
Abstract
Melatonin is thought to have the ability of antiatherogenic, antioxidant, and vasodilatory. It is not only a promising protective in acute myocardial infarction but is also a useful tool in the treatment of pathological remodeling. However, its role in myocardial hypertrophy remains unclear. In this study, we investigated the protective effects of melatonin on myocardial hypertrophy induced by lipopolysaccharide (LPS) and to identify their precise mechanisms. The cultured myocardial cell was divided into six groups: control group, LPS group, LPS + ethanol (4%), LPS + melatonin (1.5 mg/ml) group, LPS + melatonin (3 mg/ml) group, and LPS + melatonin (6 mg/ml) group. The morphologic change of myocardial cell was observed by inverted phase contrast microscope. The protein level of myocardial cell was measured by Coomassie brilliant blue protein kit. The secretion level of tumor necrosis factor-α (TNF-α) was evaluated by enzyme-linked immunosorbent assay (ELISA). Ca2+ transient in Fura-2/AM-loaded cells was measured by Till image system. The expression of Ca2+/calmodulin-dependent kinase II (CaMKII) and calcineurin (CaN) was measured by Western blot analysis. Our data demonstrated that LPS induced myocardial hypertrophy, promoted the secretion levels of TNF-α, and increased Ca2+ transient level and the expression of CaMKII and CaN. Administration of melatonin 30 min prior to LPS stimulation dose-dependently attenuated myocardial hypertrophy. In conclusion, the results revealed that melatonin had the potential to protect against myocardial hypertrophy induced by LPS in vitro through downregulation of the TNF-α expression and retains the intracellular Ca2+ homeostasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
N-Acetyl-5-methoxytryptamine (melatonin), the main product of the pineal gland (Menendez-Pelaez and Reiter 1993; Reiter and Tan 2003), is found in high concentrations in other body fluids and tissues and possesses anti-inflammatory and antioxidant actions (Manda et al. 2007; Hardeland et al. 2009; Chen et al. 2011; Galano et al. 2011). Melatonin and its metabolic derivatives are uncommonly effective direct free radical scavengers, while also stimulate the activities of several antioxidative enzymes (Reiter et al. 1995). Melatonin has been recently introduced as a promising protective in acute myocardial infarction (Dominguez-Rodriguez et al. 2012). However, recent data indicate that melatonin may also be a useful tool in the treatment of pathological remodeling within heart disease (Oliveira et al. 2013; Simko and Paulis 2013; Zhang et al. 2013).
Myocardial hypertrophy occurs in a wide variety of clinically important conditions, such as hypertension and valvular heart disease, and increases the risk of heart failure (Frey et al. 2004; Wang et al. 2012). Myocardial hypertrophy is one of the major responses of myocardial cell to mechanical and neurohormonal stimuli (Anderson et al. 2011). Whereas left ventricular (LV) pressure overload initially induces adaptive hypertrophy (Gunther and Grossman 1979), sustained pressure overload eventually results in pathological hypertrophy (Berenji et al. 2005). Pathological hypertrophy is characterized by multiple pathophysiological features, including fibrosis, cardiac dysfunction, and inflammatory responses which include macrophage infiltration, fibroblast proliferation, and secretion of inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and IL-1 (Diwan et al. 2003; Berenji et al. 2005). Previous reports using animal models demonstrated that increased proinflammatory cytokine expression and intracellular Ca2+ overload plays an important role in the pathogenesis of heart failure with pathological hypertrophy (Shioi et al. 1997; Wang et al. 2012; Lu et al. 2013). Lipopolysaccharide (LPS), the antigenic component of the gram-negative bacterial cell wall, is known as the exogenous ligand of Toll-like receptor-4 (Chow et al. 1999), and a strong stimulus for inflammatory response by upregulation or release of cytokines (Yang et al. 1998). However, recent studies have suggested that LPS is sufficient to cause myocardial hypertrophy (Liu et al. 2008; Singh et al. 2012).
In the present study, we investigated the protective effects of melatonin on myocardial hypertrophy induced by LPS and to identify their precise mechanisms.
Materials and Methods
Culture of neonatal rat ventricular myocytes and treatment protocol.
Primary cultures of myocardial cell were prepared from the ventricles of 30 neonatal SD (Sprague Dawley) rats as described by Simpson (1985). Myocardial cell were collected for culture from 1- to 2-d-old neonatal rats and maintained at 37°C in humidified air containing 5% CO2. After dissociation of the heart tissue with trypsin, cells were preplated for 1 h onto 100-mm culture dishes in DMEM with 10% fetal calf serum to reduce the number of non-myocyte cells. The cells which had not attached on the dishes in an hour were plated in six-well culture plates at a density of 1 × 106/ml. Non-myocytes in the cultures were limited to ≤10% of the total cell number by inclusion of bromodeoxyuridine (0.1 mM) (Roche, Mannheim, Germany) in the medium for the first 2 d. After 24 h, the culture medium was replaced with serum-free medium. Myocardial hypertrophy was induced by LPS. Briefly, after cultured for 24 h in serum-free medium, the myocardial cell were treated with 1 μg/ml LPS (Sigma, St. Louis, MO) for 48 h to initiate hypertrophy. Melatonin (1.5, 3, 6 mg/ml) (Sigma) were dissolved in normal sodium (NS) containing 4% ethanol and administered 30 min before LPS administration in the LPS + melatonin (1.5 mg/ml) group, LPS + melatonin (3 mg/ml) group, and LPS + melatonin (6 mg/ml) group. The same amount of NS containing 4% ethanol was administered 30 min before LPS administration in the LPS + ethanol (4%) group. The cells were subsequently cultured for an additional 72 h before further evaluation. This dose of melatonin was based on earlier studies that showed anti-inflammatory effect (Cuzzocrea et al. 1999; Ortiz et al. 2014). Photomicrographs were taken by inverted phase contrast microscope and analyzed for size of cells by CIAS Daheng image analysis system (Beijing, China).
The determination of protein level of myocardial cell.
The abovementioned cells were washed with PBS, then added with sodium dodecyl sulfate (SDS) cell lysis buffer (containing 1 μM EDTA, 2% SDS, 50 μM Tris–HCl). SDS (300 μl) was put in a 24-well plate and rock for several times to ensure that the cells were resolved sufficiently, then collected in Eppendorf. After ice bath for 30 min, they were centrifuged at 4°C and 12,000 r/min for 10 min. The supernatant were collected then detected by Coomassie brilliant blue protein kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Detection of the secretion level of TNF-α in myocardial cell by ELISA.
The supernatant of the abovementioned cultured cells was collected, centrifuged at 5000 r/min for 5 min, and immediately frozen at −70°C. The concentrations of TNF-α secreted by myocardial cell in the medium were measured using mouse-specific ELISA kits (Boatman Biotech Co., Ltd., Shanghai, China) according to the manufacturer’s protocols. Results were expressed as the mean of three assays for each condition in five different cultures.
Measurement of cytosolic calcium transient.
A spectrofluorimetric method was used to measure cytosolic Ca2+ transients using Fura-2/AM (Sigma) as the Ca2+ indicator. Cultured myocardial cell were incubated with Fura-2/AM (4 μM) in the medium for 25 min. To allow the de-esterification of Fura-2/AM in the cytosol, the loaded cells were maintained at 24–26°C for 60 min before [Ca2+]i was measured. The myocardial cell were transferred to a superfusion chamber on the stage of an inverted microscope, which was coupled to a Till imaging system (Martinsried, Germany), and the cells were superfused with Hanks buffer. The emitted light was filtered at 510 nm. Fluorescence signals at 340 nm (F340) and 380 nm (F380) were recorded in a personal computer for data processing and analysis. At the end of each experiment, the Ca2+ ionophore ionomycin (20 μM) was added to each sample to estimate maximal fluorescence. Ethylene glycol tetracetic acid (EGTA) was added to a final concentration of 20 mM for the Ca2+-free condition. Cytosolic [Ca2+] was calculated by the following formula: [Ca2+]i = K d × (Sf2 / Sb2) × (R 340/380 − R min) / (R max − R340/380) (Grynkiewicz et al. 1985), where K d is the dissociation constant of Fura-2/AM for Ca2+ and was assumed to be 225 nM at 37°C. R 340/380 is the ratio of corrected fluorescence signals. R max is the ratio obtained after ionomycin treatment. R min is the ratio of the corrected signals obtained after EGTA treatment. Sf2 and Sb2 represent the emission intensities at 380 nm excitation at saturation and under Ca2+-free conditions, respectively.
Western blot analysis.
Cells were diluted in buffer containing 65 mM Tris–HCl, pH 6.8, 3% SDS, 10% glycerol, and 6 M urea. After measurement of the protein concentration (BCA kit, Pierce, Rockford, IL), β-mercaptoethanol and bromophenol blue were added to the buffer for electrophoresis. A volume containing 60 μg protein (for Ca2+/calmodulin-dependent kinase II (CaMKII) δB or calcineurin (CaN)) was separated on a 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane (BioRad, Hercules, CA). The blots were incubated at 4°C overnight with antibodies to CaMKIIδB or CaN, and the resulting bands were detected using enhanced chemiluminescence. An antibody to CaMKIIδB at Thr-276 (1:1000 dilution; Santa Cruz, Santa Cruz, CA) was used to detect the activated form of the kinase. The activated form of the phosphatase was evaluated with an antibody to CaN (1:2000 dilution; Santa Cruz). The intensities of the resulting bands were quantified using a CAMIAS008 image analysis system.
Statistical analysis.
The experiment was repeated six times, and each time, five neonatal SD rats were prepared (n = 6). When determining the volume of the cells, cells in the field of 200 times were calculated.
Statistical analysis was performed using Statistical Package for Social Science 16.0 (SPSS 16.0). Statistical comparisons were performed using one-way analysis of variance (ANOVA), and differences at P < 0.05 were considered statistically significant.
Results
The volume of myocardial cell.
The shape of myocardial cell was fusiform, irregular triangle, or polygon. The volume of myocardial cell in the LPS group and LPS + ethanol (4%) group was obviously augmented compared with the control group. The volume of myocardial cell in the LPS + melatonin (1.5 mg/ml) group was smaller than that of the LPS group and LPS + ethanol (4%) group but larger than that of the control group. The volume of myocardial cell in the LPS + melatonin (3 mg/ml) group and LPS + melatonin (6 mg/ml) group was similar to that of the control group (Fig. 1).

The volume of myocardial cell in each group. Photomicrographs of myocardial cell cultured in each group were taken by inverted phase contrast microscope and analyzed for size of cells by CIAS Daheng image analysis system. The shape of myocardial cell was fusiform, irregular triangle, or polygon (A). The volume of myocardial cell in the LPS group and LPS + ethanol (4%) group was obviously augmented compared with the control group. The volume of myocardial cell in the LPS + melatonin (1.5 mg/ml) group was smaller than that of the LPS group and LPS + ethanol (4%) group but larger than that of the control group. The volume of myocardial cell in LPS + melatonin (3 mg/ml) group and LPS + melatonin (6 mg/ml) group was similar to that of the control group (A, B). ∆p < 0.01, versus control group; *p < 0.01, versus LPS group; #p < 0.01, versus LPS + ethanol (4%) group; @p < 0.01, versus LPS + melatonin (1.5 mg/ml) group.
The protein level of myocardial cell.
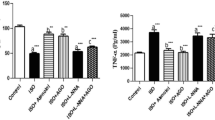
The total protein level of myocardial cell in the LPS group and LPS + ethanol (4%) group was more than that of the control group. The total protein level of myocardial cell in the LPS + melatonin (1.5 mg/ml) group was less than that of the LPS group and LPS + ethanol (4%) group but more than that of the control group. The total protein level of myocardial cell in the LPS + melatonin (3 mg/ml) group and LPS + melatonin (6 mg/ml) group was similar to that of the control group (Fig. 2).

The protein level of myocardial cell in each group. The protein level of myocardial cell was measured by Coomassie brilliant blue protein kit. The total protein level of myocardial cell in the LPS group and LPS + ethanol (4%) group was more than that of the control group. The total protein level of myocardial cell in the LPS + melatonin (1.5 mg/ml) group was less than that of the LPS group and LPS + ethanol (4%) group but more than that of the control group. The total protein level of myocardial cell in the LPS + melatonin (3 mg/ml) group and LPS + melatonin (6 mg/ml) group was similar to that of the control group. ∆p < 0.01, versus control group; *p < 0.01, versus LPS group; #p < 0.01, versus LPS + ethanol (4%) group; @p < 0.01, versus LPS + melatonin (1.5 mg/ml) group.
The secretion level of TNF-α in myocardial cell.
The level of TNF-α secreted in medium of the LPS group and LPS + ethanol (4%) group was more than that of the control group. The level of TNF-α secreted in medium of the LPS + melatonin (1.5 mg/ml) group was less than that of the LPS group and LPS + ethanol (4%) group but more than that of the control group. The level of TNF-α secreted in medium of the LPS + melatonin (3 mg/ml) group and LPS + melatonin (6 mg/ml) group was similar to that of the control group (Fig. 3). Our data also demonstrated that TNF-alpha induced myocardial hypertrophy. Administration of melatonin 30 min prior to TNF-alpha stimulation attenuated myocardial hypertrophy (Supplementary Figs. 1 and 2).

The TNF-α level of myocardial cell in each group. The secretion level of tumor necrosis factor-α (TNF-α) was evaluated by ELISA. The TNF-α level of myocardial cell in the LPS group and LPS + ethanol (4%) group was more than that of the control group. The TNF-α level of myocardial cell of in the LPS + melatonin (1.5 mg/ml) group was less than that of the LPS group and LPS + ethanol (4%) group but more than that of the control group. The TNF-α level of myocardial cell of the LPS + melatonin (3 mg/ml) group and LPS + melatonin (6 mg/ml) group was similar to that of the control group. ∆p < 0.01, versus control group; *p < 0.01, versus LPS group; #p < 0.01, versus LPS + ethanol (4%) group; @p < 0.01, versus LPS + melatonin (1.5 mg/ml) group.
The spontaneous [Ca 2+ ] i transients in myocardial cell.
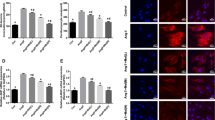
Compared with the control group, the amplitude of spontaneous [Ca2+]i transients in myocardial cell of the LPS groups and LPS + ethanol (4%) group was enhanced. The amplitude of spontaneous [Ca2+]i transients in myocardial cell of the LPS groups + melatonin (1.5 mg/ml) group was less than that of the LPS group and LPS + ethanol (4%) group but more than that of the control group. The amplitude of spontaneous [Ca2+]i transients in myocardial cell of LPS + melatonin (3 mg/ml) group and LPS + melatonin (6 mg/ml) group was similar to that of the control group (Fig. 4A ). None of the treatments had any effect on the resting [Ca2+]i (Fig. 4B ) or the frequency of spontaneous [Ca2+]i transients (Fig. 4C ).

The spontaneous [Ca2+]i transients of myocardial cell in each group. Ca2+ transient in Fura-2/AM-loaded cells was measured by Till image system. Compared with the control group, the amplitude of spontaneous [Ca2+]i transients in myocardial cell of the LPS groups and LPS + ethanol (4%) group was enhanced. The amplitude of spontaneous [Ca2+]i transients in myocardial cell of the LPS groups + melatonin (1.5 mg/ml) group was less than that of the LPS group and LPS + ethanol (4%) group but more than that of control group. The amplitude of spontaneous [Ca2+]i transients in myocardial cell of the LPS + melatonin (3 mg/ml) group and LPS + melatonin (6 mg/ml) group was similar to that of the control group (A). None of the treatments had any effect on the resting [Ca2+]i (B) or the frequency of spontaneous [Ca2+]i transients (C). ∆p < 0.01, versus control group; * p < 0.01, versus LPS group; #p < 0.01, versus LPS + ethanol (4%) group; @p < 0.01, versus LPS + melatonin (1.5 mg/ml) group.
The expression of CaMKIIδB and CaN in myocardial cell.
The expression of CaMKIIδB and CaN in myocardial cell of the LPS group and LPS + ethanol (4%) group was more than that of the control group. The expression of CaMKIIδB and CaN in myocardial cell of the LPS + melatonin (1.5 mg/ml) group was less than that of the LPS group and LPS + ethanol (4%) group but more than that of the control group. The expression of CaMKIIδB and CaN in myocardial cell of the LPS + melatonin (3 mg/ml) group and LPS + melatonin (6 mg/ml) group was similar to that of the control group (Fig. 5).

The expression of CaMKIIδB and CaN of myocardial cell in each group. The expression of CaMKIIδB and CaN in myocardial cell was measured by Western blot analysis. The expression of CaMKIIδB and CaN in myocardial cell of the LPS group and LPS + ethanol (4%) group was more than that of the control group. The expression of CaMKIIδB and CaN in myocardial cell of the LPS + melatonin (1.5 mg/ml) group was less than that of the LPS group and LPS + ethanol (4%) group but more than that of the control group. The expression of CaMKIIδB and CaN in myocardial cell of the LPS + melatonin (3 mg/ml) group and LPS + melatonin (6 mg/ml) group was similar to that of the control group. ∆p < 0.01, versus control group; *p < 0.01, versus LPS group; #p < 0.01, versus LPS + ethanol (%) group; @p < 0.01, versus LPS + melatonin (6 mg/ml) group; $p < 0.01, versus LPS + melatonin (3 mg/ml) group.
Discussion
LPS is one of the most common causes of inflammation and is sufficient to induce myocardiac hypertrophy in H9c2 myocardiac cells (Liu et al. 2008). Myocardiac hypertrophy is accompanied by inflammatory responses which include macrophage infiltration, fibroblast proliferation, and secretion of inflammatory cytokines, such as TNF-α, IL-6, and IL-1 (Diwan et al. 2003; Berenji et al. 2005). Clinical evidences suggest that chronic inflammation and heart failure are linked (Heymans et al. 2009). In this study, we found that myocardial cell cultured in LPS showed obvious hypertrophy compared with the cells cultured in the control group, which was consistent with previous studies.
Melatonin is useful tool in the treatment of pathological remodeling. It is believed that melatonin is clearly protective when damage to the heart is a result of free radicals. It can curtailed oxidative damage to the heart that resulted in an attenuation of left ventricular fibrosis (Reiter et al. 2010). However, its role in myocardial hypertrophy remains unclear. Pretreated myocardial cell with melatonin could significantly attenuate the hypertrophic responses induced by LPS, implying that melatonin may also play a protective role against inflammation-induced myocardiac hypertrophy.
TNF-α is a potent proinflammatory cytokine that is produced by several types of cells, including myocardial cell (Arras et al. 1996). It is reported that the circulating levels of TNF-α is elevated in patients with chronic heart failure, such as ischemic heart disease and dilated cardiomyopathy (Yokoyama et al. 1997). Myocardial hypertrophy is one of the principal features in these cardiac diseases (Beltrami et al. 1994). In this study, the secretion level of TNF-α in medium was elevated which is consistent with those of previous studies. Administration of melatonin prior to lipopolysaccharide stimulation dose-dependently attenuated the secretion level of TNF-α. Several studies have indicated that TNF-α is one of the most important factors in the induction of myocardiac hypertrophy (Yokoyama et al. 1997; Bozkurt et al. 1998; Nakamura et al. 1998; Sekiguchi et al. 2004). Moreover, the direct effect of TNF-α on myocardiac hypertrophy has been demonstrated in cultured myocardial cell (Wang et al. 2012). However, the molecular mechanism of TNF-α-induced myocardiac hypertrophy remains unclear. TNF-α interacts with two structurally distinct receptors, type 1 (TNFR1) and type 2 (TNFR2), which are expressed in myocardial cell and stimulates downstream signaling molecules to mediate the biological responses. So, the heart is not only a site of TNF-α synthesis but is also a target of TNF-α activity.
A number of hypertrophic stimuli increase intracellular Ca2+ levels (Zhu et al. 2000), and reports suggest that Ca2+ is involved in the generation of myocardiac hypertrophy (Bogoyevitch et al. 1996). Ca2+ plays a pivotal role in not only the process of excitation-contraction coupling but also in the activation of Ca2+-dependent signaling pathways in the myocardium. One of the main established Ca2+-dependent pathways during myocardiac hypertrophy is the CaN signaling pathway, which has been implicated in the progression of myocardiac hypertrophy induced by pressure overload, angiotensin II, and endothelin-1 (Zhu et al. 2000; Fu et al. 2001; Saito et al. 2003). The other main Ca2+-dependent pathways during myocardiac hypertrophy is CaMKII signaling pathway. It regulates expression of inflammatory genes such as TNF-α and affects adverse outcomes after myocardial infarction induced by LPS (Erickson et al. 2008; Singh et al. 2009). This can also partly explain why the secretion level of TNF-α in medium was elevated in this study. Our data showed the amplitude of spontaneous Ca2+ and the expression of CaMKIIδB and CaN in myocardial cell increased in myocardial cell induced by LPS. Administration of melatonin prior to lipopolysaccharide stimulation dose-dependently attenuated the amplitude of spontaneous Ca2+ and the expression of CaMKIIδB and CaN in myocardial cell in myocardial cell induced by LPS.
Melatonin is a natural molecule which expresses low toxicity at both physiological and pharmacological concentrations. It is easily available and inexpensive, and its formula with controlled release and long action is accessible. Melatonin with its extraordinary antioxidant and scavenging properties (Perez et al. 2007; Reiter et al. 2007) modulates the level of inflammatory cytokines (Korkmaz et al. 2009) and sympathicovagal balance (Arangino et al. 1999) and interferes with specific melatonin receptors in the brain and cardiovascular system (Reiter et al. 2007). Melatonin can protect the heart against pathological left ventricular remodeling through affecting the process of collagen cross-linking (Simko et al. 2010). The study provides evidence confirming that melatonin has the potential to protect against myocardial hypertrophy induced by LPS in vitro through downregulation of the TNF-α expression and retains the intracellular Ca2+ homeostasis. If the effect of melatonin was confirmed in the clinical setting, melatonin could become a promising tool in the struggle against myocardial hypertrophy.
References
Anderson ME, Brown JH, Bers DM (2011) CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol 51:468–473
Arangino S, Cagnacci A, Angiolucci M, Vacca AM, Longu G, Volpe A, Melis GB (1999) Effects of melatonin on vascular reactivity, catecholamine levels, and blood pressure in healthy men. Am J Cardiol 83:1417–1419
Arras M, Hoche A, Bohle R, Eckert P, Riedel W, Schaper J (1996) Tumor necrosis factor-alpha in macrophages of heart, liver, kidney, and in the pituitary gland. Cell Tissue Res 285:39–49
Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Quaini F, Sonnenblick EH, Olivetti G, Anversa P (1994) Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circ 89:151–163
Berenji K, Drazner MH, Rothermel BA, Hill JA (2005) Does load-induced ventricular hypertrophy progress to systolic heart failure? Am J Physiol Heart Circ Physiol 289:H8–H16
Bogoyevitch MA, Andersson MB, Gillespie-Brown J, Clerk A, Glennon PE, Fuller SJ, Sugden PH (1996) Adrenergic receptor stimulation of the mitogen-activated protein kinase cascade and cardiac hypertrophy. Biochem J 314(Pt 1):115–121
Bozkurt B, Kribbs SB, Clubb FJ Jr, Michael LH, Didenko VV, Hornsby PJ, Seta Y, Oral H, Spinale FG, Mann DL (1998) Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circ 97:1382–1391
Chen CF, Wang D, Reiter RJ, Yeh DY (2011) Oral melatonin attenuates lung inflammation and airway hyperreactivity induced by inhalation of aerosolized pancreatic fluid in rats. J Pineal Res 50:46–53
Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F (1999) Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem 274:10689–10692
Cuzzocrea S, Costantino G, Mazzon E, Caputi AP (1999) Regulation of prostaglandin production in carrageenan-induced pleurisy by melatonin. J Pineal Res 27:9–14
Diwan A, Tran T, Misra A, Mann DL (2003) Inflammatory mediators and the failing heart: a translational approach. Curr Mol Med 3:161–182
Dominguez-Rodriguez A, Abreu-Gonzalez P, Reiter RJ (2012) Melatonin and cardioprotection in the acute myocardial infarction: a promising cardioprotective agent. Int J Cardiol 158:309–310
Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N et al (2008) A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133:462–474
Frey N, Katus HA, Olson EN, Hill JA (2004) Hypertrophy of the heart: a new therapeutic target? Circ 109:1580–1589
Fu M, Zhang J, Xu S, Pang Y, Liu N, Tang C (2001) Role of calcineurin in angiotensin II-induced cardiac myocyte hypertrophy of rats. Chin Med Sci J 16:1–4
Galano A, Tan DX, Reiter RJ (2011) Melatonin as a natural ally against oxidative stress: a physicochemical examination. J Pineal Res 51:1–16
Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260:3440–3450
Gunther S, Grossman W (1979) Determinants of ventricular function in pressure-overload hypertrophy in man. Circ 59:679–688
Hardeland R, Tan DX, Reiter RJ (2009) Kynuramines, metabolites of melatonin and other indoles: the resurrection of an almost forgotten class of biogenic amines. J Pineal Res 47:109–126
Heymans S, Hirsch E, Anker SD, Aukrust P, Balligand JL, Cohen-Tervaert JW, Drexler H, Filippatos G, Felix SB, Gullestad L et al (2009) Inflammation as a therapeutic target in heart failure? A scientific statement from the translational research committee of the heart failure association of the european society of cardiology. Eur J Heart Fail 11:119–129
Korkmaz A, Reiter RJ, Topal T, Manchester LC, Oter S, Tan DX (2009) Melatonin: an established antioxidant worthy of use in clinical trials. Mol Med 15:43–50
Liu CJ, Cheng YC, Lee KW, Hsu HH, Chu CH, Tsai FJ, Tsai CH, Chu CY, Liu JY, Kuo WW et al (2008) Lipopolysaccharide induces cellular hypertrophy through calcineurin/NFAT-3 signaling pathway in H9c2 myocardiac cells. Mol Cell Biochem 313:167–178
Lu M, Wang H, Wang J, Zhang J, Yang J, Liang L, Maslov LN (2013) Astragaloside IV protects against cardiac hypertrophy via inhibiting the Ca2+/CaN signaling pathway. Planta Med 80(1):63–69. doi:10.1055/s-0033-1360129
Manda K, Ueno M, Anzai K (2007) AFMK, a melatonin metabolite, attenuates X-ray-induced oxidative damage to DNA, proteins and lipids in mice. J Pineal Res 42:386–393
Menendez-Pelaez A, Reiter RJ (1993) Distribution of melatonin in mammalian tissues: the relative importance of nuclear versus cytosolic localization. J Pineal Res 15:59–69
Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, Namba M (1998) Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circ 98:794–799
Oliveira LG, Kuehn CC, dos Santos CD, Miranda MA, da Costa CM, Mendonca VJ, do Prado Junior JC (2013) Protective actions of melatonin against heart damage during chronic Chagas disease. Acta Trop 128:652–658
Ortiz F, Garcia JA, Acuna-Castroviejo D, Doerrier C, Lopez A, Venegas C, Volt H, Luna-Sanchez M, Lopez LC, Escames G (2014) The beneficial effects of melatonin against heart mitochondrial impairment during sepsis: inhibition of iNOS and preservation of nNOS. J Pineal Res 56:71–81
Perez MJ, Castano B, Gonzalez-Buitrago JM, Marin JJ (2007) Multiple protective effects of melatonin against maternal cholestasis-induced oxidative stress and apoptosis in the rat fetal liver-placenta-maternal liver trio. J Pineal Res 43:130–139
Reiter RJ, Tan DX (2003) What constitutes a physiological concentration of melatonin? J Pineal Res 34:79–80
Reiter RJ, Melchiorri D, Sewerynek E, Poeggeler B, Barlow-Walden L, Chuang J, Ortiz GG, Acuna-Castroviejo D (1995) A review of the evidence supporting melatonin’s role as an antioxidant. J Pineal Res 18:1–11
Reiter RJ, Tan DX, Manchester LC, Pilar Terron M, Flores LJ, Koppisepi S (2007) Medical implications of melatonin: receptor-mediated and receptor-independent actions. Adv Med Sci 52:11–28
Reiter RJ, Manchester LC, Fuentes-Broto L, Tan D-X (2010) Cardiac hypertrophy and remodelling: pathophysiological consequences and protective effects of melatonin. J Hypertens 28:S7–S12
Saito T, Fukuzawa J, Osaki J, Sakuragi H, Yao N, Haneda T, Fujino T, Wakamiya N, Kikuchi K, Hasebe N (2003) Roles of calcineurin and calcium/calmodulin-dependent protein kinase II in pressure overload-induced cardiac hypertrophy. J Mol Cell Cardiol 35:1153–1160
Sekiguchi K, Li X, Coker M, Flesch M, Barger PM, Sivasubramanian N, Mann DL (2004) Cross-regulation between the renin-angiotensin system and inflammatory mediators in cardiac hypertrophy and failure. Cardiovasc Res 63:433–442
Shioi T, Matsumori A, Kihara Y, Inoko M, Ono K, Iwanaga Y, Yamada T, Iwasaki A, Matsushima K, Sasayama S (1997) Increased expression of interleukin-1 beta and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the hypertrophied and failing heart with pressure overload. Circ Res 81:664–671
Simko F, Paulis L (2013) Antifibrotic effect of melatonin—perspective protection in hypertensive heart disease. Int J Cardiol 168:2876–2877
Simko F, Pechanova O, Pelouch V, Krajcirovicova K, Celec P, Palffy R, Bednarova K, Vrankova S, Adamcova M, Paulis L (2010) Continuous light and L-NAME-induced left ventricular remodelling: different protection with melatonin and captopril. J Hypertens 28(Suppl 1):S13–S18
Simpson P (1985) Stimulation of hypertrophy of cultured neonatal rat heart cells through an alpha 1-adrenergic receptor and induction of beating through an alpha 1- and beta 1-adrenergic receptor interaction. Evidence for independent regulation of growth and beating. Circ Res 56:884–894
Singh MV, Kapoun A, Higgins L, Kutschke W, Thurman JM, Zhang R, Singh M, Yang J, Guan X, Lowe JS et al (2009) Ca2+/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. J Clin Invest 119:986–996
Singh MV, Swaminathan PD, Luczak ED, Kutschke W, Weiss RM, Anderson ME (2012) MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol 52:1135–1144
Wang GJ, Wang HX, Yao YS, Guo LY, Liu P et al (2012) The role of Ca2+/calmodulin-dependent protein kinase II and calcineurin in TNF-alpha-induced myocardial hypertrophy. Braz J Med Biol Res 45:1045–1051
Yang RB, Mark MR, Gray A, Huang A, Xie MH, Zhang M, Goddard A, Wood WI, Gurney AL, Godowski PJ (1998) Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nat 395:284–288
Yokoyama T, Nakano M, Bednarczyk JL, McIntyre BW, Entman M, Mann DL (1997) Tumor necrosis factor-alpha provokes a hypertrophic growth response in adult cardiac myocytes. Circ 95:1247–1252
Zhang H, Liu D, Wang X, Chen X, Long Y, Chai W, Zhou X, Rui X, Zhang Q, Wang H et al (2013) Melatonin improved rat cardiac mitochondria and survival rate in septic heart injury. J Pineal Res 55:1–6
Zhu W, Zou Y, Shiojima I, Kudoh S, Aikawa R, Hayashi D, Mizukami M, Toko H, Shibasaki F, Yazaki Y et al (2000) Ca2+/calmodulin-dependent kinase II and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. J Biol Chem 275:15239–15245
Acknowledgment
This study was supported by grants from the Innovation and demonstration of Nantong Social Science (S11954).
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: T. Okamoto
Qi Lu and Xin Yi contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure 1
The volume of myocardial cell in each group. Photomicrographs of myocardial cell cultured in each group were taken by inverted phase contrast microscope and analyzed for size of cells by CIAS Daheng image analysis system. The shape of myocardial cell was fusiform, irregular triangle or polygon (A). The volume of myocardial cell in TNF-α group and TNF-α+ethanol group was obviously augmented compared with control group. The volume of myocardial cell in TNF-α+melatonin group was smaller than that of TNF-αgroup and TNF-α+ethanol group and was similar to that of control group (A, B). ∆ p< 0.01, vs. control group; * p< 0.01, vs. TNF-αgroup; # p< 0.01, vs. TNF-α+ethanol group. (GIF 29 kb)
Figure 2
The protein level of myocardial cell in each group. The protein level of myocardial cell was measured by coomassie brilliant blue protein kit. The total protein level of myocardial cell in TNF-αgroup and TNF-α+ethanol group was more than that of control group and was was similar to that of control group. ∆ p< 0.01, vs. control group; * p< 0.01, vs. TNF-αgroup; # p< 0.01, vs. TNF-α+ethanol group. (GIF 10 kb)
Rights and permissions
About this article

Cite this article
Lu, Q., Yi, X., Cheng, X. et al. Melatonin protects against myocardial hypertrophy induced by lipopolysaccharide. In Vitro Cell.Dev.Biol.-Animal 51, 353–360 (2015). https://doi.org/10.1007/s11626-014-9844-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-014-9844-0