
Abstract
Chile is the origin of the Solanum tuberosum group Chilotanum corresponding to lowland tetraploid landraces. The most complete morphological description of the Chilean germplasm was published in 2008 in the Catalogue of Varieties of Native Potatoes from Chile, which describes 211 varieties. Two institutions of the Chilean government, INIA and SAG (Agricultural and Livestock Service of Chile), keep collections of 332 and 257 accessions, respectively; the varieties registered in the catalogue are included in the SAG collection. Thus, an important gap occurs between the number of varieties listed in the National Catalogue of Chile and the number of traditional varieties found in the Chilean collections of potatoes. In this work, two national collections of Chilean native germplasms (INIA-Remehue and SAG) were analysed via four microsatellite markers with the aims of (1) discriminating different genotypes in order to more accurately catalogue the current number of native Chilean varieties, (2) evaluating the diversity of genotypes potentially known with the same nomenclature by farmers, and (3) comparing allelic diversity of native germplasm with 44 commercial varieties. Of the 589 native accessions analysed, 320 different allelic phenotypes were found indicating that there are at least 320 different genotypes in the collections. Of these, 158 belonging to the INIA collection were not found in the SAG collection. These 158 new genotypes should increase the number of known Chilean varieties. As expected, different genotypes were known under the same popular name. In terms of allelic diversity, the group of commercial varieties contained a subset of the alleles within the collection of SAG and INIA-Remehue.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The potato (Solanum tuberosum L.) is the fourth most important food crop in the world. It originated in the Americas (Hawkes 1994). South American potato landraces are still highly diverse with a variety of tuber shapes, skin, and flesh colours. While the taxonomic treatment of Solanum species is still under discussion, Spooner (2009) suggested classification of 107 wild species and four cultivated species. Among the cultivated species, Solanum tuberosum is the most popular cultivated potato and is also known as the “common potato” in most parts of the world. Depending on taxonomic treatment, S. tuberosum has been divided into the sub-species andigena and tuberosum (Hawkes, 1990) or divided into cultivar groups: Andigenum (including diploids, triploids, and tetraploids) and Chilotanum (tetraploids) (Spooner et al. 2007). The Chilotanum group corresponds to lowland tetraploid Chilean landraces with a geographical origin in Chiloé and the Chonos Archipelago. Hawkes (1990) proposed that it evolved from Andigenum and was introduced into southern Chile followed by adaptation to longer day lengths, but the origin of the Chilean landraces is controversial (Ovchinnikova et al. 2011). Cultivated species have a base chromosome number of n = 12 and may be diploid (2n = 2x = 24), triploid (2n = 3x = 36), tetraploid, or pentaploid (Machida–Hirano 2015).
Hosaka and Hanneman (1988) have indicated that some genotypes belonging to the Chilotanum group were brought to Europe during the nineteenth century for breeding. These authors, based on the molecular markers of chloroplast DNA, suggested that Chilotanum has been the main maternal genetic source for the development of most commercial varieties in the world. Ríos et al. (2007) and Ames and Spooner (2008) supported the idea that early European potatoes were selected from Chilean introductions. However, there are still many varieties of rare landraces in southern Chile with attributes that are likely not incorporated in commercial varieties. These materials have a high value for genetic breeding and are potential sources of flavours, textures, aroma, nutrients, novel industrial compounds as well as resistance to biotic and abiotic stress. In addition, these resources are valuable for cultural patrimony and commercial development.
The diversity of the Chilotanum group in Chile is currently spread over different farmer communities in southern Chile, germplasm banks, and private collections. The most complete morphological description of the Chilean germplasm has been published in the Catalogue of Varieties of Native Potatoes from Chile, which describes 211 varieties in terms of the morphological characters suggested by UPOV (Contreras and Castro, 2008). This material was given to the National Agricultural and Livestock Service of Chile (Servicio agrícola y ganadero, SAG) to be maintained under field conditions (Contreras and Castro 2008; SAG 2015). The above-mentioned catalogue does not provide information at the genetic level about the described varieties in order to discriminate genotypes, and it is unclear how representative it is for the entire diversity of the Chilotanum group. For example, there is no comparison of this collection with another genetic pool of Chilean germplasm, such as the collection maintained in the Institute of Agricultural Research (INIA) of Chile.
It is reasonable to expect that some of the genotypes cultivated as traditional varieties by Chilean farmers have not yet been described. An updated and more sophisticated compilation of Chilean genotypes is critical for this important centre of the origin of the potato. Along with new genotypes, new alleles could be present in the non-described varieties.
Potato reproduction is mainly achieved via vegetative propagules. For this reason, clonal propagation led to perpetuation of the genotypes that are kept and used by famers because of their valuable characteristics. Therefore, the identification at the genetic level is very important with scientific, legal, productive, and commercial implications.
The Chilean program of Potato Breeding of INIA has collected and received native potatoes from different sources from southern Chile since 1970. In total, there are 332 accessions that have not yet been compared to the material conserved by the SAG. Many traditional varieties are known by common names. However, farmers give common names to potatoes according to morphological traits of the tubers and not according to the underlying genotype. The morphological characteristics of the tubers are controlled by not many genes (Mendoza and Mosquera, 2011). Thus, different genotypes can be known by the same name due to similar tuber appearance. On the other hand, one genotype could have different names because there is no traceability regarding the assignment of its name. Both of these phenomena occur in other gene pools in the world (Ghislain et al. 2004; Spanoghe et al. 2015). Therefore, it is highly necessary to rationalize collections in order to classify varieties based on attributes and characteristics and identify duplicates, so that they can be used and introduced into breeding programs. Also, the present lack of material traceability makes conservation, scientific utilization, biodiversity monitoring, breeding, agronomic development, utilization in industry, marketing, and/or intellectual property development unfeasible. Taking into account the renewed interest in native potatoes as a commercial product, the issues mentioned above appear relevant and proper identification is critical.
It is now well established that a genetic characterization based on molecular markers reveals differences in genotypes undistinguishable at the morphological level (Ghislain et al. 2004; Ruiz de Galareta et al. 2007; Rocha et al. 2010; Spanoghe et al. 2015), while phenotypic characterization needs considering several seasons of characterization due to the effect of the environment on the genotype (Cubero, 2003). For a description at the genetic level, the International Potato Centre (CIP) has developed a set of microsatellite markers based on a study of 741 potato landraces (Ghislain et al., 2009).
The objectives of this article are as follows: (a) to quantify the genotypic diversity that is found in a wide pool of collections of Solanum tuberosum Group Chilotanum, including the varieties described in the Catalogue of Varieties of Native Potatoes from Chile, currently maintained by the National Agricultural and Livestock Service of Chile (SAG), and the native germplasm kept in the INIA potato germplasm bank; (b) to investigate the diversity of genotypes included in a particular denomination; and (c) to compare allelic richness of the native Chilotanum germplasm with commercial varieties.
Material and Methods
Plant Materials
Two collections of native Chilean potatoes, respectively named SAG and INIA-Remehue, were used in this study. The first contains 257 accessions that are kept by SAG in Osorno. The second contains 332 accessions donated by farmers from southern Chile and conserved by the Potato Bank of the Breeding Program in the Regional Centre of Research INIA-Remehue. In addition, a group of 44 commercial varieties was included. These varieties were from the USA, Germany, Netherlands, Argentina, Mexico, Canada, and Chile. A list of all clones is provided in ESM 1.
DNA samples were collected from field collections at the SAG and INIA-Remehue experimental stations, located at two locations near Osorno, Chile. Accessions were planted in one row of ten plants of pure clones, at normal distance. Young leaves were taken from one plant.
DNA Extractions
The DNA from each accession or cultivar was extracted from young leaves as described by Fulton et al. (1995). Quantification was performed using the Nanoquant Spectophotometer. Integrity of DNA was verified by visualization in 1% agarose gels in TBE Buffer 0.5X and stained with GelRed. Before performing PCR, the DNA solutions were diluted to 10 ng/μl.
SSR Markers and Amplification
Four microsatellite markers developed by Ghislain et al. (2004) were selected while considering their localization in different chromosomes, discriminatory power, and clarity of band patterns. The markers STM2013 and STM1052 were previously employed by Mathias et al. (2007) in varieties used in Chile with high discriminatory power, and the markers STM1016 and STM1106 were used by Ghislain et al. (2009). These markers were chosen because they are able to distinguish the 44 commercial varieties commonly used in Chile and included in this study. Protocols used for amplification were performed as described by CIP (Ghislain et al. 2009). Briefly, 5 μl from the diluted DNA samples were used for PCR reaction in a total volume of 15 μl. PCR amplification mixtures were subjected to 94 °C for 3 min and 35 amplification cycles (94 °C for 50 s, 1 min for annealing temperature, and 72 °C for 50 s). The final step was 72 °C for 5 min for amplification of SSR STM1016, STM1106, and STM2013. The SSR STM1052 used a Touchdown PCR program (94 °C for 2 min and then 15 amplification cycles of 94 °C for 1 min, 50 °C for 2 min, 60 °C for 1.5 min, 94 °C for 1 min, 50 °C for 2 min, 72 °C for 1.5 min and a final step of 72° for 5 min (CIP, 2008). PCR reactions were performed in the ESCO swift-MaxPro Thermal Cycler.
Visualization of SSR Fragments
PCR products were run on 6% denaturing polyacrylamide gels (37.5:1 acrylamide:bis acrylamide) with 7.5 M urea in TBE 1X buffer and silver stained for visualization of amplified bands. Electrophoresis was performed at 1000 V for 3 or 5 h depending on the SSR to be analysed. A 50 bp standard DNA ladder (Fermentas) was employed during electrophoresis.
Allele Scoring, Genotype Identification, and Analysis
The alleles were scored as present (1) or absent (0) for all accessions and treated as dominant markers. For this analysis, a similarity matrix was calculated using the Jaccard’s dissimilarity index. Allele sizes and identification for SSRs STM1052 and STM1106 were determined by coincidence of gel mobility between the bands of control samples with known size (kindly provided by CIP) and bands of the analysed samples. For SSR markers, STM2013 and STM1016 allele sizes were determined by coincidence of gel-mobility between the bands of the sequencing reactions (pUC/M13 forward primer, Silver SequenceTM DNA Sequencing system) and the upper band of the SSR marker.
Data analysis included the whole data set, undistinghishable accessions were not discarded, to make the following calculations. Polymorphic index content (PIC) for each SSR locus was calculated by the formula PIC = 1 − ∑i 1 fi2 where fi is the relative frequency of the ith allelic phenotype detected of the SSR loci (Nei, 1987), as suggested by Anderson (1993) and Reid and Kerr (2007).
Principal coordinates analysis (PCoA) was conducted in Darwin 6.0 (Perrier et al. 2006), from Euclidean distances (‘D) obtained by transforming the Jaccard dissimilarity index D by multiplication by a positive constant (a = 1) and addition of a constant (b = 10), both user defined, by means of the formula D’ = a*D + b, as recommended by Perrier et al. (2003). Using a multiplication by a positive constant or an addition of a constant, this transformation produced a new dissimilarity with mathematical properties (Euclidean index) required for PCoA analysis (Perrier et al., 2003). PCoA extracts a first axis (one dimension) such that Σ i, j (dij – δij)2 is minimum (where dij is the observed distance between i and j, δij is the distance between the projections of i and j on this axis). Then it extracts a second axis, orthogonal to the previous one (independence condition) minimizing the squared differences, and so on. Solutions are given by eigenvectors and eigenvalues of the matrix W of scalar products between elements that is defined from the dij according to the Torgerson formula: Wij = − (dij2 – di.2 – d.j2 + d2)/2. Dissimilarity trees (dendrograms) in radial representation were constructed using an unweighted neighbour joining (NJ) clustering method for a dissimilarity matrix calculated with the Jaccard’s coefficient using the DARwin 6.0 software (Perrier et al., 2006; De Haan et al., 2013). Histograms of the distribution of Jaccard’s dissimilarity index were also prepared. The number of different genotypes was obtained in terms of the base genetic distance. Accessions separated by a genetic distance more than zero were considered to be different.
The relative frequencies of allelic phenotypes and allelic richness (number of alleles detected/number of loci analysed) were calculated for all the collection accession, including those of INIA-Remehue, SAG, and commercial varieties.
Results
Estimation of the Genotypic Diversity in Chilean Collections
Of the 589 native accessions analysed, 320 different allelic phenotypes were found indicating that there were at least 320 different genotypes in the whole collection. This was the maximal resolution achieved with the four SSR loci used. It is unclear if the remaining undifferentiated material corresponded to duplicated accessions or could still be discriminated by additional markers. Of these 320 differentiated genotypes, 158 corresponded to accessions from INIA-Remehue with allelic phenotypes not found in the SAG collection registered in the Catalogue of Native Potato Varieties (Contreras and Castro 2008). Therefore, a large proportion of the genotypes from the two native collections was different (47% of the accessions belonging to INIA-Remehue were not found in SAG collection). These 158 new genotypes increased the number of varieties that could be described and registered as Chilean native varieties. Table 1 indicates the amount (expressed as number of accessions) of genotypic diversity found in Chilean collections of native potato with the markers used.
As an additional antecedent, we surveyed the tuber morphology of 102 accessions from INIA. There were 18 groups of accessions that shared a similar banding pattern for the four SSRs employed but within which there were clear differences for traits such as skin colour and the presence of a secondary colour in the flesh. These are features that are highly influenced by the genotype (Mendoza and Mosquera, 2011). Therefore, this morphological diversity makes it likely that there are additional genotypes that are not yet revealed by the SSRs.
Evaluation of the Genotypic Diversity with Regard to the Collection Denominations
Results revealed that many different genotypes are known with the same popular name. As a proof of this, Table 2 shows examples of the dissimilarity index calculated between some accessions with different origins but with the same common name. Table 3 presents some accessions sharing the same common name while presenting distinct allelic phenotypes. Cases presented here correspond to some examples from the collections.
Comparison of the Allelic Diversity of Native Germplasm with Commercial Varieties
In total, 32 alleles were detected for the accessions included. From these, 21 alleles were found in more than 10% of the accessions. Nineteen of these alleles were shared in the three collections (INIA, SAG, and commercial varieties). The INIA-Remehue and SAG shared 31 alleles: 21 of them were found in more than 10% of the accessions in both collections, ten of them observed in less than 10% of the accessions, and one of the 32 alleles was missing in INIA-Remehue. In the collection of commercial varieties, the varieties did not contain six alleles from the total number of 32 detected for the whole analysed germplasm (see details in Table 4 and ESM 2).
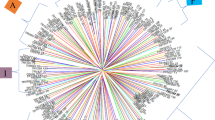
In terms of allelic diversity, a principal coordinate analysis indicated that clouds of points corresponding to INIA-Remehue, SAG, and commercial varieties were superimposed (Fig. 1). The dendrogram tree shows that accessions from INIA-Remehue and SAG clustered together (Fig. 2). Therefore, the three sub-collections shared a large proportion of the genetic material analysed and the commercial varieties were circumscribed to a narrow range of coordinates that appeared as a subset inside the collection of SAG and INIA-Remehue. In the vertical axis of PCoA (Fig. 1), the distribution of commercial varieties is on the left side, putting in the right side a considerable number of native accessions from INIA and SAG. This fact is reinforced by the NJ dendrogram tree where commercial varieties are included in some branches closely related with native material, while some native accessions from SAG and INIA Remehue appear separated in a more distant branch (Fig. 2).

Principal Coordinate Analysis (PCoA) showing two axes. Each point corresponds to one accession. Green, blue, and red points indicate accessions belonging to the SAG, INIA-Remehue, and commercial varieties collections, respectively

Tree constructed by unweighted neighbour joining (NJ) clustering method for a dissimilarity matrix calculated with the Jaccard’s coefficient using the DARwin 6.0 software. Each point corresponds to one accession. Green, blue, and red points indicate accessions belonging to SAG, INIA-Remehue, and commercial varieties collections, respectively
Despite the low number of SSR employed, it was possible to detect 320 different genotypes in native material with no coincidence with commercial varieties. This indicates that there is a considerable diversity in the genotypes. Each collection contributed to new allelic combinations rather than different alleles. A distribution of dissimilarity values between all the accessions showed that the mean of the dissimilarity values was around 0.5–0.6 (Fig. 3) indicating that in most of the comparisons between accessions there was about 50% chance of similar alleles. A similar distribution was found for the two native collections with similar mean values. For pairwise comparison between natives only, the maximum value of dissimilarity reached 0.94 while the mean was 0.52. For comparison including native (INIA-Remehue + SAG) and commercial varieties, the maximum value was also 0.94 and the mean 0.53. Within the group of commercial varieties, the maximum value was 0.75, with a mean of 0.45.

Distribution of Jaccard’s dissimilarity index in 589 native accessions and 44 commercial varieties of potatoes (633 analysis units). Scale ranges from 0 to 60.000 pairwise comparisons
It is important to notice the high proportion of the closely related cultivars pairs within the Chilean pool. Near to 10,000 pairs of comparison were below a threshold of 0.3 (Fig. 3). This could be also visualized in Fig. 1, where numerous points appear closely positioned in the graphic of PCoA.
Discussion
Southern Chile is a hub of potato diversity and the origin of the Solanum tuberosum varietal group Chilotanum (Spooner, 2007). Hitherto, the work of Contreras and Castro (2008) is the major source of data about the number and diversity of native varieties present in Chile. This is a reference for the inventories of native potatoes made up by the Agriculture and Livestock Service of Chile (SAG). The data presented in the present work show that there are many more genotypes than those reported and described thus far despite the vegetative propagation of this species.
When comparing the allelic richness among the three collections, we detected only one allele that was exclusive to the SAG collection. In general, the same alleles were present in the INIA-Remehue and the SAG collections indicating that the allelic richness was similar for the loci under study despite the fact that these collections had different sources of origin. This suggests that the high diversity of the genotypes detected in the Chilean germplasm was produced by combining a group of alleles rather than by introduction of new alleles from external sources into these collections. It is possible to speculate that the genotypic diversity was produced via true botanical seeds during the domestication of the potato in southern Chile. De Haan et al. (2013) compared a collection of Solanum tuberosum subsp tuberosum kept ex situ with diverse in situ potatoes from the Peruvian Andes; they found that 74% of the alleles were shared between the ex situ and in situ collections. However, in the cited study, both collections shared just a few cultivars and each collection had predominantly unique cultivars indicating that the landrace diversity is very dynamic and is necessary to understand the drivers underlying the diversity in traditional cultivars of potato existing in a region.
At the moment, from the 32 detected alleles, six observed in the native collections were not found in the 44 commercial varieties. Therefore, these six alleles could be considered for diagnostic purposes to identify native genotypes as suggested by Fu et al. (2009) for another collection. On the other hand, these alleles could be linked to genomic regions associated with undesirable traits from an agronomic point-of-view; they could have been eliminated during potato breeding. The first axis of PCoA is grouping commercial varieties in the left side, while many native accessions from SAG and INIA appear in the right side indicating that breeding process has probably chosen some part of the Chilean allelic pool and the genetic distance became lower within the varieties belonging to the commercial pool. This can also be observed in the NJ dendrogram tree (Fig. 2) where some branches did not contain commercial varieties. In support of this, the maximum and mean values of dissimilarity in pairwise comparison within the group of commercial varieties were lower than those observed in the Chilean native pool.
The allelic richness is also referred to as the allelic diversity or mean number of alleles per locus. It is one of the most commonly reported measures of genetic variation (Leberg, 2002). In this work, the mean number of alleles per locus was 8.0, similar or not so far of that reported by Solano et al. (2013) and Perez and Riegel (2004) who reported a mean of 9.16 and 9.6 alleles per locus using 7 SSR.
Despite the low number of (four) SSR markers used in this work, it was sufficiently informative to identify 320 different allelic phenotypes and thus 320 potential varieties. In several works, few SSR informative markers were useful for distinguishing a big number of genotypes. For example, Reid and Kerr (2007) used six SSR markers to identify 400 genotypes of potatoes. Rocha et al. (2010) used three SSR markers to distinguish 16 varieties commercialized in Brazil. Mathias et al. (2007) found that three SSRs were able to discriminate the 15 commercial varieties registered in Chile; two of these markers were used in the current study.
We expected that intraclonal variants could not be detected by SSR. Hale et al. (2005) reported that materials with consistently different phenotypic expression could not be differentiated via 112 AFLP primer combinations. This was probably due to the tetraploid nature of the potato. The allele dosage could play a role in the phenotype, or the influence of epigenetic changes can produce new phenotypes, so, in addition to diversity detected by SSR markers, phenotypic differences between traditional cultivars could be due to epigenetic factors or dosage of alleles adding a new source of variability to be elucidated in future work.
There is a broad interest in unique potato varieties including diverse colours, shapes, and flavours. These traditional landraces are known by farmers who use common names as a traditional way to identify varieties, but these names also offer ethno-botanical and ethno-agricultural information. However, the molecular analysis used offers additional genetic information to discriminate between accessions with the same common name. For example, traditional varieties from the Canary Islands including the variety Corralera were composed of four different genotypes (Ruiz de Galareta et al. 2007). In the collections analysed here, there were six accessions with the name Michuñe negro, but three different allelic phenotypes. This indicates that this traditional variety corresponds to at least three different varieties. Rubilar et al. (2012) evaluated the chemical and nutritional compositions of the variety Michuñe negro, but the lack of a genetic profile makes it impossible to identify the genotype that corresponds to these chemical and nutritional attributes. Consequently, there is a large heterogeneity within a particular native variety given that it is sold using only the common name. This common name is based on the origin or appearance of the tubers, e.g., Murta Ojuda or Tres Meses de Llicaldad (the names are alluding to deep eyes and 3 months of cultivation cycle, respectively). The incorporation of native varieties in the national registry of described varieties should consider also the identification of genotypes by molecular markers and the existence of a large number of varieties in addition to the 211 that have been described by Contreras and Castro (2008).
The need to identify and trace native cultivars for presentation in marketplaces can be addressed with molecular markers. The CIP kit offers molecular characterization of potatoes (Ghislain et al. 2009), and molecular patterns obtained via SSR amplification could be used as a DNA barcode to identify accessions with exclusive molecular patterns or specific bands. On the other hand, access to next generation sequencing methods and genotyping by sequencing (GBS) makes it possible to generate enormous quantities of molecular data to aid in the differentiation of genotypes as well as to the study of genetic diversity (Uitdewilligen et al. 2013). These technologies could be used more broadly in the future when methods for bioinformatics analysis and data management are improved (Kaminsky et al., 2015).
Abbreviations
- CIP:
-
International Potato Centre
- DNA:
-
Deoxyribonucleic acid
- GBS:
-
Genotyping by sequencing
- INIA:
-
Instituto de Investigaciones Agropecuarias de Chile (Institute of Agricultural Research of Chile)
- PCR:
-
Polymerase chain reaction
- SAG:
-
Servicio Agrícola y Ganadero de Chile (Agricultural and Livestock Service of Chile)
- SSR:
-
Simple sequence repeats
- UPOV:
-
International Union for the Protection of New Varieties in Plants
References
Ames M, Spooner D (2008) DNA from herbarium specimens settles a controversy about origins of the European potato. Am J Bot 95(2):252–257
Anderson JA, Churchill GA, Autrique JE, Tanksley SD, Sorrells ME (1993) Optimizing parental selection for genetic linkage maps. Genome 36(1):181–6
Contreras A, Castro I (2008) Catálogo de variedades de papas nativas de Chile. Universidad Austral de Chile, Chile, p 234
Cubero J (2003) Introducción a la Mejora Genética Vegetal. Ediciones Mundi_Prensa, Madrid España, p 556
De Haan S, Nuñez J, Bonierbale M, Ghislain M, van der Maesen J (2013) A Simple Sequence Repeat (SSR) Marker Comparison of a Large In- and Ex-situ Potato Landrace Cultivar Collection from Peru Reaffirms the Complementary Nature of both Conservation Strategies. Diversity 5:505–521
Fu Y, Peterson G, Richards K, Tam R, Percy J (2009) Genetic diversity of Canadian and exotic potato germplasm revealed by simple sequence repeat markers. Am J Pot Res 86:38–48
Fulton TM, Chunwongse J, Tanksley SD (1995) Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol Biol Report 13(3):207–209
Ghislain M, Spooner DM, Rodriguez F, Villamón F, Núñez J, Vásquez C et al (2004) Selection of highly informative and user-friendly microsatellites (SSRs) for genotyping of cultivated potato. Theor Appl Genet 108:881–890
Ghislain M, Núñez J, Herrera M, Pignataro J, Guzman F, Bonierbale M, Spooner D (2009) Robust and highly informative microsatellite-based genetic identity kit for potato. Mol Breeding 23:377–388
Hale A, Miller C, Rengayanaki K, Fritz A, Coombs J, Frank L, Douches S (2005) Suitability of AFLP and microsatellite marker analysis for discriminating intraclonal variants of the potato cultivar Russet Norkotah. J Amer Soc Hort Sci 130(4):624–630
Hawkes JG (1990) The potato: evolution, biodiversity and genetic resources. Belhaven Press, London, p 259
Hawkes JG (1994) Origins of cultivated potatoes and species relationships. In: Bradshaw JE, Mackay GR (eds) Potato genetics. CAB International, Wallingford, pp 3–42
Hosaka K, Hanneman RE (1988) The origin of the cultivated tetraploid potato based on chloroplast DNA. Theor Appl Genet 76:172–176
International Potato Center (CIP) (2008) In: Herrera M d R, Ghishlain M (eds) Potato Genetic Identity (PGI) Kit. Protocols and reference ladders. Applied Biotechnology laboratory training manual, Lima, Perú
Kaminsky KP, Korup K, Andersen MN, Sonderkaer M, Kirk AG, Nielsen KL (2015) Cytosolic glutamine synthetase is important for photosynthetic efficiency and water use efficiency in potato as revealed by high‑throughput sequencing QTL analysis. Theor. Appl. Genet. DOI 10.1007/s00122-015-2573-2
Leberg PL (2002) Estimating allelic richness: effects of sample size and bottlenecks. Mol Ecol 11:2445–2449
Machida-Hirano R (2015) Diversity of potato genetic resources. Breed Sci 65:26–40
Mathias M, Sagrado B, Kalazich J (2007) Uso de Marcadores SSR para Identificación de Germoplasma de Papa en el Programa de Mejoramiento de INIA de Chile. Agric Téc 67(1):3–15
Mendoza H, Mosquera V (2011) Selección de variedades de papa. Héctor Alcántara Santillán, Lima – Perú, p 108
Nei M (1987) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci U S A 70(12):3321–3323
Ovchinnikova A, Krylova E, Gavrilenko T, Smekalova T, Zhuk M, Knapp S, Spooner D (2011) Taxonomy of cultivated potatoes (Solanum section Petota: Solanaceae). Bot J Linn Soc 165:107–155
Perez J, Riegel R (2004). Evaluación de la diversidad genética de papas nativas Solanum tuberosum L. spp. tuberosum (Hawkes) silvestres y cultivadas del sur de Chile, mediante el uso de marcadores microsatélites. Thesis, Universidad Austral de Chile: Valdivia, Chile, p 68
Perrier X, Jacquemoud-Collet JP (2006) DARwin Software, version 5.0.158. CIRAD, Montpellier, France
Perrier X, Flori A, Bonnot F (2003) Data analysis methods. In: Hamon P, Seguin M, Perrier X, Glaszmann JC (eds) Genetic diversity of cultivated tropical plants. Enfield, Science Publishers, Montpellier, pp 43–76
Reid A, Kerr EM (2007) A rapid simple sequence repeat (SSR)-based identification method for potato cultivars. Plant Genet Resour Characterizat Util 5:7–13
Rios D, Ghislain M, Rodriguez F, Spooner D (2007) What is the origin of the European potato. Evidence from Canary Island Landraces. Crop Sci 47:1271–1280
Rocha E, Paiva L, de Carvalho H, Guimarães C (2010) Molecular characterization and genetic diversity of potato cultivars using SSR and RAPD markers. Crop Breed Appl Biotechnol 10(3):204–210. doi:10.1590/S1984-70332010000300004, Retrieved August 11, 2015, from http://www.scielo.br/scielo.php?script=sci_arttext&pid=S198470332010000300004&lng=en&tlng=en
Rubilar M, Villaroel M, Paillacar M, Moenne Locóz D, Shene C, Biolley E, Acevedo F (2012) Native black Michuñe potato variety: characterization, frying conditions and sensory evaluations. Arch Latinoam Nutr 62(1):87–93
Ruiz de Galareta J, Barandalla L, Lorenzo R, Gonzalez J, Rios D, Ritter E (2007) Microsatellite variation in potato landraces from the island of La Palma. Span J Agric Res 5(2):186–192
SAG, 2015. Servicio Agrícola y Ganadero SAG, registro histórico de variedades oficialmente descritas. http://www.sag.gob.cl/ambitos-de-accion/lista-de-variedades-oficialmente-descritas. Retrieved August 10, 2015.
Solano J, Mathias M, Esnault F, Brabant P (2013) Genetic diversity among native varieties and commercial cultivars of Solanum tuberosum ssp. tuberosum L. present in Chile. Electronic Journal Of Biotechnology, 16(6). Retrieved August 11, 2015, fromhttp://www.ejbiotechnology.info/index.php/ejbiotechnology/article/view/v16n6-15/1791
Spanoghe M, Marique T, Rivière J, Lanterbecq D, Gadenne M (2015) Investigation and Development of Potato Parentage Analysis Methods Using Multiplexed SSR Fingerprinting. 2015. Potato Res 58:43–65
Spooner DM (2009) DNA barcoding will frequently fail in complicated groups: an example in wild potatoes. Amer J Bot 96:1177–1189
Spooner DM, Núñez J, Trujillo G, Del Rosario HM, Guzmán F, Ghislain M (2007) Extensive simple sequence repeat genotyping of potato landraces supports a major reevaluation of their gene pool structure and classification. Proc Natl Acad Sci U S A 104:19398–19403
Uitdewilligen JGAML, Wolters A-MA, D’hoop BB, Borm TJA, Visser RGF, van Eck HJ (2013) A Next-Generation Sequencing Method for Genotyping-bySequencing of Highly Heterozygous Autotetraploid Potato. PLoS One 8(5):e62355. doi:10.1371/journal.pone.0062355
Acknowledgments
This study was funded by the CONICYT PBCT Program PDA09 Fortalecimiento de capacidades regionales en biotecnología para explotar la diversidad del genoma de la papa, Chilean Minister of Agriculture and Project 501453–70 Conservación de Recursos Genéticos.
We greatly acknowledge the Agricultural and Livestock Service (SAG) for kindly providing potato clones from their field collection.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Muñoz, M., Folch, C., Rodriguez, F. et al. Genotype Number and Allelic Diversity Overview in the National Collection of Chilean Potatoes. Potato Res. 59, 227–240 (2016). https://doi.org/10.1007/s11540-016-9329-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11540-016-9329-5