowe JH (1989) An infrared spectroscopic study of the interactions of carbohydrates with dried proteins. Biochemistry 28(9):3916–3922
Carpenter JF, Crowe LM, Crowe JH (1987a) Stabilization of phosphofructokinase with sugars during freeze-drying: characterization of enhanced protection in the presence of divalent cations. Biochim Biophys Acta 923(1):109–115
Carpenter JF, Hand SC, Crowe LM, Crowe JH (1986) Cryoprotection of phosphofructokinase with organic solutes: characterization of enhanced protection in the presence of divalent cations. Arch Biochem Biophys 250(2):505–511
Carpenter JF, Martin B, Crowe LM, Crowe JH (1987b) Stabilization of phosphofructokinase during air-drying with sugars and sugar/transition metal mixtures. Cryobiology 24(5):455–464
Coger R, Toner M (1995) Preservation techniques for biomaterials In: Biomedical Engineering Handbook. CRC Press, Boca Raton, pp 1557–1566
Crowe JH, Carpenter JF, Crowe LM, Anchordoguy TJ (1990) Are freezing and dehydration similar stress vectors? A comparison of modes of interaction of stabilizing solutes with biomolecules. Cryobiology 27(3):219–231
Crowe JH, Carpente JF, Crowe LM (1998) The role of vitrification in anhydrobiosis. Annu Rev Physiol 60:73–103
Crowe JH, Crowe LM, Carpenter JF, Rudolph AS, Aurell Wistrom C, Spargo BJ, Anchordoquy TJ (1988) Interactions of sugars with membranes. Biochim Biophys Acta 947(2):367–384
Crowe JH, Crowe LM, Carpenter JF, Aurell Wistrom C (1987) Stabilization of dry phospholipid bilayers and proteins by sugars. Biochem J 242(1):1–10
Crowe JH, Crowe LM, Hoekstra FA (1989a) Phase transitions and permeability changes in dry membranes during rehydration. J Bioenerg Biomembr 21(1):77–91
Crowe JH, Crowe LM, Oliver AE, Tsvetkova N, Wolkers W, Tablin F (2001) The trehalose myth revisited: introduction to a symposium on stabilization of cells in the dry state. Cryobiology 43(2):89–105
Crowe JH, McKersie BB, Crowe LM (1989b) Effects of free fatty acids and transition. temperature on the stability of dry liposomes. Biochim Biophys Acta 979(1):7–10
de Boer F, Drager AM, Pinedo HM, Kessler FL, van der Wall E, Jonkhoff AR, van der Lelie J, Huijgens PC, Ossenkoppele GJ, Schuurhuis GJ (2002) Extensive early apoptosis in frozen-thawed CD34-positive stem cells decreases threshold doses for haematological recovery after autologous peripheral blood progenitor cell transplantation. Bone Marrow Transplant 29(3):249–255
DeLoecker W, Koptelov VA, Grischenko VI, De Loecker P (1998) Effects of cell concentration on viability and metabolic activity during cryopreservation. Cryobiology 37:103–109
Eroglu A, Russo MJ, Bieganski R, Fowler A, Cheley S, Bayley H, Toner M (2000) Intracellular trehalose improves the survival of cryopreserved mammalian cells. Nat Biotechnol 18(2):163–167
Estrada E, Rivera Del Alamo MM, Rodriguez-Gil JE, Yeste M (2017) The addition of reduced glutathione to cryopreservation media induces changes in the structure of motile subpopulations of frozen-thawed boar sperm. Cryobiology S0011-2240(17):30149–30149
Fu T, Guo D, Huang X, O'Gorman MR, Huang L, Crawford SE, Soriano HE (2001) Apoptosis occurs in isolated and banked primary mouse hepatocytes. Cell Transplant 10(1):59–66
Fuhram GJ, Fuhram FA (1959) Oxygen consumption of animals and tissues as a function of temperature. J Gen Physiol 42:215
Fujita R, Hui T, Chelly M, Demetriou AA (2005) The effect of antioxidants and a caspase inhibitor on cryopreserved rat hepatocytes. Cell Transplant 14(6):391–396
Fuller BJ (2003) Gene expression in response to low temperatures in mammalian cells: a review of current ideas. Cryo Letters 24(2):95–102
Greco NJ, Seetharaman S, Kurtz J, Lee WR, Moroff G (2006) Evaluation of the reactivity of apoptosis markers before and after cryopreservation in cord blood CD34(+) cells. Stem Cells Dev 15(1):124–135
Heng BC, Richards M, Cao T (2009) Are stem cells inherently more prone to cryopreservation-induced apoptosis compared to ordinary somatic cells? Hum Reprod 24(2):492 author reply 492-493
Heng BC, Ye CP, Liu H, Toh WS, Rufaihah AJ, Yang Z, Bay BH, Ge Z, Ouyang HW, Lee EH, Cao T (2006) Loss of viability during freeze-thaw of intact and adherent human embryonic stem cells with conventional slow-cooling protocols is predominantly due to apoptosis rather than cellular necrosis. J Biomed Sci 13(3):433–445
Kaity A, Ashmore SE, Drew RA, Dulloo ME (2008) Assessment of genetic and epigenetic changes following cryopreservation in papaya. Plant Cell Rep 27(9):1529–1539
Karimi-Busheri F, Rasouli-Nia A, Weinfeld M (2016) Key issues related to cryopreservation and storage of stem cells and cancer stem cells: protecting biological integrity. Adv Exp Med Biol 951:1–12
Karlsson JOM, Toner M (1996) Long-term storage of tissues by cryopreservation: critical issues. Biomaterials 17:243–255
Karow AM (1981) Biophysical and chemical considerations in cryopreservation. In: Karow AM, Pegg DE (eds) Organ preservation for transplantation. Dekker, New York, p 113
Lovelock JE, Bishop MWH (1959) Prevention of freezing damage to living cells by dimethyl sulfoxide. Nature 183:1394
Matsushita T, Yagi T, Hardin JA, Cragun JD, Crow FW, Bergen HR 3rd, Gores GJ, Nyberg SL (2003) Apoptotic cell death and function of cryopreserved porcine hepatocytes in a bioartificial liver. Cell Transplant 12(2):109–121
Mazur P (1984) Freezing of living cells: mechanisms and implications. Am J Phys 247:125
Miyazaki T, Suemori H (2016) Slow cooling cryopreservation optimized to human pluripotent stem cells. Adv Exp Med Biol 951:57–65
Neyzen S, Van de Leur E, Borkham-Kamphorst E, Herrmann J, Hollweg G, Gressner AM, Weiskirchen R (2006) Cryopreservation of hepatic stellate cells. J Hepatol 44(5):910–917
O'Flaherty C, Beconi M, Beorlegui N (1997) Effect of natural antioxidants, superoxide dismutase and hydrogen peroxide on capacitation of frozen-thawed bull spermatozoa. Andrologia 29(5):269–275
Pegg DE, Diaper MP, Skaer HB, Hunt CJ (1984) The effect of cooling rate and warming rate on the packing effect in human erythrocytes frozen and thawed in the presence of 2M glycerol. Cryobiology 21:491–502
Pegg DE, Hunt CJ, Fong LP (1987) Osmotic properties of the rabbit corneal endothelium and their relevance to cryopreservation. Cell Biophys 10:169–189
Pegg DE, Jacobsen IA, Armitage WJ, Taylor MJ (1979) Mechanisms of cryoinjury in organs. In: Pegg DE, Jacobsen IA (eds) Organ Preservation II. Churchill Livingstone, Edinburgh, pp 132–146
Polge C, Smith AY, Parkes AS (1949) Revival of spermatozoa after vitrification and de-hydration at low temperatures. Nature 164:666
Raison JK (1973) The influence of temperature-induced phase changes on the kinetics of respiratory and other membrane-associated enzyme systems. Bioenergetics 4:285–309
Rudolph AS, Crowe JH (1985) Membrane stabilization during freezing: the role of two natural cryoprotectants, trehalose and proline. Cryobiology 22(4):367–377
Slade L, Levine H (1991) A food polymer science approach to structure-property relationships in aqueous food systems: non-equilibrium behavior of carbohydrate-water systems. Adv Exp Med Biol 302:29–101
Snyder KK, Van Buskirk RG, Baust JM, Mathew AJ, Baust JG (2004) Biological Packaging for the Global Cell and Tissue Therapy Markets. Bioprocessing 3(3):39–45
Sosef M, Baust JM, Fowler A, Toner M (2005) Cryopreservation of isolated primary rat hepatocytes: elevated survival and enhanced long-term function. Ann Surg 241(1):123–133
Stylianou J, Vowels M, Hadfield K (2005) CryoStor significantly improves cryopreservation of haematopoetic stem cells (HSC). Cryotherapy 7(Suppl 1):117
Sugimachi K, Sosef MN, Baust JM, Fowler A, Tompkins RG, Toner M (2004) Long-term function of cryopreserved rat hepatocytes in a coculture system. Cell Transplant 13(2):187–195
Taylor MJ (2007) Biology of cell survival in the cold: the basis for biopreservation of tissues and organs. In: Baust JG, Baust JM (eds) Advances in Biopreservation. Taylor & Francis, New York, pp 15–62
Taylor MJ, Campbell LH, Rutledge RN, Brockbank KGM (2001) Comparison of Unisol with Euro-Collins solution as a vehicle solution for cryoprotectants. Trans Proc 33:677–679
Van Buskirk RG (2007) Viability and functional assays used to assess preservation efficacy: the multiple endpoint/tier approach. In: Baust JG, Baust JM (eds) Advances in Biopreservation. Francis Publishing, New York, pp 123–141
Van Buskirk RG, Baust JG, Baust JM (2006) Navigating the post-preservation viability fog. Bioprocess Tutorial Genet Eng News 26(19):38–38
Worsham DN, Reems JA, Szczepiorkowski ZM, McKenna DH, Leemhuis T, Mathew AJ, Cancelas JA (2017) Biomedical Excellence for Safer Transfusion (BEST). Clinical methods of cryopreservation for donor lymphocyte infusions vary in their ability to preserve functional T-cell subpopulations. Transfusion 57(6):1555–1565
Yang B, Parsha K, Schaar K, Satani N, Xi X, Aronowski J, Savitz SI (2016b) Cryopreservation of bone marrow mononuclear cells alters their viability and subpopulation composition but not their treatment effects in a rodent stroke. Stem Cells Int 2016:5876836
Yang J, Diaz N, Adelsberger J, Zhou X, Stevens R, Rupert A, Metcalf JA, Baseler M, Barbon C, Imamichi T, Lempicki R, Cosentino LM (2016a) The effects of storage temperature on PBMC gene expression. BMC Immunol 17:6
Yong KW, Choi JR, Wan Safwani WK (2016) Biobanking of human mesenchymal stem cells: future strategy to facilitate clinical applications. Adv Exp Med Biol 951:99–110
Acknowledgements
The author would like to thank the following for their invaluable contributions and review of the manuscript—Gertrude Case Buehring, Eugene Elmore, Raymond W. Nims, Paul Price, Yvonne Reid, and Frank Simione.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: Tetsuji Okamoto
Glossary of Terms
- Air time
-
the time between removing a sample form storage and placement in to a water bath or other thawing device.
- Apoptosis
-
gene-regulated cell death.
- Aqueous
-
of or containing water, typically as a solvent or medium
- Cooling rate
-
the rate at which temperature of a sample decreases with time usually measured in °C/min.
- Cryopreservation
-
a process where organelles, cells, tissues, organs or any other biological construct are preserved by cooling to very low temperatures (typically − 80°C using solid carbon dioxide or − 196°C using liquid nitrogen) without causing additional damage caused by the formation of ice during freezing.
- Cryopreservation induced delayed onset cell death (CIDOCD)
-
cell death which occurs in a delayed manner 24–72 h post-thaw due to the apoptotic and necrotic activity.
- Cryoprotective agent/cryoprotectant
-
a substance used to protect biological tissue from freezing damage. An example would by dimethyl sulfoxide or glycerol.
- Cytotoxicity
-
the degree to which an agent has a specific destructive action on cells.
- Dewar
-
an insulated container used especially to store liquefied gases, having a double wall with a vacuum between the walls and silvered surfaces facing the vacuum.
- Disaccharide sugar
-
sugar formed when two monosaccharides (simple sugars) are joined by glycosidic linkage. Like monosaccharides, disaccharides are soluble in water. Three common examples are trehalose, sucrose, and maltose.
- Equilibrium freezing point
-
the temperature at which a given solution will freeze. This can change based on the composition of the solution that is being cooled.
- Gel phase
-
the state of a lipid bilayer or membrane in which the lipids are packed more orderly and remain relatively immobile. This usually occurs below the transition temperature, Tm.
- Hypothermia
-
a condition where the core temperature of an organism falls below a critical threshold. In humans, hypothermia occurs when the body temperature drops below 35°C.
- Hypoxia
-
a decrease in the availability of oxygen to cells tissues or organs or a state of below normal oxygen levels.
- Ice nucleation
-
the point at which the formation of ice begins in a given solution.
- Ischemia
-
is a restriction in blood supply to tissues, causing a shortage of oxygen and glucose needed for cellular metabolism
- Latent heat of fusion
-
the energy released, usually observed as heat, when water changes to ice in a biological system
- Liquid crystalline phase
-
the state of a lipid bilayer or membrane in which the membrane is fluidic; lipids can diffuse freely inside the dual layer of the membrane. This usually occurs above the transition temperature, Tm.
- Oncotic force
-
also called colloidal osmotic pressure, it is a form of osmotic pressure exerted by proteins or in the case of cryopreservation macromolecules in the solution, which may pull water from cells and have protective effects on cell membranes.
- Osmotic equilibrium
-
is used to indicate that the concentration of a solute in water is the same on both sides of a semi-permeable membrane. While the water still passes through the membrane, there is no gain or loss on either side.
- Osmotic pressure
-
the minimum pressure which needs to be applied to a solution to prevent the inward flow of water across a semi-permeable membrane
- Recrystallization
-
when very small (microscopic) ice crystals that may have formed during freezing are given the opportunity to grow during the re-warming of a sample.
- Sample agitation
-
the process of gently mixing a sample during thawing to reduce/eliminate the formation of step thermal gradients within a sample which can be highly lethal.
- Supercooling
-
also known as under-cooling; is the process of lowering the temperature of a liquid or a gas below its freezing point without it becoming a solid.
- Thermal gradient
-
the rate of temperature change with through a body or across a surface.
- Tg
-
glass transition temperature; the temperature region at which a substance undergoes a phase transition from a liquidous to a glassy state.
- Tm
-
also known as the transition temperature of a lipid bilayer; it is the temperature at which a membrane shifts from being in a liquid crystalline phase to a gel phase.
- Vehicle solution/carrier medium
-
a base solution in which a cryoprotectant is added and is usually designed to help facilitate cell maintenance under adverse conditions such as cooling below physiological temperature or freezing.
- Viability assessment tiers
-
the strategy of utilizing multiple assays prior to or following cryopreservation to more effectively determine sample quality (viability and function).
Rights and permissions
About this article

Cite this article
Baust, J.M., Campbell, L.H. & Harbell, J.W. Best practices for cryopreserving, thawing, recovering, and assessing cells. In Vitro Cell.Dev.Biol.-Animal 53, 855–871 (2017). https://doi.org/10.1007/s11626-017-0201-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-017-0201-y