
Abstract
The olivine-type LiFe1-x Y x PO4/C (x = 0, 0.01, 0.02, 0.03, 0.04, 0.05) products were prepared through liquid-phase precipitation reaction combined with the high-temperature solid-state method. The structure, morphology, and electrochemical performance of the samples were characterized by X-ray diffraction (XRD), scanning electron microscope (SEM), transmission electron microscope (TEM), energy-dispersive spectroscopy (EDS), galvanostatic charge-discharge, cyclic voltammetry, and electrochemical impedance spectroscopy (EIS). We found that the small amount of Y3+ ion-doped can keep the microstructure of LiFePO4, modify the particle morphology, decrease charge transfer resistance, and enhance exchange current density, thus enhance the electrochemical performance of the LiFePO4/C. However, the large doping content of Y3+ ion cannot be completely doped into LiFePO4 lattice, but existing partly in the form of YPO4. The electrochemical performance of LiFePO4/C was restricted owing to YPO4. Among all the doped samples, LiFe0.98Y0.02PO4/C showed the best electrochemical performance. The LiFe0.98Y0.02PO4/C sample exhibited the initial discharge capacity of 166.7, 155.8, 148.2, 139.8, and 121.1 mAh g−1 at a rate of 0.2, 0.5, 1, 2, and 5 C, respectively. And, the discharge capacity of the material was 119.6 mAh g−1 after 100 cycles at 5 C rates.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Olivine-structured LiFePO4 is one of the most promising cathode materials for next-generation lithium-ion batteries in applications of large-size and high-power devices [1]. Compared with commercial layered LiCoO2 material with lower capacity and poorer safety, it possesses more excellent chemical and thermal stability, acceptable flat voltage plateau (3.4 V vs Li+/Li) and a high theoretical rate capacity of 170 mAh g−1, and it offers economic and environmental advantages because of its low-cost and nontoxicity. However, the undesirable high-rate performance of LiFePO4 resulting from its poor electronic conductivity nature and sluggish ionic diffusion restricts its applications in large power devices [2]. The key issues are how fast Li+ ion inserted/extracted, and how fast electrons can be transported during charge/discharge process.
Enormous efforts have been made to improve the electronic and ionic conductivity of the bulk as well as surface of the LiFePO4 material. Up to now, surface coating of LiFePO4 with carbon is one popular method to obtain good electrochemical performance [3–12]. However, the effectiveness of carbon coating in enhancing electrochemical performances of LiFePO4 can be affected by many factors [13–18]. The factors influencing the effect of the coating carbon include the following: carbon content and carbon thickness, carbon morphology and distribution, and so on. If the coating layer carbon has thin layer, complete graphitization and appropriate content on the surface of LiFePO4, the electron conductivity of LiFePO4 bulk will be greatly improved. Except the direct contribution to electron conductivity, the carbon coating can also bring some conductive by-product of metal phosphides, which is beneficial to the increased electronic conductivity of LiFePO4 [11, 19].
Another key method is to dope metal ion, which has been considered as an efficient way for the enhancement of the electrochemical performance of LiFePO4. In the past several years, significant progress has been made in this filed by doping various metal cations [20–29], such as Cu2+, Mn2+, Nd3+, Nb5+ into LiFePO4, and their improved electrochemical performances have been reported. For example, Huang et al. [30] prepared Na+ ion-doped LiFePO4/C and obtained improved reversible capacity of 142 mAh g−1 at a rate of 1 C. The improved electrochemical performance of Na+ ion-doped LiFePO4/C sample can be attributed to its larger lattice parameters in a and c axis direction. Cho et al. [31] prepared LiFe1-x La x PO4/C by a solid-state reaction. It was indicated that these La-ion dopants had no effect on the structure of the material. Instead, it considerably enhanced its high rate performance and cyclic stability. Otherwise, to better understand the relationships between the electrochemical performance and crystal structure, Hong et al. [32] synthesized V-modified LiFePO4 by different methods. They found that the vanadium was substituted into the lattice occupying iron sites in the FeO6 octahedron for the V-modified LiFePO4 samples prepared by the conventional solid-state reaction method and a solution method. This structural modification enhances the electrochemical performance by increasing the Li+ effective cross-sectional area of the LiO6 octahedral face and thereby reducing the bottleneck for Li+ migration.
Based on above analysis, doping appropriate amount of metal ions and coating carbon is a very effective method to improve the performance of LiFePO4. Rare earth Y element has a special electronic structure (the different arrangements of electrons give rise to abundant energy levels). The influence of Y3+ ion on the electrochemical properties of LiFePO4 has rarely been reported. In this paper, we have synthesized LiFe1-x Y x PO4/C products by liquid-phase precipitation reaction combined with the high-temperature solid-state method. We expected that doping rare earth element Y3+ and coating carbon will be beneficial to improve the high-rate performance of LiFePO4.
Experimental
Olivine-type LiFe1-x Y x PO4/C (x = 0, 0.01, 0.02, 0.03, 0.04, 0.05) composites were prepared by liquid-phase precipitation reaction combined with the high-temperature solid-state method. A total of 0.05 mol NH4H2PO4 (AR) and 0.05 mol FeSO4·7H2O (AR) were first separately dissolved in 50 mL distilled water. The two aqueous solutions were pumped into a continuously stirred tank reactor. A 2.86-g H2O2 solution (30 wt%) was also pumped into the reactor to generate FePO4·xH2O precipitation (the pH value for the reaction system was about 2). The suspension was then further stirred for 15 min. To ensure suitable reaction condition, temperature (25 °C) and stirring speed of the resulting mixture were precisely controlled throughout the synthesis process. The resulting FePO4 hydrate powders were filtered, washed, and dried at 80 °C for 12 h, then heat-treated at 500 °C for 3 h in air atmosphere to obtain crystalline anhydrous FePO4 powders. LiFe1-x Y x PO4/C composites were synthesized by mixing the prepared FePO4 powders with a stoichiometric amount of Li2CO3 (AR), a certain amount of Y2O3, and superfluous citric acid. The mixture was then ball-milled 6 h and finally calcined at 700 °C for 8 h under nitrogen atmosphere. The carbon content is ca. 5 % in the obtained powders. The method we adopted to measure the carbon content is dissolving the samples into hydrochloric acid solution. After filtrated and dried, we get the residual carbon.
The XRD measurement of the as-synthesized products was carried out on a RigakuSmart Lab X-ray diffractometer operated at 40 kV using a Cu Kα radiation at scan rate of 5° min−1. The morphology and microstructure of the powders were observed by field-emission scanning electron microscopy (FE-SEM) on S-4800 FE-SEM and high-resolution transmission electron microscope (HRTEM) with model JEM2010.
The electrochemical performances of the as-synthesized products were measured in a simulative cell consisted of a working electrode and a lithium foil electrode separated by a Celgard 2400 microporous membrane. The working electrode was prepared by spreading the mixed slurry with 80 wt.% active materials, 10 wt.% acetylene black, and 10 wt.% polyvinylidene fluoride (PVDF) binder in the N-methylpyrrolidione (NMP) onto an Al foil substrate, and then dried at 120 °C in a vacuum drying oven for 12 h. The electrolyte was 1 M LiPF6/EC+DEC (1:1, v/v). The cells were assembled in an argon-filled glove box and tested by galvanostatic charge-discharge cycling in the voltage ranges of 2.4–4.2 V on a battery testing system (LAND, Wuhan, China). The area of the electrode is 0.785 cm2, with the active material loading of 4–5 mg on each one. The electrochemical impedance spectra (EIS) test (the frequency range of 0.01–100,000 Hz) was performed on an electrochemical workstation CHI660A (Chenhua, Shanghai, China).
Results and discussion
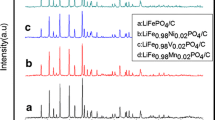
The phase purity and crystal structure of the products obtained were examined by X-ray diffraction (XRD). Illustrated in Fig. 1 are the XRD patterns of LiFe1-x Y x PO4/C (x = 0, 0.01, 0.02, 0.03, 0.04, 0.05) samples. All the XRD patterns can be indexed as orthorhombic Pnma space group (JCPDS card No. 83-2092). In addition, when the doping content of Y3+ ion is low, the impurity peaks cannot be detected. To clarify this phenomenon, the Rietveld refinement of the XRD pattern for LiFe0.98Y0.02PO4/C composite is shown in Fig. 2. It can be seen that the measured pattern and calculated pattern match well, indicating that the Y ions tend to occupy Fe sites of the olivine LiFePO4 instead of forming another phase. With the increase of the Y3+ ion content, a tiny amount of YPO4 appears, which brings lattice defects in LiFePO4 (the XRD pattern for LiFe0.97Y0.03PO4/C sample around 22–23°), resulting in the enhancement of the electrochemical performance of LiFePO4/C [33]. However, when the doping content of Y3+ ion reaches 5 mol%, more YPO4 come into being, which causes sever lattice distortions, leading to the declined electrochemical properties. Based on the above analysis, we conclude that Y3+ ion can enter into the lattice of olivine LiFePO4 occupying Fe sites when the doping amount is relatively low. However, when the doping amount is too high, Y3+ ion cannot totally enter into LiFePO4 lattice and the extra Y3+ ion exist in the form of YPO4.

XRD patterns of LiFe1-x Y x PO4/C (0≤x≤0.05) samples

Rietveld refinement of XRD pattern for LiFe0.98Y0.02PO4/C composite
The particle size and morphology of the LiFePO4/C and LiFe0.98Y0.02PO4/C products are provided by scanning electron microscope (SEM), as shown in Fig. 3. It can be clearly seen form Fig. 3 that the dispersibility of the Y3+ ion doping LiFePO4/C sample becomes significantly better than that of the undoped LiFePO4/C sample, which suggests that an appropriate doping amount of Y3+ ion can markedly increase the interface between the nanoparticles and electrolyte, thus improving the electrochemical performance of LiFePO4/C electrode. To further analyze LiFe0.98Y0.02PO4/C, energy-dispersive X-ray spectrometry (EDS) mappings were performed, as shown in Fig. 4. Figure 4a shows a typical SEM image of LiFe0.98Y0.02PO4/C nanoparticles, and Fig. 4b–f gives recorded maps of C-K, O-K, P-K, Fe-K, and Y-K signals. According to the result, we can see that the Y–K and C-K signals are detected all through the particle, demonstrating that Y element and carbon are homogeneously distributed on the entire detection area for LiFe0.98Y0.02PO4/C sample.

SEM images of LiFePO4/C (a, c) and LiFe0.98Y0.02PO4/C products (b, d)

Energy-dispersive X-ray spectrometry (EDS) mappings of LiFe0.98Y0.02PO4/C product
The surface morphology and microstructure of the prepared material were further analyzed by transmission electron microscopy (TEM). Figure 5 indicates the TEM images of samples LiFePO4/C and LiFe0.98Y0.02PO4/C. It is seen From Fig. 5a, c and b, d that both the nanoparticles of LiFePO4/C and LiFe0.98Y0.02PO4/C are composed of sphere-like nanoparticles. The TEM images from the edge of these nanoparticles reveal that they have a relatively smooth and closed structure, covered with a coating layer. The perfect carbon coating layer is beneficial for improving the electrochemical performance of the LiFePO4 materials.

TEM images of LiFePO4/C (a, c) and LiFe0.98Y0.02PO4/C products (b, d)
To test the electrochemical lithium storage performance of the synthesized products, simulative cells using a lithium anode and LiFe1-x Y x PO4/C (x = 0.00, 0.01, 0.02, 0.03, 0.04, 0.05) composites are charged-discharged between 2.4 and 4.2 V. Figure 6a shows voltage versus specific capacity for all electrodes at the first discharge-charge cycles at rate of 0.2 C. It can be found that all the cells exhibit one charge or discharge plateau around 3.45 V, which suggests that the intercalation/deintercalation behavior of Li+ ion is smooth in every case. For the discharge capacity, interestingly, the LiFe0.98Y0.02PO4/C sample exhibits a discharge capacity of 166.7 mAh g−1 at 0.2 C rate, which is much higher than those of LiFe1-x Y x PO4/C (145.6, 158.7, 152.4, 134.1, and 129.9 mAh g−1 for x = 0.00, 0.01, 0.03, 0.04, and 0.05, respectively). We speculate that the low doping content of Y3+ ion can bring lattice defects in LiFePO4, which can enhance the electrochemical performance of LiFePO4/C. However, with the increase of Y3+ ion content, the impurity phases YPO4 come into being and the lattice distortions gets severer, resulting in the declined electrochemical properties. To further test the electrochemical performances of LiFePO4/C and LiFe0.98Y0.02PO4/C electrodes at various rates. Figure 6b, c illustrates typical voltage-capacity curves of LiFePO4/C and LiFe0.98Y0.02PO4/C electrodes at 0.2, 0.5, 1, 2, and 5 C rates, respectively. It is seen that LiFe0.98Y0.02PO4/C electrode achieves discharge capacities of 166.7, 155.8, 148.2, 139.8, and 121.1 mAh g−1 at a rate of 0.2, 0.5, 1, 2, and 5 C, which is much higher than that of LiFePO4/C electrode which gives capacities of 145.6, 139.9, 131.2, 118.8, and 71.8 mAh g−1, respectively. These results are outstanding superior to results obtained in the literature [34–36]. Moreover, we can see from Fig. 6d that LiFe0.98Y0.02PO4/C sample exhibits excellent electrochemical Li+ storage performance with higher capacity and rate capability than that of LiFePO4/C. Additionally, to further test the potential applicability of LiFe0.98Y0.02PO4/C electrode at high rate, the reversible capacity of LiFe0.98Y0.02PO4/C electrode achieves 119.6 mmAh g−1 at the high rate of 5 C after 100 cycles, as shown in Fig. 7. The corresponding coulombic efficiency is ca. 98.8 %. The remarkable rate and cycle performance can be attributed to the appearance of the defects and the increasing disorder of the lattice in LiFePO4/C by doping Y3+ ion.

Typical galvanostatic curves of all the electrodes at a rate of 1 C (a). Typical voltage-capacity curves of LiFePO4/C (b) and LiFe0.98Y0.02PO4/C (c) electrodes at rates of 0.2, 0.5, 1, 2, and 5 C, respectively. The cycling performance of LiFePO4/C and LiFe0.98Y0.02PO4/C electrodes at various rates (d)

The charge and discharge capacity and coulombic efficiency of LiFe0.98Y0.02PO4/C sample for 100 cycles at a rate of 5 C
Figure 8 gives the CV curves of the as-synthesized LiFePO4/C and LiFe0.98Y0.02PO4/C electrodes a low scan rate of 0.5 mV s−1. It can be seen from Fig. 8 that the potential interval between the cathodic and anodic peaks for LiFePO4/C and LiFe0.98Y0.02PO4/C electrode are comparable. However, the redox peak of LiFe0.98Y0.02PO4/C electrode is more sharper and symmetric than that of LiFePO4/C electrode, indicating that the redox kinetics of LiFe0.98Y0.02PO4/C electrode are improved by doping Y3+ ion.

CV curves of the prepared LiFePO4/C and LiFe0.98Y0.02PO4/C electrodes at a scan rate of 0.5 mV s−1
The electrochemical impedance spectra (EIS) is used to further analyze the effect of doping Y3+ ion on the electrode reaction impedance. Electrochemical impedance spectrum was carried out at 50 % of discharge state, as shown in Fig. 9a. An EIS spectrum is composed of a semicircle at high-frequency range and an inclined line within the low-frequency range. The resistance of the semicircle is attributed to the charge transfer process. The Nyquist plots are fitted using the equivalent circuit (the inset of Fig. 9a), and the derived impedance parameters are listed in Table 1. It is obvious that the R ct of LiFe0.98Y0.02PO4/C is lower than that of the LiFePO4/C electrode. The charge transfer resistance of the LiFe0.98Y0.02PO4/C electrode is 90.7 Ω. The value of the charge transfer resistance is also lower than that of literature report [37]. The smaller charge transfer resistance indicates the more feasible transfer of lithium-ion and electron on the electrode, which is beneficial to overcome the restriction of kinetics in the charge/discharge process and improve the electrochemical performance of the LiFePO4 material. An exchange current density (I 0) is a very important parameter of kinetics for an electrochemical reaction, which can be used to measure the catalytic activity of electrodes. It is calculated by the following formula (1), and the results are also listed in Table 1.

EIS for LiFePO4/C and LiFe0.98Y0.02PO4/C electrodes (a). The plot of Z re vs the reciprocal root square of the lower angular frequencies (ω −1/2) for LiFePO4/C and LiFe0.98Y0.02PO4/C electrodes (b). The inset image is the corresponding equivalent circuit
where R is the gas constant, T is the absolute temperature, n is the charge transfer number, R ct is the charge transfer resistance, and F is the Faraday constant.
It is apparently seen in Table 1 that the value of exchange current density (I 0) for LiFe0.98Y0.02PO4/C electrode is remarkably higher than that of LiFePO4/C. This result implies that the LiFe0.98Y0.02PO4/C electrode has better electrochemical activity, which leads to the enhanced high rate performance of LiFePO4/C material.
In addition, the inclined line in EIS spectrum can be attributed to the lithium-ion diffusion into the bulk of the electrode material, the so-called Warburg diffusion. The Warburg coefficient σ can be calculated by equation [38] (2):
where R e is the resistance of the electrolyte, R ct is the charge transfer resistance, and ω is the angular frequency in the low-frequency region and Z re is the real axis resistance in the low-frequency region. Both R e and R ct are kinetics parameters independent of frequency. Then, σ is the slope for the plot of Z re vs the reciprocal root square of the lower angular frequencies (ω −1/2). The plot of Z re vs the reciprocal root square of the lower angular frequencies (ω −1/2) for the LiFePO4/C and LiFe0.98Y0.02PO4/C electrodes are shown in Fig. 9b. The slope of the fitted line is the Warburg coefficient σ. However, the Li+-ion diffusion coefficient is obtained by the following equation [39] (3):
where R is the gas constant, T is the absolute temperature (K), F is the Faraday constant, A is the surface area of the LiFePO4 cathode, n is the number of electrons during the process of Li+-ion transportation, C is the molar concentration of Li+-ion in the LiFePO4 cathode, and σ is the Warburg coefficient. The Li+-ion diffusion coefficient of the two electrodes is also listed in Table 1. The calculated lithium diffusion coefficient of the LiFePO4/C and LiFe0.98Y0.02PO4/C is 3.2 × 10−14 cm2 s−1 and 9.03 × 10−13 cm2 s−1, respectively. The higher D obtained by doping Y3+ ion can be attributed to creating the defect and increasing disorder of the lattice in LiFePO4/C and leads to the improvement of the electrochemical performance. These results are in good agreement with the charge-discharge characteristics. The above analysis suggests that the improvement of high performance for LiFe0.98Y0.02PO4/C materials can be attributed to the enhancement of the lithium-ion diffusivity in the bulk LiFePO4.
Conclusions
Olivine-structured LiFe1-x Y x PO4/C (x = 0, 0.01, 0.02, 0.03, 0.04, 0.05) composites have been synthesized by liquid-phase precipitation reaction combined with the high-temperature solid-state method. Y3+ ions can enter into the lattice of the olivine LiFePO4 at the Fe sites when the doping content is low. Meanwhile, it can modify the particle morphology, decrease polarization overpotential and charge transfer resistance, increase exchange current density, and thus improve the electrochemical performance of the LiFePO4/C. However, the large doping content of Y3+ ion can form more YPO4 impurity phases, which can weaken the electrochemical performance of LiFePO4/C. The LiFe0.98Y0.02PO4/C showed the best rate capacity and cycling stability among all the samples. The initial discharge capacity of the as-prepared material can reach up to 166.7, 155.8, 148.2, 139.8, and 121.1 mAh g−1 at a rate of 0.2, 0.5, 1, 2, and 5 C, respectively. And, the material retains about 98.8 % of its initial capacity after 100 cycles at 5 C rate.
References
Padhi AK, Nanjundaswamy KS, Goodenough JB (1997) J Electrochem Soc 144:1188–1194
Yuan LX, Wang ZH, Zhang WX, Hu XL, Chen JT, Huang YH, Goodenough JB (2011) Energy Environ Sci 4:269–284
Cheng F, Wang S, Lu AH, Li WC (2013) J Power Sources 229:249–257
Avci E, Mazman M, Uzun D, Bicer E, Sener T (2013) J Power Sources 240:328–337
Xu G, Li F, Tao Z, Wei X, Liu Y, Li X, Ren Z, Shen G, Han G (2014) J Power Sources 246:696–702
Yao B, Ding Z, Zhang J, Feng X, Yin L (2014) J Solid State Chem 216:9–12
Huang G, Li W, Sun H, Wang J, Zhang J, Jiang H, Zhai F (2013) Electrochim Acta 97:92–98
Chen Z, Du B, Xu M, Zhu H, Li L, Wang W (2013) Electrochim Acta 109:262–268
Qin G, Ma Q, Wang C (2014) Electrochim Acta 115:407–415
Zhou Y, Wang J, Hu Y, OHayre R, Shao Z (2010) Chem Commun 46:7151–7153
Zhao B, Yu X, Cai R, Ran R, Wang H, Shao Z (2012) J Mater Chem 22:2900–2907
Zhang X, Zhang X, He W, Yue Y, Liu H, Ma J (2012) Chem Commun 48:10093–10095
Wang J, Yang J, Tang Y, Li R, Liang G, Sham TK, Sun X (2013) J Mater Chem A 1:1579–1586
Yang J, Wang J, Tang Y, Wang D, Li X, Hu Y, Li R, Liang G, Sham TK, Sun X (2013) Energy Environ Sci 6:1521–1528
Ji H, Zhang L, Pettes MT, Li H, Chen S, Shi L, Piner R, Ruoff RS (2012) Nano Lett 12:2446–2451
Qin G, Wu Q, Zhao J, Ma Q, Wang C (2014) J Power Sources 248:588–595
Gong C, Xue Z, Wang X, Zhou XP, Xie XL, Mai YW (2014) J Power Sources 246:260–268
Li J, Qu Q, Zhang L, Zheng H (2013) J Alloys Compd 579:377–383
Kang B, Ceder G (2009) Nature 458:190–193
Yang R, Song X, Zhao M, Wang F (2009) J Alloys Compd 468:365–369
Zhang Q, Wang S, Zhou Z, Ma G, Jiang W, Guo X, Zhao S (2011) Solid State Ionics 191:40–44
Li C, Hu N, Wang C, Kang X, Wumair T, Han Y (2011) J Alloys Compd 509:1897–1900
Wang Y, Yang Y, Hu X, Yang Y, Shao H (2009) J Alloys Compd 481:590–594
Sun CS, Zhou Z, Xu ZG, Wang DG, Wei JP, Bian XK, Yan J (2009) J Power Sources 193:841–845
Bilecka I, Hintennach A, Rossell MD, Xie D, Novak P, Niederberger M (2011) J Mater Chem 21:5881–5890
Yue H, Wu Z, Li L (2014) J Alloys Compd 583:1–6
Huang Y, Xu Y, Yang X (2013) Electrochim Acta 113:156–163
Ma Z, Fan Y, Shao G, Wang L, Song J, Wang G, Liu T (2014) Electrochim Acta 139:256–263
Ma Z, Shao G, Wang G, Zhang Y, Du J (2014) J Solid State Chem 210:232–237
Yin X, Huang K, Liu S, Wang H, Wang H (2010) J Power Sources 195:4308–4312
Cho YD, Fey GTK, Kao HM (2008) J Solid State Electrochem 12:815–823
Hong J, Wang XL, Wang Q, Omenya F, Chernova NA, Whittingham MS, Graetz J (2012) J Phys Chem C 116:20787–20793
Saiful Islam M, Driscoll DJ, Fisher CAJ, Slater PR (2005) Chem Mater 17:5085–5092
Huang Y, Liu H, Lu YC, Hou Y, Li Q (2015) J Power Sources 284:236–244
Miao C, Bai P, Jiang Q, Sun S, Wang X (2014) J Power Sources 246:232–238
Vu A, Stein A (2014) J Power Sources 245:48–58
Liu H, Li C, Cao Q, Wu YP, Holze R (2008) J Solid State Electrochem 12:1017–1020
Yan C, Zhao X, Guo R (2010) Electrochim Acta 55:922–926
Wang L, Ma P, Zhang Y, Gao C, Yan C (2009) J Salt Lake Res 17:52–55
Acknowledgments
We are grateful for the financial support from the Natural Science Foundation of Hebei Province (B2012203069) and support from education department of Hebei province on natural science research key projects for institution of higher learning (ZH2011228).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Chen, J., Wang, X., Ma, Z. et al. Effects of yttrium ion doping on electrochemical performance of LiFePO4/C cathodes for lithium-ion battery. Ionics 21, 2701–2708 (2015). https://doi.org/10.1007/s11581-015-1467-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-015-1467-2