
Abstract
Introduction
Congenital anomalies of the gastrointestinal tract are a significant cause of morbidity in children and less frequently in adults. In rare cases, they may run undetected during childhood and may present during adolescence. These abnormalities include developmental obstructive defects of the duodenum and the small intestine, anomalies of rotation and fixation, intestinal duplications, and anomalies of the colon and rectum.
Discussion
When detected in adulthood, they may require different evaluation and surgical correction. Some of these anomalies should be managed surgically as soon as they cause symptoms. Others may cause persistent problems in adulthood requiring medical management for years. Herein, we present a comprehensive review of all the different ways of diagnosis and management of these anomalies reported in the literature.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital anomalies of the gastrointestinal (GI) tract commonly present in the neonatal period or early infancy. Some of them can have life-threatening consequences if diagnosis is delayed. Therefore, the prognosis is largely dependent upon early diagnosis and appropriate management and surgical treatment. However, in rare cases, they can present de novo during adolescence. These lesions may form an important part of pathology in patients along time out of the cradle. When diagnosed in adulthood, they may require a different perspective than in children regarding management and surgical correction. In this study, we present descriptions of the most common anomalies which may be encountered in the adult. We propose management options according to the current medical literature.
Congenital Anomalies
Adult Idiopathic Hypertrophic Pyloric Stenosis
Congenital hypertrophic pyloric stenosis is a benign disease caused by hypertrophy of the circular muscle fibers of the pylorus. It is manifested usually during the first 2 months of life. Its incidence is approximately 0.25–0.5% of all births. A mild form of congenital pyloric stenosis may occasionally present later in adult life.1 The exact occurrence of this form cannot be estimated accurately, since the majority of these patients are asymptomatic for years. Zavala et al.2 reported on a family with both congenital and adult type of hypertrophic pyloric stenosis. This hypothesis is further supported by the fact that approximately 80% of patients with the adult form of the disease are men, which is in accordance with the male predominance of congenital pyloric stenosis.3 In addition, this form of primary pyloric stenosis should be differentiated from the secondary form, which is caused by other diseases of the gastrointestinal tract, such as peptic ulcer disease, hypertrophic gastritis, or malignancy.4
In the adult idiopathic hypertrophic pyloric stenosis (AIHPS), the pylorus is bulbous or fusiform, with its thickest portion at the pyloroduodenal junction5 (Fig. 1). Microscopically, there is a substantial hypertrophy of the circular and occasionally of the longitudinal muscle in a focal or diffuse fashion.6,7

In AIHPS, the pylorus is bulbous or fusiform, with its thickest portion at the pyloroduodenal junction (A = pyloroduodenal junction, B = pylorogastric junction).
Clinical diagnosis depends primarily on the specific symptomatology, which consists of symptoms of delayed gastric emptying, without the presence of pain. Other diseases which may cause delayed gastric emptying such as diabetes mellitus, scleroderma, gastroparesis, or secondary pyloric stenosis should be ruled out.8
AIHPS can coexist with other rare congenital anomalies, such as congenital hypothyroidism, congenital diaphragmatic hernia, congenital short bowel, jejunal atresia, and recessive polycystic kidney disease.2,9
The radiologic and endoscopic studies are often nonspecific. In upper GI series, the diagnosis should be suspected in case there is elongation of the pyloric canal accompanied often by a marked dilatation of the stomach.7,9 In AIHPS, the narrowed pyloric canal may be seen as an extremely thin line of barium (string sign).10 A marked thickening of the pyloric muscle may produce a convex indentation at the base of the duodenal bulb, causing a mushroom-like deformity (Kirklin’s sign).11 In addition, the presence of a barium-filled cleft between the hypertrophied muscle and the fibers of the pylorus itself can project into either one or both sides of the pylorus proximal to the base of the bulb (Twining’s sign).12 However, none of the above signs are pathognomonic, whereas the presence of two or more of them strengthens the radiologic diagnosis.7 Endoscopy is also useful for diagnosis: a fixed narrowed pylorus with a smooth border is the classic finding and is described as a “donut” or as the cervix sign.5,13 Another feature is the failure of the pylorus to close completely.14 Moreover, intubation of the duodenum with the endoscope can be unfeasible.
As for therapy, the first aim is to rule out carcinoma histologically, with a full thickness biopsy.15 Endoscopic dilatation of the pylorus has been suggested for patients with a high surgical risk but carries a high recurrence rate.6,14 The Ramstedt pyloromyotomy, which is often used in children, is troublesome in adults because it may cause mucosal ulceration or pyloric scarring with only partial relief.4,14
Pyloroplasty is an acceptable option (Fig. 1); however, there is a trend for recurrent obstruction. Brahos et al.16 first performed the double pyloroplasty technique, a posterior myotomy combined with the classical Weinberg modification of the Heineke-Miculicz pyloroplasty. This technique enables the surgeon to obtain biopsies, and it helps to avoid a gastric resection, especially in high-risk patients.
However, a distal gastric resection with Billroth I and II anastomosis is the most recommended procedure,5,15 especially for patients with a thick pylorus, which renders a pyloroplasty technically difficult (Table 1).
The introduction of laparoscopic pyloromyotomy by Alain et al. in 199117 has opened new horizons in the treatment of hypertrophic pyloric stenosis. The laparoscopic approach carried the general advantages of laparoscopic surgery, but it had a considerable complication rate when it was first applied in children.18 There are scattered reports of adult cases with AIHPS, which were treated laparoscopically with satisfactory results.14,19
Congenital Duodenal Anomalies
Congenital duodenal anomalies are defects in the embryologic development of the foregut. While the primitive foregut undergoes lengthening and rotation in the early embryonic life, the hepatobiliary and pancreatic anlagen begin as buds at the middle of the duodenum and gradually grow and rotate. During this period, duodenal atresias, intraluminal webs, annular pancreata, and various malrotations may develop. The annulus may completely or partially encircle the duodenum and may loosely be attached to the duodenal serosa or occasionally may be interdigitated with the muscularis mucosa of the duodenum. As for the webs, there are complete duodenal atresias or imperforate webs, intraluminal imperforate webs, and perforated webs with eccentric or central apertures. Most of the webs occur in areas of fusion or visceral outgrowth, hence their propensity to occur in the ampullary region.20 The incidence of these anomalies in the pediatric population is estimated to be one in 20,000 to 40,000 births, with incomplete obstructive lesions accounting for only 2% of them.20,21 Duodenal stenosis from annular pancreas is also very rare in adults. In fact, Ravitch22 found only three cases of this anomaly in 20,000 autopsies in John Hopkins Hospital.
The delayed presentation of these anomalies in the adolescent or adult period can hardly be explained. Total duodenal obstruction is not compatible with long-term survival. However, there are numerous reports describing annular pancreata and duodenal webs in the second, third, and later decades of life. These isolated cases may survive with liquid diet for years.21 In such cases, the presence of a dilated stomach with a proximal duodenal bulb suggests a gradual loss of peristaltic action to overcome a narrowing of the descending portion of the duodenum.
Clinical symptomatology consists of postprandial epigastric fullness, nausea, vomiting, and weight loss. Duodenal web patients may suffer from peptic ulcer disease, but this is not the case in patients with annular pancreata.21 In the latter group of patients, the development of acute pancreatitis in the annulus causes acute duodenal obstruction, with subsequent resolution of the pancreatic inflammation allowing relief of the obstruction.23 In some patients with annular pancreata, true duodenal stenosis may gradually develop, due to repeated attacks of pancreatitis in the annular tissue or from reactive fibrosis in the duodenal wall.24
Diagnosis of both lesions is efficiently done with upper GI contrast studies. A transverse diaphragm in the descending duodenum with an eccentric or central aperture may be seen in patients with webs. The annular pancreas can be diagnosed with computed tomography (CT) and magnetic resonance imaging, whereas in upper GI series a smooth or tapered narrowing of the second part of the duodenum with dilatation of the proximal bulb can be seen.24 In addition, endoscopic retrograde cholangiopancreatography can delineate the ductal system of the annulus and its junction with the ventral pancreatic and biliary ducts.20
In the treatment of duodenal webs, bypass procedures were used in the past but have been largely abandoned. The most widely used procedure now is longitudinal duodenotomy followed by excision of the web, mucosal reapproximation, and transverse closure of the duodenum. During the excision of the web, intubation of the bile duct and the ampulla with a probe should be done, as to avoid damage in the biliopancreatic sphincteric mechanism.20 As for the annular pancreas, resection of the annulus was done frequently in the past and was often followed by pancreatic fistula formation. In addition, division of the annulus did not necessarily relieve the obstruction, since the narrowed duodenum might be permanently stenotic due to the intermingling of the annular tissue with the duodenal wall.24 Nowadays, the annular constriction of the duodenum could be bypassed by duodenoduodenostomy or duodenojejunostomy to the first duodenal portion.20 A retrocolic duodenojejunostomy, favored by Warren and associates,24 is the procedure of choice in adults, as it offers decompression of the proximal duodenum and a satisfactory bypass.
The laparoscopic approach has been introduced in the treatment of congenital duodenal anomalies in adults. There has been a report of two cases of annular pancreas treated successfully with laparoscopic gastrojejunostomy.25
Intestinal Malrotation
The term “intestinal malrotation” refers to a spectrum of anomalies involving the position and peritoneal attachments of the small and the large bowel. A wide diversity of anatomic anomalies, ranging from a not-quite-normal intestinal position to complete nonrotation or to reverse rotation, results in a wide variety of clinical manifestations. Progress in the understanding of the pathogenesis of intestinal malrotation was done only after normal embryology of the gastrointestinal tract was described, first by Meckel in 1817 and then more extensively by Mall in 1987.26,27
However, the embryological analysis of the disease was not completely understood until 1923, when Dott28 described the anatomic variations of malrotation and correlated them with points of aberrant or failed embryologic development. The incidence of malrotation in the general population is not known. It is estimated to range from one in 6,000 to one in 200 of live births.29,30 Most cases (90%) present during the first year of life. However, a small percentage of individuals with malrotation enter adulthood with it undetected.29,31
The most significant anomalies of rotation and fixation are nonrotation, malrotation, and reverse fixation. In nonrotation, the most frequently encountered anomaly, the midgut returns to the peritoneal cavity after rotating only 180° instead of the normal 270°. The postarterial limb returns to the abdominal cavity first instead of last. Therefore, the small intestine lies on the right side of the abdomen and the colon on the left. The ileum crosses the midline from right to left to enter the cecum. Adult patients with nonrotation are usually asymptomatic, but often volvulus may accompany this anomaly.32 In malrotation, the prearterial segment, which returns to the abdomen first, is usually in a normal position, and the degree of malrotation is indicated by the position of the cecum. Thus, the cecum may be on the left side, higher than normal on the right side, or in an intermediate position (Fig. 2). Reverse rotation occurs when the postarterial segment of the midgut returns to the abdomen first. The cecum begins its migration and passes to the right behind the superior mesenteric artery. Finally, the transverse colon lies behind the duodenum and is separated from it by the superior mesenteric artery.31,32

In malrotation, the cecum most frequently lies on the left side of the abdomen; the anomaly is frequently associated with the presence of Ladd bands, which may compress the duodenum.
Clinical presentation varies according to the degree and type of malrotation. In itself, abnormal positioning of the intestine does not cause symptoms. These arise from the abnormal position and fixation of the bowel, which allow the bowel to twist. The obstruction may be complete or partial and results from midgut volvulus or, less frequently, Ladd bands or internal hernia. Therefore, the clinical manifestations of rotation anomalies may be identical to those of proximal small-bowel obstruction (complete or partial), with or/without symptoms secondary to vascular occlusion.32
Moreover, there are patients with chronic abdominal complaints, including pain and intermittent obstruction or with atypical symptoms similar to those of common abdominal conditions.31 Patients presenting with bilious vomiting, acute abdominal pain, fever, tachycardia, and peritoneal tenderness on physical examination should be suspected of having an abdominal disaster. Confirmatory laboratory tests include an elevated white blood cell count and acidosis with an elevated base deficit and lactate level. In addition, abdominal pain usually cannot be relieved with nasogastric decompression. These patients require immediate exploratory laparotomy, which usually reveals midgut volvulus and vascular compromise with resultant intestinal ischemia. Patients with chronic abdominal complaints usually present with recurrent episodes of crampy abdominal pain, nausea, and bilious vomiting. These episodes may have been present or may have a more recent onset. Delay in diagnosis is common in the majority of these patients.31,33 Anatomic finding at surgery in these patients include mixed rotation or nonrotation with intermittent or partial midgut volvulus, partial intestinal obstruction due to Ladd bands (Fig. 3), or internal hernias. A third group of patients may be asymptomatic or have atypical complaints and suddenly come to our attention with an acute abdominal condition unrelated to malrotation but with bizarre abdominal findings due to their unusual gastrointestinal anatomy. For example, we may have cecal perforation in a patient with nonrotation with pain localized to the left abdominal quadrant or appendicitis in a patient with a subhepatic cecum with pain localized in the right upper quadrant.31 Preoperative diagnosis may be difficult in these patients and usually is done intraoperatively.

Complete dissection of the Ladd bands (LB), before division, on both the lateral and medial aspect of the duodenum (D).
The diagnosis of these anomalies in adults usually requires a variety of diagnostic modalities, such as plain abdominal radiographs, upper GI series, and CT scan.
Plain abdominal radiography may show signs of abnormally located bowel, i.e., small-bowel markings mainly on the right and large-bowel markings mainly on the left. Such X-rays may prompt further evaluation. Upper GI series is the standard of reference in the diagnosis of malrotation. It can indicate the level and nature of obstruction. They may show a duodenal–jejunal junction which fails to cross midline and is located below the level of the duodenal bulb or malposition of the right colon and the cecum. When a volvulus is present, the small bowel usually has a “corkscrew” appearance because it twists around the superior mesenteric artery.32 CT scan may reveal right-sided small intestine and a left-sided cecum, with/without an inverse relationship of the superior mesenteric artery and superior mesenteric vein, a highly indicative sign of malrotation. Midgut volvulus is seen as a dilated fluid-filled stomach, thick-walled loops of ischemic right-sided small intestine, and often free intraperitoneal fluid.34 Three-dimensional helical CT may reveal twisting of the superior mesenteric vein about the superior mesenteric artery. In general, identification of an abnormality requires meticulous diagnostic evaluation with additional diagnostic modalities.
Treatment should be addressed to each individual patient according to the degree and severity of malrotation and the timing of diagnosis. Patients with intestinal obstruction should undergo an explorative laparotomy. At surgery, the volvulus should be reduced and any nonviable bowel should be resected. Treatment of the underlying malrotation includes lysis of all adhesions and abnormal bands, straightening of the duodenum so it descends directly into the right lower quadrant, appendectomy, and widening of the small-bowel mesentery (Ladd procedure)35,36 (Table 1). Cecopexy is not recommended since there is no proof it has anything to offer in terms of perioperative or postoperative morbidity.31,33,37 Patients with chronic abdominal complaints should undergo a thorough radiologic evaluation before proceeding to surgical intervention. At surgery, when the obstruction is found secondary to Ladd bands, all abnormal intestinal attachments should be lysed. When an internal hernia is found, the hernia should be reduced; the sac should be resected; and the hernia defect should be closed. When there is reverse rotation, any adhesions between the mesentery of the small and the large intestine should be lysed, and the intestine should be rotated clockwise 360°, so as to create a condition of nonrotation. A Ladd procedure should be added in all above cases31 (Table 1).
There is an increasing use of laparoscopy for the treatment of malrotation in children as well as in adults.38 The Mayo Clinic series,39 the largest one reported for adults, confirmed that laparoscopic Ladd procedure is safe, feasible, and effective. In addition, it offers the patients the benefits of laparoscopy, such as early recovery and short hospital stay. Laparoscopy also is helpful when the diagnosis of malrotation is not certain. Mazziotti et al.40 have developed a method to determine whether laparoscopic Ladd procedure is indicated: if the length between the duodenojejunal junction and the ileocecal valve is less than half the transverse diameter of the peritoneal cavity, the procedure is indicated to prevent volvulus. However, when laparoscopy is used, conversion to open procedure is common because of the difficulties encountered. Nevertheless, some authors41 support that laparoscopy is not suitable for acute conditions, such as midgut volvulus, due to the excessive intestinal distension.
Malrotation that is discovered at the time of exploratory laparotomy/laparoscopy for an emergency condition, such as appendicitis or hollow viscus perforation, should be left to treat on an elective basis. If malrotation subsequently gives rise to symptoms or if the patient decides its correction, then malrotation can be treated on a later date.
There is a substantial controversy regarding the surgical correction of malrotation in case it is discovered incidentally on radiologic examination, while the patient is asymptomatic. Since many patients remain asymptomatic for their entire life, certain authors support that operation is not justified unless the patients develops symptoms directly related to malrotation.42,43 However, others advocate that surgical intervention is required anyway, due to the significant risk of midgut volvulus, even years after a symptom-free period.44,45
Several studies have indicated that certain anatomic characteristics, such as an abnormal position of the duodenal–jejunal junction or isolated malrotation of the cecum, may have some predictive value regarding the possibility of volvulus.46 However, there are no official criteria by which the risk of volvulus can be predicted. In general, the decision for surgery in patients with asymptomatic malrotation should be made by the surgeon and the patient, after having discussed extensively the risks and benefits of the operative and nonoperative approaches.
Gastrointestinal Duplication
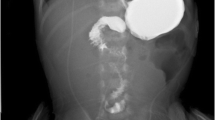
Gastrointestinal duplications are congenital malformations that can be encountered anywhere throughout the gastrointestinal tract, from the mouth to the anus. They occur mostly in pediatric patients but may occasionally run asymptomatic for years and present later in the adult life. They can be spherical cysts (Fig. 4) or tubular structures. They are attached to the wall of an adjacent part of the gastrointestinal tract and possess at least one exterior layer of smooth muscle and are lined with various types of gastrointestinal mucosa.47 The lumen of the duplication cysts usually lies between the leaves of the mesentery but may be entirely separated, with a mesentery of its own, formed by splitting of the original. There have been many theories regarding its pathogenesis. The most dominant one supports that duplication cysts appear at an early stage of embryological development due to incomplete separation of the primitive gut from the notochord. This theory is supported by the fact that these formations often coexist with osseous anomalies of the axial skeleton.48,49

A spherical type of duplication cyst in the small bowel of an adolescent boy.
Gastric duplications are noncommunicating cysts usually located along the greater curvature of the stomach. Gastric cysts are very rare as they account for 2% to 7% of all gastrointestinal duplications.50 They most often present with pain and vomiting.51,52 They are usually diagnosed as a result of a complication, such as gastrointestinal bleeding.51 Bleeding within the gastrointestinal tract may result from intussusception, peptic ulceration of the cyst lining, or necrosis of the cystic mucosa due to continuous pressure from the expanding cyst.53,54 Gastric duplications communicating with the pancreatic duct may be a cause of recurrent pancreatitis or peptic ulceration.55,56 Computed tomography and magnetic resonance imaging are common methods for evaluating tumors along the upper GI tract and may raise the suspicion for gastric duplication.57 99Tc scintigraphy is also a valuable diagnostic tool because of its high specificity to identify gastric mucosa.58 Very few reports so far have presented the use of laparoscopic surgery in the treatment of gastric duplications.59 In the laparoscopic approach, the goal is to excise completely the gastric duplication without violation of the gastric lumen, whenever possible.
The small intestine is a more common site for these formations. Ileum is the most common site for gastrointestinal duplication, accounting for over 60% of cases.60 Duplication in the small intestine can be asymptomatic for years but can present abruptly with lower gastrointestinal bleeding. These cases should undergo the routine diagnostic evaluation for gastrointestinal bleeding, but usually the source of bleeding is identified at laparotomy.61 At surgery, the lesions should be resected along with a portion of the adjacent intestine. The mucosa of the duplication should be completely removed because, when mucosa remains, acid peptic ulceration from ectopic gastric mucosa may lead to bleeding and often requires reoperation.53
Colonic duplications are rare, accounting for 4–18% of all gastrointestinal duplications, with the cecum being the most common location. Colonic duplications are also asymptomatic for prolonged periods but may result in significant morbidity and mortality in case of complications, such as acute bowel obstruction or severe gastrointestinal hemorrhage.61 Colonic cysts can be asymptomatic for years but usually present with pain and/or obstructive symptoms.62 Occasionally, they may present with a complication, such as obstruction, bleeding, or constipation. Abrupt hemorrhage with hemodynamic instability may occur in case of a cyst lined with ulcerated mucosa which erodes into adjacent organs and/or vessels.53,63 Cystic duplications can cause obstruction of the colon as a result of direct compression, intussusception, or volvulus whereas tubular duplications of the rectum may have direct communication with the perineum.61,63 There are several reports of intestinal carcinomas found within duplication cysts.62,64 Preoperative diagnosis is often difficult, and the radiographic findings may be nonspecific. Ultrasonography and computed tomography may be useful in obtaining a diagnosis.53,63
Treatment is reserved for symptomatic cases and usually includes excision of the cyst with the neighboring part of the intestine because of the intimate attachment of the common wall or because isolated resection of the cyst would compromise blood flow to the adjacent segment of the colon.61,63,65 Cyst excision is mandatory for the possibility of malignant degeneration. Postoperative complications are nonspecific and include postoperative bleeding, infection, and bowel obstruction. However, in case of large duplication segments, injury to the normal intestine with resultant short bowel syndrome is also a possibility.53 Despite these problems, the overall outcome after surgery for intestinal duplications is generally favorable.
The laparoscopic approach has also been introduced in the treatment of duplications of the small and the large bowel in children60 as well as in adults66–68 with promising results.
Jejunoileal Atresia and Stenosis
Atresia and stenosis are common birth effects affecting the small intestine. The duodenum is the most common site, followed by the jejunum and the ileum; however, it can also affect multiple sites in the small intestine.69 The approximate incidence in the Western societies has been reported to be approximately three to four per 10,000 births in the general population.69–71 The mechanism responsible for its pathogenesis is not well clarified. Although in the 1950s the dominant theory was that intestinal atresia was the result of vascular accidents during the fetal life,72 today most researchers support the hypothesis of the pivotal role of genetic and embryogenetic factors which lead to the development of this anomaly.73
This defect may take several forms, such as the apple-peel atresia, multiple intestinal atresia with short-gut syndrome, proximal jejunal atresia with megaduodenum, and colon atresia.73,74 All above formations are usually discovered during the neonatal or infantile life and require immediate correction. However, long-term complications in these patients may present later during childhood or adolescence and may require reintervention.75
Meckel’s Diverticulum
Meckel’s diverticulum is a congenital blind pouch in the small bowel, which results from an incomplete obliteration of the vitelline duct during the fifth week of gestation. This entity took its name after Johann Friedrich Meckel the younger who first studied the diverticulum’s anatomy and embryology.76 For Meckel’s diverticulum, the common rule found in every review book for general surgery is the “rule of twos,” i.e., it is found in 2% of the population, twice as common in males, most frequently found in those less than 2 years of age, and usually 2 ft from the ileocecal valve.77 For that reason, surgical residents begin searching for this anomaly with their first surgical exploration. However, a true diverticulum is rarely discovered in the adult (Fig. 5), and complications related to this anomaly rarely occur with increasing age.78

A Meckel’s diverticulum discovered incidentally in a 16-year-old boy during an exploratory laparotomy for acute pain in the right iliac fossa.
The development of this anomaly occurs with the failure of obliteration of the proximal vitelline duct during the seventh to eighth week of gestation. This diverticulum is categorized as a true diverticulum because it contains the normal layers of the small bowel. It may contain ectopic mucosa, such as gastric mucosa which is responsible for the adjacent ulceration of the ileum.77 Pancreatic mucosa is the second most common ectopic mucosa found in this diverticulum. Occasionally, the diverticulum may be the site of a malignant tumor. Weinstein et al.78 published the most thorough description of malignancy in Meckel’s diverticulum: in a series of 80 malignant cases, there were 35 sarcomas, 29 carcinoids, and 16 adenocarcinomas.
Meckel’s diverticulum usually does not give symptoms. A small percentage of patients may give symptoms, with the obstructive symptoms being the most prevalent in the adult, and they are most commonly due to intussusception, volvulus, inflammatory adhesions, diverticular strictures, enteroliths, or incarcerated hernia.78–81 Inflammation (diverticulitis) and/or perforation are found in up to 20–30% of symptomatic patients and are often indistinguishable from acute appendicitis.80 Bleeding is the most common complication in children and is reported in over 50% of cases.82,83 In adults, bleeding is the presenting complaint in only 11.8% of cases.79 Bleeding is usually minor, resulting in chronic anemia. Occasionally, it may cause massive lower gastrointestinal bleeding in adults, as several case reports describe in the literature.77,84 However, it occurs less often than obstruction. Yamaguchi et al.79, in a series comprising almost 50% adults, showed hemorrhage as being less common than obstruction at a rate of approximately 5:1 (54%:12%). Preoperative evaluation of Meckel’s diverticulum is difficult, and routine radiological studies, such as plain abdominal radiograph, arteriography, upper GI series, and computed tomography, are often nondiagnostic and theretofore of limited diagnostic value.85 In suspected symptomatic Meckel’s diverticulum, preoperative evaluation includes 99mTc (technetium-99m pertechnetate) scanning, which relies on the presence of ectopic gastric mucosa.86 In this study, 99mTc is injected intravenously, and over time it accumulates in the gastric mucosa. Because complications such as bleeding are caused by the ectopic gastric tissue, diagnosis may be helped in symptomatic cases. In children, the scan has a sensitivity of 85% and specificity of 95%, but in adults, the sensitivity drops to 62.5% and the specificity to 9%. The accuracy of the scan can be improved with the use of pentagastrin or cimetidine.85,87 When the scan is nondiagnostic or in patients with nonbleeding presentation, ultrasonography is perhaps the most useful noninvasive method of achieving diagnosis.88
The choice of treatment of Meckel’s diverticulum depends on whether it was discovered incidentally or it caused symptoms. Ileal resection including the diverticulum is the treatment of choice for asymptomatic diverticulum because the extent of heterotopic tissue cannot be determined safely by palpation, and ulcerations may recur in case the ectopic tissue persists. Ileal resection permits the removal of any inflamed or heterotopic tissue.89
The most controversial issue in the management of Meckel’s diverticulum is the decision of surgical resection in asymptomatic cases discovered incidentally. Postoperative morbidity and mortality depend on the indication for removal. While the mortality after surgical excision is almost zero, morbidity in asymptomatic patients is 8.6%, which is close to the 8.3% morbidity found in symptomatic patients (8.5% overall).77 Those who support incidental excision believe the relatively high mortality and morbidity rates with symptomatic disease justify the associated morbidity of elective excision. The opponents of incidental excision quote the low (4.2%) lifetime risk of symptoms development.90 In addition, a recent systematic review has shown that there is no compelling evidence to support prophylactic resection.91 In fact, resection of incidentally detected Meckel’s diverticulum has a significantly higher early morbidity rate than leaving the diverticulum in situ (5.3% vs 1.3%, P < 0.0001; Table 1).92
Nowadays, the advent of laparoscopy may have changed this scenario. Laparoscopy permits a complete abdominal exploration, increasing the number of incidentally found diverticula. The laparoscopic approach is extremely useful as it permits the removal of an incidentally found diverticulum with a gastrointestinal stapling device.93 Nevertheless, laparoscopy has been used to treat patients with Meckel’s diverticulum complicated by intestinal obstruction or bleeding caused by heterotopic gastric mucosa.94,95
Conclusions
Diagnosis of a congenital anomaly of the GI tract in an adult is not a rare event. Some of these anomalies may remain undetected for years and are diagnosed on the basis of incidental findings at routine examination in the adult life. Indeed, congenital anomalies can form an important part of the daily practice of the general surgeon on patients in the adult life. A thorough knowledge of basic embryology is necessary to understand these lesions. Management considerations in the adult patients are often different compared to the pediatric group. Some of these patients may have persistent problems in adulthood requiring medical attention for years. In addition, there is a large variety of available therapeutic options to offer to adult patients with congenital anomalies of the GI tract. For most of the reviewed diseases, evidence-based management directions are difficult, due to a lack of randomized trials and long-term follow-up.
References
Ikenaga T, Honmyo U, Takano S, et al. Primary hypertrophic pyloric stenosis in the adult. J Gastroenterol Hepatol 1992;7:524–526.
Zavala C, Bolio A, Montalvo R. Hypertrophic pyloric stenosis: adult and congenital types occurring in the same family. J Med Genet 1969;6:126–128.
van Roggen JFG, van Krieken JH. Adult hypertrophic pyloric stenosis: case report and review. J Clin Pathol 1998;51:479–480.
Quigley RL, Pruitt SK, Pappas TN, Akwari O. Primary hypertrophic pyloric stenosis in the adult. Arch Surg 1990;125:1219–1221.
Go TS, Morse WH. Hypertrophic pyloric stenosis in adults. Am J Gastroenterol 1973;60:400–405.
Dye TE, Vidals VG, Lockhart CE, Snider WR. Adult hypertrophic pyloric stenosis. Am Surg 1979;45:478–484.
Hellan M, Lee T, Lerner T. Diagnosis and therapy of primary hypertrophic pyloric stenosis in adults: case report and review of the literature. J Gastrointest Surg 2006;10:265–269.
Navab F, Flores L. Multinodular adult hypertrophic pyloric stenosis. J Clin Gastroenterol 1989;11:667–670.
Papaziogas B, Lazaridis C, Souparis A, et al. Idiopathic hypertrophic pyloric stenosis combined with left paraduodenal hernia. Med Princ Pract 2007; 16:151–154.
Kleitsch WP. Pyloric hypertrophy in the adult. Nebr State Med J 1953;38:87–89.
Kirklin BR, Harris MT. Hypertrophy of the pyloric muscle of adults: a distinctive roentgenologic sign. Am J Roentgenol 1933;29:437–442.
Twining EW. Chronic hypertrophic stenosis of the pylorus in adults. Br J Radiol 1933;6:644–655.
Schuster MM, Smith VM. The pyloric “cervix sign” in adult hypertrophic pyloric stenosis. Gastrointest Endosc 1970;16:210–211.
Danikas D, Geis WP, Ginalis EM, Gorcey SA, Stratoulias C. Laparoscopic pyloroplasty in idiopathic hypertrophic pyloric stenosis in an adult. JSLS 2000; 4:173–175.
Simson JN, Thomas AJ, Stoker TA. Adult hypertrophic pyloric stenosis and gastric carcinoma. Br J Surg 1986;73:379–380.
Brahos GJ, Mack E. Adult hypertrophic pyloric stenosis managed by double pyloroplasty. JAMA 1980;243:1928–1929.
Alain JL, Grousseau D, Terrier G. Extramucosal pyloromyotomy by laparoscopy. Surg Endosc 1991;5:174–175.
van der Bilt JDW, Kramer WLM, van der Zee DC, Bax NMA. Laparoscopic pyloromyotomy for hypertrophic pyloric stenosis. Impact of experience on the results in 182 cases. Surg Endosc 2004;18:907–909.
Selzer D, Croffie J, Breckler F, Rescorla F. Hypertrophic pyloric stenosis in an adolescent. J Laparoendosc Adv Surg Tech 2009;19:451–452.
Ladd AP, Madura JA. Congenital duodenal anomalies in the adult. Arch Surg 2001;136:576–584.
Ross JA. Congenital abnormalities in adult surgery. J R Coll Surg Edinb 1980;25:8–16.
Ravitch MM. The pancreas in infants and children. Surg Clin North Am 1975;55:377–385.
Sperazza JC, Flanagan RA Jr, Katlic MR. Annular pancreas and intermittent duodenal obstruction in an alcoholic adult. Clev Clin J Med 1992;59:208–210.
Lloyd-Jones W, Mountain JC, Warren KW. Annular pancreas in the adult. Ann Surg 1972;176:163–170.
De Ugarte DA, Dutson EP, Hiyama DT. Annular pancreas in the adult: management with laparoscopic gastrojejunostomy. Am Surg 2006;72:71–73.
Touloukian RJ, Smith EI. Disorders of rotation and fixation. In O’Neill JA, Rowe MI, Grosfeld JL, et al, eds. Pediatric surgery. 5th ed. St. Louis: Mosby, 1998, pp 1199–1214.
Skandalakis JE, Gray SW, Ricketts R, Richardson DD. The small intestines. In Skandalakis JE, Gray SW, eds. Embryology for surgeons: the embryological basis for the treatment of congenital anomalies. 2nd ed. Baltimore: Williams & Wilkins, 1994, pp 184–236.
Dott NM. Anomalies of intestinal rotation: their embryology and surgical aspects with report of five cases. Br J Surg 1923;11:251–286.
Clark LA, Oldham KT. Malrotation. In Ashcraft KW, Murphy JP, Sharp RJ, et al, eds. Pediatric surgery. 3rd ed. Philadelphia: Saunders, 2000, pp 425–442.
Donnellan WL, Kimura K. Malrotation, internal hernias, congenital bands. In Donnelan WL, Burrington JD, Kimura K, eds. Abdominal surgery of infancy and childhood. New York: Harwood Academic Publishers, 1996, 43:1–27.
Kapfer SA, Rappold JF. Intestinal malrotation—not just the pediatric surgeon’s problem. J Am Coll Surg 2004;199:628–635.
Berrocal T, Lamas M, Gutierez J, et al. Congenital anomalies of the small intestine, colon and rectum. Radiographics 1999;19:1219–1236.
Firor HV, Steiger E. Morbidity of rotational abnormalities of the gut beyond infancy. Clev Clin Q 1983;50:303–309.
Bodard E, Monheim P, Machiels F, et al. CT of midgut malrotation presenting in an adult. J Comput Assist Tomogr 1994;18:501–504.
Ladd WE. Congenital obstruction of the duodenum in children. N Engl J Med 1932;206:277–283.
Ladd WE, Gross RE. Abdominal surgery of infancy and childhood. Philadelphia: Saunders, 1941, pp 53–70.
von Flue M, Herzog U, Ackermann C, et al. Acute and chronic presentation of intestinal nonrotation in adults. Dis Colon Rectum 1994;37:192–198.
Seymour NE, Andersen DK. Laparoscopic treatment of intestinal malrotation in adults. JSLS 2005;9:298–301.
Matzke GM, Dozois EJ, Larson DW, Moir CR. Surgical management of intestinal malrotation in adults: comparative results for open and laparoscopic Ladd procedures. Surg Endosc 2005;19:1416–1419.
Mazziotti MV, Strassberg SM, Langer JC. Intestinal rotation abnormalities without volvulus: the role of laparoscopy. J Am Coll Surg 1997;185:172–176.
Fu T, Tong WD, He YJ, et al. Surgical management of intestinal malrotation in adults. World J Surg 2007;31:1797–1803.
Gohl ML, Demeester TR. Midgut nonrotation in adults: an aggressive approach. Am J Surg 1975;129:319–323.
McVay MR, Kokoska ER, Jackson RJ, Smith SD. The changing spectrum of intestinal malrotation: diagnosis and management. Am J Surg 2007;194:712–719.
Maxson RT, Franklin PA, Wagner CW. Malrotation in the older child: surgical management, treatment, and outcome. Am Surg 1995;61:135–138.
Praise P, Flageole H, Shaw KS, et al. Should malrotation in children be treated differently according to age? J Pediatr Surg 2000;35:756–758.
Schey WL, Donaldson JS, Sty JR. Malrotation of bowel: variable patterns with different surgical considerations. J Pediatr Surg 1993;28:96–101.
Macpherson RI. Gastrointestinal duplications: clinical, pathologic, etiologic, and radiologic considerations. Radiographics 1993;13:1063–1080.
Bentley JFR, Smith JR. Developmental posterior enteric remnant and spinal malformations. Arch Dis Child 1960;35:76–86.
Johnstone MJ, Clegg JF. Gastrointestinal haemorrhage from small bowel duplication. Postgrad Med J 1977;53:700–702.
Coit DG, Mies C. Adenocarcinoma arising within a gastric duplication cyst. J Surg Oncol 1992;50:274–277.
Youngblood P, Blumenthal BI. Enteric duplication cyst. South Med J 1983;76:670–672.
Klimopoulos S, Gialvalis D, Marougas M, et al. Unusual case of massive hemorrhage of a gastric hemorrhage of a gastric duplication cyst in a very advanced age. Langenbecks Arch Surg 2008;394:745–747.
Holcomb GW 3rd, Gheissari A, O’Neill JA, et al. Surgical management of alimentary tract duplications. Ann Surg 1989;209:167–174.
Faerber EN, Balsara R, Vinocur CD, de Chadarevian JP. Gastric duplication with hemoptysis: CT findings. Am J Roentgenol 1993;161:1245–1246.
Longmire WP, Rose AS. Haemoductal pancreatitis. Surg Gynecol Obstet 1973;136:246–250.
Rao KLN, Pimpalwar A, Vaiphei K, Chowdhary S. Intrapancreatic gastric duplication cyst presenting as lower gastrointestinal bleeding. J Pediatr Surg 2003;38:243–244.
Zeebregts CJ, Slot B, Brinkhuis M, Gerritsen JJ. Gastric duplication cyst. Arch Surg 2004;139:687–688.
Dittrich JR, Spottswood SE, Jolles PR. Gastric duplication cyst: scintigraphy and correlative imaging. Clin Nucl Med 1997;22:93–96.
Machado MAC, Santos VR, Martino RB. Laparoscopic resection of gastric diverticulum. Surg Laparosc Endosc Perc Tech 2003;13:268–270.
Puliglanda PS, Nguyen LT, St-Vil D, et al. Gastrointestinal duplications. J Pediatr Surg 2003;38:740–744.
Fotiadis C, Genetzakis M, Papandreou I, et al. Colonic duplication in adults: Reports of two cases presenting with rectal bleeding. World J Gastroenterol 2005;11:5072–5074.
Choong CK, Frizelle FA. Giant colonic diverticulum: report of four cases and review of the literature. Dis Colon Rectum 1998;41:1178–1185.
Frittelli P, Costa G, Zanella L, et al. Intestinal duplication in the adult. A case report of a colonic duplication and a review of the literature. Chir Ital 2002;54:721–728.
Horie H, Iwasaki I, Takahashi H. Carcinoid in a gastrointestinal duplication. J Pediar Surg 1986;21:902–904.
Robert J, Ambrosetti P, Widgren S, Rohner A. Perforated tubular duplication of the sigmoid colon in adults. Gastronerol Clin Biol 1990;14:776–779.
Jimenez M, Cadiere GB, Dapri G, et al. Duodenal duplication cyst in an adult: first simultaneous laparoscopic and endoscopic surgery. J Laparoendosc Adv Surg Tech A 2009;19:207–710.
Salameh JR, Votanopoulos KI, Hilal RE, et al. Rectal duplication cyst in an adult: the laparoscopic approach. J Laparoendosc Adv Surg Tech A 2002;12:453–456.
Kabay S, Yucel M, Yaylak F, et al. Combined duplication of the colon and vermiform appendix in an adult patient. World J Gastroenterol 2008;28:641–643.
Cragan JD, Martin ML, Moore CA, Khoury MJ. Descriptive epidemiology of small intestinal atresia, Atlanta, Georgia. Teratology 1993;48:441–450.
Ethen MK, Canfield MA. Impact of including elective pregnancy terminations before 20 weeks gestation on birth defect rates. Teratology 2002;66:S32–S35.
Harris J, Kallen B, Robert E. Descriptive epidemiology of alimentary tract atresia. Teratology 1995;52:15–29.
Louw JH, Barnard CN. Congenital intestinal atresia: observations on its origin. Lancet 1955;2:1065–1067.
Shorter NA, Georges A, Perenyi A, Garrow E. A proposed classification system for familial intestinal atresia and its relevance to the understanding of the etiology of jejunoileal atresia. J Pediatr Surg 2006;41:1822–1825.
Touloukian RJ. Diagnosis and treatment of jejunoileal atresia. World J Surg 1993;17:310–317.
Escobar MA, Ladd AP, Grosfeld JL, et al. Duodenal atresia and stenosis: long-term follow-up over 30 years. J Pediatr Surg 2004;39:867–871.
Edmonson JM. Johann Friedrich Meckel the younger: Meckel’s diverticulum. Gastrointest Endosc 2001;54:19A–20A.
Stone PA, Hofeldt MJ, Cambell JE, et al. Meckel diverticulum: ten-year experience in adults. South Med J 2004;97:1038–1041.
Weinstein EC, Cain JC, Remine W. Meckel diverticulum: 55 years of clinical and surgical experience. JAMA 1962;182:251–253.
Yamaguchi M, Takeuchi S, Awazu S. Meckel diverticulum: investigation of 600 patients in Japanese literature. Am J Surg 1978;136:247–249.
Park JJ, Wolff BG, Tollefson MK, et al. Meckel diverticulum. The Mayo Clinic experience in 1476 patients (1950–2002). Ann Surg 2005;241:529–533.
Dumper J, Mackenzie S, Mitchell P, et al. Complications of Meckel’s diverticula in adults. Can J Surg 2006;49:353–357.
Rutherford RB, Akers DR. Meckel diverticulum: a review of 148 pediatric patients with specific reference to the pattern of bleeding and to mesodiverticular vascular bands. Surgery 1966;59:618–626.
Mackey WC, Dineen P. A fifty-year experience with Meckel diverticulum. Surg Gynecol Obstetr 1983;156:56–64.
Lichtstein DM, Herskowitz B. Massive gastrointestinal bleeding from Meckel’s diverticulum in a 91-year-old man. South Med J 1998;91:753–754.
Martin JP, Connor PD, Charles K. Meckel’s diverticulum. Am Fam Physician 2000;44:1037–1042.
Lin S, Suhocki PV, Ludwig KA, et al. GI bleeding in adult patients with Meckel diverticulum: the role of technetium 99m pertechnetate scan. South Med J 2002;95:1338–1341.
Sfakianakis GN, Conway JJ. Detection of ectopic gastric mucosa in Meckel’s diverticulum and in other aberrations by scintigraphy: I. Pathophysiology and 10-year clinical experience. J Nucl Med 1981;22:647–654.
Daneman A, Lobo E, Alton DJ, Shuckett B. The value of sonography, CT and air enema for detection of complicated Meckel diverticulum in children with non-specific clinical presentation. Pediatr Radiol 1998;28:928–932.
Williams RS. Management of Meckel’s diverticulum. Br J Surg 1981;68:477–480.
Soltero MJ, Bill AH. The natural history of Meckel diverticulum and its relation to incidental removal. Am J Surg 1976;132:168–173.
Zani A, Eaton S, Rees CM, Pierro A. Incidentally detected Meckel diverticulum: to resect or not to resect? Ann Surg 2008;247:276–281.
Cullen JJ, Kelly KA, Moir CR, et al. Surgical management of Meckel’s diverticulum: an epidemiologic, population-based study. Ann Surg 1994;220:564–568, discussion 568–569.
Bona D, Schipani LS, Nencioni M, Rubino B, Bonavina L. Laparoscopic resection for incidentally detected Meckel diverticulum. World J Gastroenterol 2008;14:4961–4963.
Ishigami S, Baba K, Kato K, et al. Small bowel obstruction secondary to Meckel diverticulum detected and treated laparoscopically-case report. Surg Laparosc Endosc Percutan Tech 2006;16:344–346.
Rivas H, Cacchione RN, Allen JW. Laparoscopic management of Meckel’s diverticulum in adults. Surg Endosc 2003;17:620–622.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vaos, G., Misiakos, E.P. Congenital Anomalies of the Gastrointestinal Tract Diagnosed in Adulthood—Diagnosis and Management. J Gastrointest Surg 14, 916–925 (2010). https://doi.org/10.1007/s11605-009-1124-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11605-009-1124-z