
Abstract
Cytoplasmic microinjection (CI) of foreign gene into in vivo fertilized zygotes has emerged as a useful tool for transgenic pig production. In the current study, we investigated factors affecting transgenic efficiency and developmental potential of parthenogenetic (PA) and in vitro-fertilized (IVF) porcine embryos produced by CI. These factors included adding of RNase inhibitor, DNA or RNA concentration, injection time, and different structures of plasmids. Our results showed that adding of 1–4 U/μL of RNase inhibitor did not have negative effect on development potential of CI-PA embryos, and RNase inhibitor injection significantly increased EGFP expressing rate of CI-PA embryos. High injection DNA concentration and long injection interval after PA significantly reduced blastocyst formation. Different molecular structures such as DNA or RNA affected CI-PA embryos development, and RNA had little harmful effect on pig’s early embryonic development. EGFP expression rate of CI-IVF embryos was improved following the increase of foreign DNA concentration, but blastocyst formation rate was decreased. Injection time after IVF did not show any significant difference on embryonic development, but longer interval resulted in a significantly lower EGFP expressing rate. Cas9 mRNA and myostatin (GDF-8) sgRNA co-injection indicated that the mutation rate of CI-IVF group was significantly higher than that of CI-PA. The CI-IVF-generated embryos were then transferred to six recipient pigs, but no live piglets were obtained. The following pronuclear formation assessment showed more than 76.1% IVF zygotes were polyspermy. These results demonstrate that CI-PA and CI-IVF were effective methods for production of transgenic pig embryos. However, polyspermic fertilization and poor quality of porcine IVF blastocysts are still the main problem of resulting in pregnancy failure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mammalian transgenesis as an important tool of biological research has been used for xenotransplantation research (Cibelli et al. 2013), production of animal models for human diseases (Fan and Lai 2013), analyzing tissue-specific cis-regulatory elements of developmental genes (Sumiyama and Ruddle 2003), and recognizing activities of evolutionary conserved elements on the genome in large scales (Urasaki and Kawakami 2009). Transgenesis in pig, most commonly achieved by pronuclear DNA injection (PNI) or by somatic cell nuclear transfer (SCNT), is an inefficient and expensive process, hampered by poor predictability of levels and patterns of transgene expression (Kues and Niemann 2011; Whyte and Prather 2011). In addition to PNI and SCNT, intracytoplasmatic sperm injection (ICSI) has been used for pig transgenesis. While a functional method in rodent species, ICSI has been proven more challenging in production of transgenic livestock. To date, the production efficiency of transgenic pigs by ICSI was very low, and the likelihood of producing transgenic fetuses and live piglets is less than 1% (Yong et al. 2006; Garcia-Vazquez et al. 2010).
Different from that of PNI and ICSI, cytoplasmic microinjection (CI) was first reported by Brinster et al. in 1985, who demonstrated that the production efficiency of transgenic mouse by CI was very low, and of 224 fetuses examined after CI, only 2 were positive (Brinster et al. 1985). Page et al. applied polylysine which is known to bind DNA by electrostatic interaction to CI, and about 12.8% of the pups were transgenesis (Page et al. 1995). It is well known that messenger RNA (mRNA) was first transcribed and edited in the nucleus and then transferred to the cytoplasm for protein translation; thus, CI maybe a suitable method for RNA injection. Recent advances in genome editing technology involving transcription activator-like effector nucleases (TALENs) and clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) 9 system offered a novel opportunity for application of CI in transgenic animal production.
TALENs are naturally occurring proteins from the plant pathogenic bacteria genus Xanthomonas consisting of DNA-binding domains composed of a series of 33–35 amino acid repeat domains that each recognizes a single base pair, and tethered to FokI endonuclease (Deng et al. 2012). When TALENs bind their target in an appropriate orientation, FokI can introduce a DNA double strand break (DSB) (Bogdanove and Voytas 2011). These DSB sites stimulate the cellular DNA repair mechanisms, including error-prone non-homologous end joining (NHEJ) and homology-directed repair (HDR) (Wyman and Kanaar 2006). By microinjecting zygotes with TALEN mRNA and single-stranded DNA oligonucleotides (Bedell et al. 2012) or donor plasmid with extended homology arms (Zu et al. 2013), TALENs have enabled the generation of loxP-engineered chromosomes and the possibility for conditional gene activation in zebrafish.
Distinct from the site-specific nuclease described above, the CRISPR/Cas system has recently emerged as a potentially facile and efficient alternative to TALENs for inducing targeted genetic alterations. In bacteria, the CRISPR system provides acquired immunity against invading foreign DNA via RNA-guided DNA cleavage (Wiedenheft et al. 2012). The Cas9 endonuclease from Streptococcus pyogenes type II CRISPR/Cas system can be engineered to produce targeted genome modification under the guidance of a synthetic single guide RNA (sgRNA) with simple base pair complementarities with a target genomic DNA sequence (Cong et al. 2013). Compared with the complicated design and assembly of TALENs, redirecting Cas9 to a new target site requires only the alteration of a gene-specific 20-nt DNA sequence in sgRNA, which can be synthesized on a large scale (Deltcheva et al. 2011). The combination of CI and in vivo matured oocytes with TALENs and CRISPR/Cas system has been used for transgenic animal production in various animals recently (Wang et al. 2013; Hsu et al. 2014; Liu et al. 2014a).
To our knowledge, there have been no studies performed in the factors affecting foreign gene introduction efficiency and developmental potential of parthenogenetic (PA) and vitro-fertilized (IVF) porcine embryos by CI. The current study was conducted to evaluate the effect of adding RNase inhibitor, injection DNA or RNA concentration, injection time, and different molecular structures on the efficiency of producing transgenic pig embryos by CI-PA and CI-IVF. The feasibility of using CI-IVF embryos to product transgenic pig was also evaluated.
Materials and Methods
Chemicals
All chemical reagents used for oocyte maturation, activation, and embryo culture were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Ovary collection and maturation
Ovaries were collected from a local abattoir and stored at 28–30°C during transportation. When ovaries arrived at the laboratory, they were washed one time with 75% ethanol in less than 15 s and three times with warmed Dulbecco PBS (DPBS), then stored in a water bath at 39°C before use. Cumulus oocyte complexes (COCs) were aspirated from ovarian follicles 3–6 mm in diameter using a 10-mL syringe fitted with an 18-gauge needle. Follicular fluid was collected in 10-mL round bottom centrifuge tubes and left for 10 min at 39°C. Subsequently, COCs were washed three times with cell culture medium (CCM) containing 2%–3% FBS. Only COCs with uniform cytoplasm and at least three layers of compact cumulus cells were selected for maturation. Oocytes were normally matured in groups of 30 per 150 μL of maturation medium. The base maturation medium was TCM 199 supplemented with 10% (v/v) porcine follicular fluid, 0.6 mM cysteine, 50 ng/mL EGF, and 10 ng/mL IGF-I. This medium was supplemented with 15 IU/mL eCG and 10 IU/mL hCG during the first 22 h of culture only, after which culture was hormone free for another 20 h at 38.5°C, 5% CO2 in air.
Oocyte activation
The matured COCs were denuded by using 0.1% hyaluronidase, and oocytes with polar body were selected for activation. Oocytes were rinsed twice in electrical activation medium (0.3 M mannitol, 0.1 mM Mg2+, 5.0 mM Hepes, and 0.1 mM Ca2+). For activation, oocytes were transferred between electrodes of a fusion chamber (BTX microslide 0.5-mm fusion chamber, model 450; BTX, San Diego, CA) covered by electrical activation medium in a chamber connected to an electrical pulsing machine (FC-150/B; BLS Ltd., Budapest, Hungary). Then oocytes were activated with three direct current (DC) of 1.00 kV/cm for 80 μsec. After that, oocytes were incubated in the porcine zygote medium-3 (PZM-3) (Yoshioka et al. 2002) medium supplemented with 7.5 μg/mL cytochalasin B and 10 μg/mL cycloheximide for 4 h.
In vitro fertilization
IVF was performed as Gil et al. (2007) with some modifications. Briefly, the matured COCs were denuded by slightly pipetting with 0.1% hyaluronidase and left about one layer of cumulus cells. Oocytes with polar body were selected for IVF. Semen-rich fractions (30 to 50 mL) from three boars of Landrace breed were collected and diluted with modified sperm preservation solution. The diluted spermatozoa was cooled down from room temperature to 17°C for 4 h and then kept at the same temperature no more than 4 d. Just before use, the stored spermatozoa was placed at room temperature for 15 to 20 min and 100 μL of spermatozoa were washed three times by centrifugation at 200g for 3 min with modified Tris-buffered medium (mTBM) which consisted of 113.1 mM NaCl, 3 mM KCl, 7.5 mM CaCl2·2H2O, 20 mM Tris (Trizma Base), 11 mM d-glucose, 5 mM sodium pyruvate, and 0.2% BSA, and then resuspended with IVF medium (mTBM supplemented with 2 mM caffeine). About 10 μL of spermatozoa was introduced into 90 μL of IVF medium containing about 20 COCs. For the final sperm, oocyte ratio was adjusted to 1000–1500 and co-incubated for 10 min. The oocytes were washed by slightly pipetting three times to remove spermatozoa not bound to the zona pellucida, and transferred to fresh IVF medium containing no sperm for another 2 h, and then washed and transferred to PZM-3 medium for culture. In the final experiment, sperm and oocyte co-incubated for different time durations, and the presumptive zygotes were washed and directly moved to PZM-3 medium for culture.
Cytoplasmic microinjection and embryo culture
PA oocytes and IVF zygotes according to the experiment design were used to cytoplasmic microinjection transgene (CI-Tr). CI was performed in a 4-μL drop of CCM under mineral oil, using a Nikon inverted microscope (Eclipse-Ti; Nikon, Tokyo, Japan). Foreign gene was first aspirated from a 4-μL DNA or RNA drop by a 5.5-μm inner diameter injection pipette until aspirating about three oocyte diameters long, and then moved to the oocytes containing drop. An oocyte was captured by a holding pipette and immobilizing with polar body at either 6 or 12 o’clock position. The injection pipette was pushed through zona pellucida and oolemma into the cytoplasm at 3 o’clock position. A small amount of ooplasm was aspirated into the injection pipette in order to ensure oocyte membrane penetration. Subsequently, the foreign gene was released into the cytoplasm.
In all experiments, the injected oocytes were washed three times with PZM-3 culture medium and then were cultured in groups of 4 per 30 μL of culture medium under mineral oil for 168 h at 38.5°C, 5% CO2 in air.
Observation of nuclear status and blastocyst cells
Thirteen hours after insemination, fertilized oocytes were pipette repeatedly to remove all the attached cumulus cells and spermatozoa with 0.1% hyaluronidase. Glass slides were prepared by laying two parallel thin rails of paraffin (4%)-vaseline (96%) mixture along the length of slide. Denuded oocytes (10–15) were placed on the slide with minimum medium. Cover slip was placed on the paraffin-vaseline mixture rails and pushed down until fluid contacted the cover slip. Oocytes were watched carefully under a stereo-microscope while pressing further, so as to get a good squash without breaking the zona pellucida of the oocytes. The slides were immersed in fixative (3:1 ethanol:acetic acid). After 48 h, slides were gently removed from the fixative. The excess fixative was dried and the oocytes were stained with 1% (w/v) orcein stain for 10 min. The slides were examined under phase contrast microscope (Eclipse-Ti; Nikon, Tokyo, Japan) at ×400 to evaluate the status of the nucleus.
The quality of blastocysts was assessed by Hoechst 33342 staining of total cell number and differential staining of ICM and TE cells (Liu et al. 2014b). Differential staining and cell counting were performed as Thouas et al. (Thouas et al. 2001). Cell counting was performed either directly or from digital photographs of images obtained on a microscope fitted with an ultraviolet lamp and excitation filters (460 nm for blue and red fluorescence and 560 nm for red only).
Transgene vectors
For the injected DNA vectors, EGFP-N1 (4.7 kb; Clontech Laboratories, Inc., Mountain View, CA, Supplementary information Fig. 1a) was linearized with ApaI. EGFP expression vector with prolactin open reading frame (PRL-EGFP, Laboratory Storage, St. Peters, MO, Supplementary information Fig. 1b) was linearized with SalI and SacI.
For the injected RNA vectors, TALEN plasmids were designed for knocking-out bone morphogenetic protein15 (TALEN-BMP15) and forkhead box O3 (TALEN-FoxO3) of porcine (Laboratory Storage, Supplementary information Fig. 1c). The target sequences were listed in the Supplementary information Table 1. The mRNA of TALENs and EGFP was obtained by transcription in vitro using mMESSAGE mMACHINE®KIT SP6 (Thermo Fisher Scientific, Waltham, MA) according to the protocol.
The vector encoding hSpCas9 was ordered from ViewSolid Biotech (Beijing, China). The Cas9 sequence was human codon optimized and cloned into pcDNA3.1 (Invitrogen, Carlsbad, CA, Supplementary information Fig. 1d). The T7 promoter, target sequence (BMP15, FoxO3, and GDF8), and gRNA scaffold, which included a restriction endonuclease site PsiI for linearized plasmid to transcript in vitro, were ordered Sangon Biotech (Shanghai, China), and cloned into pSicoR-GFP (Addgene, Cambridge, MA, Supplementary information Fig. 1e). The sequences of hSpCas9, gRNA and target sequence are listed in Supplementary information Tables 2–3. The hSpCas9 expression plasmid and the gRNA plasmid were linearized by restriction endonuclease DraIII and PsiI, respectively, for transcription in vitro. The Cas9 mRNA and the sgRNA (Cas-BMP15, Cas-FoxO3, and Cas-GDF8) were transcribed in vitro using mMESSAGE mMACHINE® T7 Kit (Thermo Fisher Scientific) and MEGAshortscript™ T7 Kit (Thermo Fisher Scientific) with recommended protocols.
EGFP expression and GDF8 mutation detection
EGFP expression embryos were evaluated by direct visualization of embryos through fluorescence microscopy with maximum excitation of 488 nm.
Mutations of embryos were identified by alignment of sequenced alleles to wild-type allele. Briefly, each embryo at 168 h was collected in a tube with ultrapure water. The collected embryo was directly subjected to PCR by utilizing the primers listed in Supplementary information Table 4. Polyacrylamide gel electrophoresis of 12% was applied for mutation detections about PCR product from each embryo. The suspected PCR product of embryo was TA cloned and sequenced for gene mutation detecting.
Embryo transfer
Ten-month crossbred (Duroc × LongLin) gilts, weighing from 100 to 105 kg, were used as recipients of the CI-IVF Cas-GDF8 and EGFP-N1 embryos. Gilts were classified as being in estrus when they actively approach mature boar and exhibit immobilization, or the standing reflex, in response to pressure placed on their backs. The injected oocytes were in vitro cultured for 24 to 48 h. Embryos that morphologically normal were selected for transfer. They were loaded into a Tom Cat catheter™ (Kendall Co, Mansfield, MA) which was kept in a portable incubator (Minitube, Verona, WI) during transportation. Embryos (ranging in number from 88 to 202 for each recipient sow) were transferred into one oviduct of the recipient gilts approximately 48 h after spontaneous estrus.
Experimental Design
Study 1: Cytoplasmic microinjection of parthenogenetic oocytes
In experiment 1, to determine the effect of RNase inhibitor to embryo development and the protection efficiency of RNA in RNase inhibitor-injected embryo, different concentrations (0, 1, 2, and 4 U/μL) of RNase inhibitor (RI; Takara; 2313A, Lot #k6801EA) was added to TALEN-BMP15 RNA solution. A total of 552 oocytes were randomly injected, and development status was investigated. Also, oocytes were randomly co-injected of EGFP RNA with or without of RNase inhibitor, and embryo development status and EGFP expression rate were evaluated. In experiment 2, a total of 800 oocytes were randomly injected with different concentrations (50 ng/μL, 100 ng/μL, or Sham injection) of EGFP-N1 DNA or TALEN-BMP15 RNA. Embryo development efficiency and EGFP expression rate were evaluated. In experiment 3, to determine an appropriate time for injection, a total of 775 oocytes were randomly divided into six groups for injection with EGFP-N1 DNA. The oocytes were first activated and cultured in PZM-3 medium for 5–10 min, 5–6 h, 8–9 h, 11–12 h, 14–15 h, and 17–18 h respectively and then injected. Experiment 4 was conducted to evaluate the effect of different transgene vectors and molecular structure on CI-Tr. A total of 948 oocytes were randomly divided into eight groups and injected with EGFP-N1 (circle or linearized) DNA, PRL-EGFP (linearized) DNA, TALEN-FoxO3 RNA, TALEN-BMP15 RNA, Cas-FoxO3 RNA, and Cas-BMP15 RNA respectively.
Study 2: Cytoplasmic microinjection of IVF zygotes
In experiment 1, to determine an appropriate injection concentration, a total of 912 IVF zygotes after fertilized for 10 h were injected with 0, 10, 20, 30, 40, 50, and 100 ng/μL linearized EGFP-N1 DNA, respectively. In experiment 2, to determine an appropriate time for injection, a total of 588 oocytes were fertilized, and then cultured in PZM-3 for 5–6, 8–9, 11–12, 14–15, and 17–18 h respectively, and then injected.
Study 3: Cas9 mRNA and GDF8 sgRNA cytoplasmic microinjection
Experiment 1 was conducted to evaluate the development potential and GDF8 mutation efficiency of embryos produced by CI-PA and CI-IVF injected with Cas9 mRNA and GDF8 sgRNA. A total of 342 oocytes were injected and 80 CI-PA blastocysts and 52 CI-IVF morulae (including blastocysts) were used to detect GDF8 mutation efficiency. In experiment 2, a total of 621 CI-IVF Cas-GDF8 embryos were transplanted into 5 gilts, and 103 CI-IVF EGFP-N1 embryos were transplanted into 1 gilt. The pregnancy status was detected. In experiment 3, a total of 522 oocytes were co-incubated with sperm for 10 min, 1 h, 2 h, 3 h, 4 h, 5 h, 6 h, 7 h, and 8 h respectively and the nuclear status was evaluated.
Statistical analysis
At least four replicate trials were conducted for each experiment. Cleavage, blastocyst rate, and positive rate were modeled according to the binomial model of parameters by arcsine transformation of the data and were analyzed by one-way ANOVA and Duncan’s multiple-range test. A value of P < 0.05 was considered to be statistically significant. All statistical analyses were performed using SPSS software version 17.0 (SPSS, Portsmouth, UK). Data are presented as means ± SEM.
Results
Study 1: Cytoplasmic microinjection of parthenogenetic oocytes
In experiment 1, there was no significant difference in embryo development and blastocyst quality (P > 0.05) when oocytes injected with different concentrations of RNase inhibitor (Table 1). The EGFP expression rate of EGFP RNA and RNase inhibitor co-injection group (10.3 ± 0.8%) was significantly higher (P < 0.001) than EGFP RNA injection group (Table 2). In experiment 2, as shown in Fig. 1A and supplementary information Table 5, when oocytes were injected with different concentrations of EGFP-N1 DNA, the blastocyst rate of 50 ng/μL group (46.4 ± 2.1%) was significantly higher (P < 0.01) than 100 ng/μL (33.9 ± 1.1%), but no significant difference from the shame injection group. However, the EGFP expression rate of 50 ng/μL group was significantly lower (P < 0.001) than 100 ng/μL group. In the three different concentrations of TALEN-BMP15 RNA injection groups, the cleavage rate, blastocyst rate, and cell number per blastocyst displayed no significant difference (P > 0.05). Results of experiment 3 are presented in Fig. 1B and Supplementary information Table 6. A significantly higher proportion (P < 0.01) of oocyte reached blastocyst stage when they activated after injection with 5–10 min (51.1 ± 3.1%). There was no significant difference on the EGFP expression rate among the experimental groups (P > 0.05). In experiment 4, the EGFP expression rate of injection of two linearized DNA plasmids was significantly higher (P < 0.05) than that of circle DNA (Table 3). In the further, DNA injection groups of linearized EGFP-N1 (47.9 ± 0.9%) and PRL-EGFP (42.2 ± 1.7%) tended to be of lower blastocyst rates (P < 0.1) than RNA injection groups of TALEN-FoxO3 (52.6 ± 1.8%), TALEN-BMP15 (52.2 ± 0.9%), and Cas-BMP15 (55.0 ± 2.9%, Table 3).

Effects of different concentrations of EGFP-N1 DNA and TALEN-BMP15 RNA plasmid and time duration on cytoplasmic microinjected PA and IVF porcine embryos development and EGFP expression. The four figures were analyzed: sham injection ( ), 10 (
), 10 ( ), 20 (
), 20 ( ), 30 (
), 30 ( ), 40 (
), 40 ( ), 50 (
), 50 ( ), and 100 ng/μL (
), and 100 ng/μL ( ) EGFP-N1 DNA and Talen-BMP15 RNA plasmid on CI-PA (A) and CI-IVF (B) embryos; 5–10 min (
) EGFP-N1 DNA and Talen-BMP15 RNA plasmid on CI-PA (A) and CI-IVF (B) embryos; 5–10 min ( ), 5–6 h (
), 5–6 h ( ), 8–9 h (
), 8–9 h ( ), 11–12 h (
), 11–12 h ( ), 14–15 h (
), 14–15 h ( ), and 17–18 h (■) time duration from activation or sperm oocyte co-incubation to injection on CI-PA (C) and CI-IVF (D) embryos. Different letters within contiguous bars represent a significant difference (P < 0.05).
), and 17–18 h (■) time duration from activation or sperm oocyte co-incubation to injection on CI-PA (C) and CI-IVF (D) embryos. Different letters within contiguous bars represent a significant difference (P < 0.05).
Study 2: Cytoplasmic microinjection of IVF zygotes
In experiment 1, as shown in Fig. 1C and Supplementary information Table 7, there is a significant lower proportion of blastocysts in 50 and 100 ng/μL linearized EGFP-N1 DNA injection groups than in 10 and 20 ng/μL groups (P < 0.05), but had no significant difference with 30 and 40 ng/μL and sham injection groups (P < 0.05). EGFP expression rate of 40 (9.1 ± 0.4%), 50 (11.8 ± 1.5%), and 100 (13.6 ± 2.0%) ng/μL was significantly higher than that in other groups. In experiment 2, there was no significant difference on blastocyst rate of different injection time groups (P > 0.05). But, EGFP expression rate of 5–6 h (12.6 ± 0.6%), 8–9 h (11.8 ± 0.5%), and 11–12 h (13.6 ± 0.6%) was significantly higher (P < 0.01) than that of the other two groups (Fig. 1D , Supplementary information Table 8).
Study 3: Cas9 mRNA and GDF8 sgRNA cytoplasmic microinjection
Results of experiment 1 are presented in Table 4, and when PA oocytes and IVF zygotes were injected with Cas9 mRNA and GDF8 sgRNA, the blastocyst rate of CI-PA group was significantly higher than that of CI-IVF group (P < 0.001). On the contrary, the mutation efficiency in the targeting site of IVF zygote group (25%) was significantly higher than that of PA oocytes (3.8%, Fig. 2 and Supplementary information Fig. 2, P < 0.001). In experiment 2, three of the six transferred gilts were not re-estrus, but no live piglet was born (Supplementary information Table 9). In order to find the reason, sperm and oocytes were co-incubated for different time and the rate of penetration, monospermy, and two pronucleus (PN) formation were evaluated. The result illustrated that penetration rate of 6 h (87.5 ± 2.8%), 7 h (90.0 ± 1.1%), and 8 h (89.0 ± 3.1%) was significantly higher (P < 0.01) than that of 10 min, 1 h, and 2 h, but had no significant difference from 3, 4, and 5 h. Similarly, two PN formation rates of 10 min, 3 h, 5 h, 6 h, 7 h, and 8 h was significantly higher (P < 0.05) than that of 1 and 2 h. However, co-incubation time of 10 min (23.9±1.4%), 1 h (22.2 ± 4.8%), 2 h (22.5 ± 5.1%), 3 h (18.5 ± 2.5%), and 4 h (14.0 ± 2.2%) resulted in significantly higher monospermy rates than that of 5, 6, 7 and 8 h groups (P < 0.05), and it displayed no significant difference between 4 and 6 h groups (Fig. 3, Supplementary information Table 10).

Sanger sequencing of the targeting site on mutant embryos. The PAM sequence is bold and labeled in red. On the right side of each allele, the sizes of insertions (+) or deletions (Δ) are indicated.

Period of oocyte sperm co-incubation (10 min (■), 1 h ( ), 2 h (
), 2 h ( ), 3 h (
), 3 h ( ), 4 h (
), 4 h ( ), 5 h (
), 5 h ( ), 6 h (■), 7 h (
), 6 h (■), 7 h ( ), and 8 h (
), and 8 h ( )) during IVF on different fertilization parameters. Different letters within the same figure represent a significant difference (P < 0.05).
)) during IVF on different fertilization parameters. Different letters within the same figure represent a significant difference (P < 0.05).
Discussion
The mechanism of classic transgenesis methods relies on integration of exogenous DNA to host genome, which could activate continuous expression of nucleases, and had harmful effects on embryos (Ivics et al. 2014). On the contrary, the effect of microinjected foreign RNA in the fertilized ovum and the growing oocyte was extremely limited (Brinster et al. 1980; Paynton et al. 1983). But, the single-stranded RNA was easy to be degraded; in the study of mouse, the half-life of exogenous mRNA in the fertilized ovum was only 3 h (Ebert et al. 1984). Thus, for the first time, we added RNase inhibitor to the exogenous RNA vector. Our results indicated that RNase inhibitor could significantly increase EGFP expressing rate of EGFP RNA injected embryo, and it did not have significantly negative effect on CI-PA embryo development (Fig. 4).

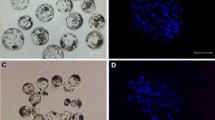
IVF and PA porcine blastocysts expressing EGFP produced by cytoplasmic microinjection with EGFP-N1 DNA plasmid. (a) PA blastocyst under bright light; (a′) the same blastocyst was shown under blue light. (b) IVF blastocyst under bright light; (b′) the same blastocyst was shown under blue light. (original magnification ×200).
When pronuclear injection was used to create transgenic animals, transgene concentration was known to influence embryogenesis and gene-transfer efficiency (Brinster et al. 1985). Exogenous DNA concentration has been shown to affect transgenic efficiency by ICSI in mice, rats, and pigs (Perry et al. 1999; Hirabayashi et al. 2005; Garcia-Vazquez et al. 2010). In our experiments, poor embryo development potential for CI was observed with high foreign DNA concentration. Moreover, there was a significantly higher proportion of EGFP expression embryos in 40 ng/μL foreign DNA injection group than that of 10, 20, and 30 ng/μL, but no significant difference from 50 and 100 ng/μL. These observations consist with previous report in embryos and tissue culture cells, in which stable integration frequency reaches a plateau with a few molecules of microinjected, linearized DNA (Brinster et al. 1985). Correspondingly, we found that most of the blocked pig embryos at day 7 showed unusually intense fluorescence, and EGFP expression blastocyst tend to be poorer embryo quality than no EGFP expression blastocyst. These data suggest that if a mass of exogenous DNA is transferred into an oocyte, considerable toxicity occurs during embryonic development, and a concentration-dependent process limits nuclear import and enzymatic reactions in the integration of injected DNA.
When the same concentration of exogenous RNA as DNA by CI was injected, we did not observe such lack of development on IVF and PA embryos. It means that exogenous RNA should bring less toxicity to embryos than exogenous DNA vectors. One possibility is that a mass of foreign DNA is brought to the ooplasm and induces the alteration and death of the embryo by endonuclease activation in an apoptotic-like process, but because abundance of maternal RNA is sorted in oocyte and easy to be degraded, RNA will result in less the same process. Our data also suggested that integration of linearized plasmids was more efficient than circular molecules, and the structure of DNA and RNA molecules does not seem to be an important parameter to CI. That was consistent with the previous studies in mouse and rats (Brinster et al. 1985; Hirabayashi et al. 2005).
To find the optimal injection time of exogenous gene introduction into porcine PA oocytes and IVF zygotes, various time spots after activation and fertilization were chosen for microinjection. Our results indicated that EGFP expression rate significantly reduced after fertilization for 14 h. However, in PA embryos, it displayed no significant difference. Previous studies have suggested that DNA integration occurred at an early cell stage for either pronuclear or cytoplasmic injection of polylysine DNA mixtures in mice (Page et al. 1995). Iqbal et al. reported that translocation of the plasmids into the nucleus happens in <12 h in mouse embryos (Iqbal et al. 2009). Thus, the possible integration mechanism is that in the pronuclei fusion, nuclear membrane disassembled, and the plasmid molecules could be easily taken up in the newly forming nucleus. At the same time, we found that long time duration significantly reduced the development potential of parthenogenetic embryos, but the same phenomenon did not happen in IVF embryos. These can be associated with porcine embryos which are highly sensitive to temperature and micro-environment changes; the fully developed PA embryos (55.0%) were more affected than poor development IVF embryos (14.6%).
We further examined the in vitro embryo developmental and mutation efficiency by direct cytoplasmic injection of Cas9 mRNA and GDF8 sgRNA into porcine zygotes. The results indicated that development of CI-PA and CI-IVF embryos was not affected, and GDF8 mutation efficiency of IVF zygotes was significantly higher than that of PA oocytes. The result further evidences the conclusion above: (1) exogenous RNA vectors have little harmful effect on pig early embryonic development and (2) pronuclei fusion might be the optimal window of opportunity that exogenous gene integrates into the host genome. To investigate the possibility of exploiting CI-IVF zygotes to generate transgenic pigs, 621 CI-IVF Cas-GDF8 embryos were transplanted into 5 gilts and 103 EGFP-N1 embryos were transplanted into 1 gilt, but no live offspring was delivered.
Data examination of injected IVF zygotes for all plasmid concentrations and different injection time indicated that the rate of blastocyst formation for IVF embryos is considerably low. It has been reported confounds the problem of polyspermic fertilization in porcine embryo IVP systems is that polyspermic embryos are still able to develop to the blastocyst stage (Grupen 2014). Han et al. demonstrated that porcine IVP zygotes containing three or four pronuclei developed to the blastocyst stage at the same rate as those containing two normal pronuclei (Han et al. 1999a). Depending on the location of the pronuclei, polyploid zygotes formed triploid, aneuploidy, or mixoploid embryos, of which only the latter developed to term (Han et al. 1999b). These reports mean that blastocyst formation of IVF zygotes will drop to an even lower level. From the Supplementary Table 5, the result indicated that with very low oocyte sperm co-incubation time, the polyspermic fertilization rate was still high (76.1%, 77.8%, 77.5%, 81.5%, 86%, 90.3%, 89.9%, 91.5%, and 91.0%) and two PN formation rate was extremely low (29.0%, 16.5%, 20.1%, 28.5%, 26.2%, 33.5%, 30.5%, 33.1% and 28.5%). It was consistent with the previous studies in pig (Gil et al. 2007). More attention should be paid to that cell number of blastocysts (around 50) and that still has large discrepancy between in vivo-derived porcine blastocysts (KIKUCHI 2004). Therefore, the most likely reason is that the polyspermic fertilization and poor quality of IVF blastocysts for CI are contributing to the failure of the transgenic pig production. Polyspermic fertilization is associated with inadequate oocyte cytoplasmic maturation, and poor quality blastocysts is associated with suboptimal embryo culture condition (Grupen 2014), which means that the deficiencies of in vitro mature (IVM) and in vitro culture (IVC) media are the main problem to CI-Tr pig production.
In conclusion, the present paper demonstrates that cytoplasmic microinjection of exogenous DNA and RNA vectors to PA oocytes and IVF zygotes is a simple, reliable, and highly efficient method for transgenic embryos production. With the progress of pig embryo IVP system, and together with easily designed, assembled, and high efficiency of the TALENs or CRISPR/Cas9 system, we anticipate that the ease of use CI combined with IVF zygotes will become a powerful tool for transgenic pig production.
References
Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG II, Tan W, Penheiter SG, Ma AC, Leung AY (2012) In vivo genome editing using a high-efficiency TALEN system. Nature 491:114–118
Bogdanove AJ, Voytas DF (2011) TAL effectors: customizable proteins for DNA targeting. Science 333:1843–1846
Brinster RL, Chen HY, Trumbauer ME, Avarbock MR (1980) Translation of globin messenger RNA by the mouse ovum. Nature 283:499–501
Brinster RL, Chen HY, Trumbauer ME, Yagle MK, Palmiter RD (1985) Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proc Natl Acad Sci U S A 82:4438–4442
Cibelli J, Emborg ME, Prockop DJ, Roberts M, Schatten G, Rao M, Harding J, Mirochnitchenko O (2013) Strategies for improving animal models for regenerative medicine. Cell Stem Cell 12:271–274
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607
Deng D, Yan C, Pan X, Mahfouz M, Wang J, Zhu J-K, Shi Y, Yan N (2012) Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 335:720–723
Ebert KM, Paynton BV, McKnight GS, Brinster RL (1984) Translation and stability of ovalbumin messenger RNA injected into growing oocytes and fertilized ova of mice. J Embryol Exp Morphol 84:91–103
Fan N, Lai L (2013) Genetically modified pig models for human diseases. J Genet Genomics 40:67–73
Garcia-Vazquez FA, Ruiz S, Matas C, Izquierdo-Rico MJ, Grullon LA, De Ondiz A, Vieira L, Aviles-Lopez K, Gutierrez-Adan A, Gadea J (2010) Production of transgenic piglets using ICSI-sperm-mediated gene transfer in combination with recombinase RecA. Reproduction 140:259–272
Gil MA, Alminana C, Cuello C, Parrilla I, Roca J, Vazquez JM, Martinez EA (2007) Brief coincubation of gametes in porcine in vitro fertilization: role of sperm:oocyte ratio and post-coincubation medium. Theriogenology 67:620–626
Grupen CG (2014) The evolution of porcine embryo in vitro production. Theriogenology 81:24–37
Han Y-M, Abeydeera LR, Kim J-H, Moon H-B, Cabot RA, Day BN, Prather RS (1999a) Growth retardation of inner cell mass cells in polyspermic porcine embryos produced in vitro. Biol Reprod 60:1110–1113
Han Y-M, Wang W-H, Abeydeera LR, Petersen AL, Kim J-H, Murphy C, Day BN, Prather RS (1999b) Pronuclear location before the first cell division determines ploidy of polyspermic pig embryos. Biol Reprod 61:1340–1346
Hirabayashi M, Kato M, Ishikawa A, Kaneko R, Yagi T, Hochi S (2005) Factors affecting production of transgenic rats by ICSI‐mediated DNA transfer: effects of sonication and freeze‐thawing of spermatozoa, rat strains for sperm and oocyte donors, and different constructs of exogenous DNA. Mol Reprod Dev 70:422–428
Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278
Iqbal K, Barg-Kues B, Broll S, Bode J, Niemann H, Kues W (2009) Cytoplasmic injection of circular plasmids allows targeted expression in mammalian embryos. Biotechniques 47:959–968
Ivics Z, Garrels W, Mates L, Yau TY, Bashir S, Zidek V, Landa V, Geurts A, Pravenec M, Rulicke T, Kues WA, Izsvak Z (2014) Germline transgenesis in pigs by cytoplasmic microinjection of Sleeping Beauty transposons. Nat Protoc 9:810–827
Kikuchi K (2004) Developmental competence of porcine blastocysts produced in vitro. J Reprod Dev 50:21–28
Kues WA, Niemann H (2011) Advances in farm animal transgenesis. Prev Vet Med 102:146–156
Liu H, Chen Y, Niu Y, Zhang K, Kang Y, Ge W, Liu X, Zhao E, Wang C, Lin S (2014a) TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell 14:323–328
Liu, S., Cui, K., Li, H. L., Sun, J. M., Lu, X. R., Shen, K. Y., Liu, Q. Y. and Shi, D. S. (2014b) Comparison of chemical, electrical, and combined activation methods for in vitro matured porcine oocytes. In Vitro Cellular & Developmental Biology-Animal, 1–10.
Page RL, Butler SP, Subramanian A, Gwazdauskas FC, Johnson JL, Velander WH (1995) Transgenesis in mice by cytoplasmic injection of polylysine/DNA mixtures. Transgenic Res 4:353–360
Paynton B, Ebert K, Brinster R (1983) Synthesis and secretion of ovalbumin by mouse-growing oocytes following microinjection of chick ovalbumin mRNA. Exp Cell Res 144:214–218
Perry AC, Wakayama T, Kishikawa H, Kasai T, Okabe M, Toyoda Y, Yanagimachi R (1999) Mammalian transgenesis by intracytoplasmic sperm injection. Science 284:1180–1183
Sumiyama K, Ruddle FH (2003) Regulation of Dlx3 gene expression in visceral arches by evolutionarily conserved enhancer elements. Proc Natl Acad Sci 100:4030–4034
Thouas G, Korfiatis N, French A, Jones G, Trounson A (2001) Simplified technique for differential staining of inner cell mass and trophectoderm cells of mouse and bovine blastocysts. Reprod Biomed Online 3:25–29
Urasaki, A. and Kawakami, K. (2009) Analysis of genes and genome by the tol2-mediated gene and enhancer trap methods. In Zebrafish Springer, pp. 85–102
Wang H, Hu Y-C, Markoulaki S, Welstead GG, Cheng AW, Shivalila CS, Pyntikova T, Dadon DB, Voytas DF, Bogdanove AJ (2013) TALEN-mediated editing of the mouse Y chromosome. Nat Biotechnol 31:530–532
Whyte JJ, Prather RS (2011) Genetic modifications of pigs for medicine and agriculture. Mol Reprod Dev 78:879–891
Wiedenheft B, Sternberg SH, Doudna JA (2012) RNA-guided genetic silencing systems in bacteria and archaea. Nature 482:331–338
Wyman C, Kanaar R (2006) DNA double-strand break repair: all’s well that ends well. Annu Rev Genet 40:363–383
Yong HY, Hao Y, Lai L, Li R, Murphy CN, Rieke A, Wax D, Samuel M, Prather RS (2006) Production of a transgenic piglet by a sperm injection technique in which no chemical or physical treatments were used for oocytes or sperm. Mol Reprod Dev 73:595–599
Yoshioka K, Suzuki C, Tanaka A, Anas IM, Iwamura S (2002) Birth of piglets derived from porcine zygotes cultured in a chemically defined medium. Biol Reprod 66:112–119
Zu Y, Tong X, Wang Z, Liu D, Pan R, Li Z, Hu Y, Luo Z, Huang P, Wu Q (2013) TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat Methods 10:329–331
Acknowledgments
This work was funded by the Hebei Provincial National Natural Science Fund (H2016314002) and the China 863 high-tec Project (2011AA100607).
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: Tetsuji Okamoto
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 13 kb)
ESM 2
(DOCX 14 kb)
ESM 3
(DOCX 13 kb)
ESM 4
(DOCX 13 kb)
ESM 5
(DOCX 15 kb)
ESM 6
(DOCX 15 kb)
ESM 7
(DOCX 15 kb)
ESM 8
(DOCX 15 kb)
ESM 9
(DOCX 14 kb)
ESM 10
(DOCX 15 kb)
Fig. 1
Transgene vector constructions. pcDNA-EGFP-N1 (GIF 7 kb)
PRL-EGFP: EGFP Expression Vector with PRL ORF Controled by BCN promoter (GIF 11 kb)
TALEN-BMP15 and TALEN- FoxO3: The TALEN expression plasmid for knocking-out BMP15 and FoxO3 of porcine (GIF 9 kb)
hSpCas9 expression plasmid with a NLS at the N-terminal (GIF 9 kb)
Cas-BMP15, Cas-FoxO3 and Cas-GDF8 gRNA expression vector with a PsiI restriction endonuclease site for linearized (GIF 8 kb)
Fig. 2
Sequence alignment of GDF8 IVF mutant embryos (GIF 187 kb)
(GIF 244 kb)
(GIF 231 kb)
Rights and permissions
About this article

Cite this article
Liu, S., Liu, X., Huang, H. et al. Factors affecting efficiency of introducing foreign DNA and RNA into parthenogenetic or in vitro-fertilized porcine eggs by cytoplasmic microinjection. In Vitro Cell.Dev.Biol.-Animal 52, 713–722 (2016). https://doi.org/10.1007/s11626-016-0025-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-016-0025-1