
Abstract
Mitochondrial dysfunction, oxidative stress and neuroinflammation have been implicated as key mediators contributing to the progressive degeneration of dopaminergic neurons in Parkinson’s disease (PD). Currently, we lack a pharmacological agent that can intervene in all key pathological mechanisms, which would offer better neuroprotective efficacy than a compound that targets a single degenerative mechanism. Herein, we investigated whether mito-apocynin (Mito-Apo), a newly-synthesized and orally available derivative of apocynin that targets mitochondria, protects against oxidative damage, glial-mediated inflammation and nigrostriatal neurodegeneration in cellular and animal models of PD. Mito-Apo treatment in primary mesencephalic cultures significantly attenuated the 1-methyl-4-phenylpyridinium (MPP+)-induced loss of tyrosine hydroxylase (TH)-positive neuronal cells and neurites. Mito-Apo also diminished MPP+-induced increases in glial cell activation and inducible nitric oxide synthase (iNOS) expression. Additionally, Mito-Apo decreased nitrotyrosine (3-NT) and 4-hydroxynonenol (4-HNE) levels in primary mesencephalic cultures. Importantly, we assessed the neuroprotective property of Mito-Apo in the MPTP mouse model of PD, wherein it restored the behavioral performance of MPTP-treated mice. Immunohistological analysis of nigral dopaminergic neurons and monoamine measurement further confirmed the neuroprotective effect of Mito-Apo against MPTP-induced nigrostriatal dopaminergic neuronal loss. Mito-Apo showed excellent brain bioavailability and also markedly attenuated MPTP-induced oxidative markers in the substantia nigra (SN). Furthermore, oral administration of Mito-Apo significantly suppressed MPTP-induced glial cell activation, upregulation of proinflammatory cytokines, iNOS and gp91phox in IBA1-positive cells of SN. Collectively, these results demonstrate that the novel mitochondria-targeted compound Mito-Apo exhibits profound neuroprotective effects in cellular and pre-clinical animal models of PD by attenuating oxidative damage and neuroinflammatory processes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a common neurodegenerative movement disorder characterized by motor and non-motor symptoms (Dauer and Przedborski 2003; Vila and Przedborski 2004). The pathological hallmarks of PD are the loss of dopaminergic neurons and their terminals in the nigrostriatal axis and the appearance of Lewy bodies in the surviving neurons in the substantia nigra pars compacta (SNpc). The underlying cellular and molecular mechanisms that cause the disease are not yet known, but studies suggest that mitochondrial dysfunction, oxidative stress, and glia-mediated inflammation increase the risk of developing PD (Dauer and Przedborski 2003; Goldman 2014; Blesa et al. 2015; Klingelhoefer and Reichmann 2015; Mullin and Schapira 2015). Sustained microglial and astroglial activation near dying dopaminergic neurons in the SNpc is evident in PD patients and animal models of the disease (McGeer et al. 1988; Hirsch and Hunot 2009). Several in vitro cell culture and in vivo animal studies have demonstrated the elevation of key enzymes involved in the production of reactive oxygen species (ROS) due to mitochondrial impairment and reactive nitrogen species (RNS) in dopaminergic neurons of the SN, including microglial gp91phox (Nox2) and inducible nitric oxide synthase (iNOS) (Gao et al. 2003b; Wu et al. 2003; Hunter et al. 2007; Hirsch et al. 2012; Murakami and Hirano 2012; Strowig et al. 2012; Yan et al. 2014; Rocha et al. 2015). In addition, different pro-inflammatory cytokines, including tumor necrosis factor alpha (TNF-α), interleukins IL-1β and IL-6, interferon gamma (IFN-γ), as well as other immune neurotoxins, are found either in the CSF or affected regions of PD brains (Nagatsu et al. 2000). However, the mechanism of glial cell activation in the diseased state is not completely known. Collectively, these findings suggest that targeting multiple pathogenic mechanisms, including mitochondrial impairment, oxidative stress, and inflammatory processes, could provide an advantage over targeting a single disease process, which is the strategy often tested by others for PD.
Several inhibitors of NADPH oxidase have been tested for their anti-inflammatory and antioxidant effects in dopaminergic cells, including apocynin (4-hydroxy-3-methoxyacetophenone), a plant-derived molecule that is well-tolerated in cell culture and animals models of PD (Gao et al. 2003a; Anantharam et al. 2007; Cristovao et al. 2009). We recently demonstrated diapocynin to be neuroprotective and anti-neuroinflammatory in the MPTP animal model as well as in the progressively degenerative LRRK2R1441G transgenic mouse model (Ghosh et al. 2012; Dranka et al. 2013). In these studies, both apocynin and diapocynin were orally administered at 300 mg/kg body weight. Although these high doses were tolerated in animals, more efficacious apocynin analogs are needed. To this effect, we designed a new class of apocynins (mito-apocynins) designed to enhance their cellular and mitochondrial uptake. Mito-apocynins were synthesized by the hybrid technique of attaching a mitochondria-targeting group to apocynin (Jin et al. 2014).
In this study, we synthesized Mito-Apo (apocynin conjugated to the mitochondria-targeting triphenylphosphonium cation moiety TPP+) (Fig. 1) and investigated its preclinical efficacy in both cell culture and animal models of PD by determining its antioxidant and anti-inflammatory properties. The presence of a highly lipophilic and delocalized cationic moiety in Mito-Apo makes it more cell-permeable and enhances its availability to mitochondria. Here, we demonstrate that Mito-Apo attenuates MPP+-induced dopaminergic neurodegeneration in primary mesencephalic neuronal cultures by attenuating glia-mediated inflammatory reactions and subsequent oxidative damage to dopaminergic neurons. Notably, oral administration of Mito-Apo reaches to the nigrostriatal region in mice where it protects dopaminergic neurons and terminals in an MPTP mouse model of PD by mitigating glial activation and glia-mediated inflammatory reactions and oxidative stress.

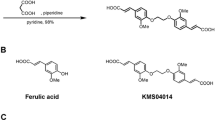
The synthetic pathway involved in the preparation of Mito-apocynin
Materials and Methods
Chemicals and Biological Reagents
1-Methyl-4-phenylpyridinium (MPP+ iodide) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP-HCl) were purchased from Sigma (St. Louis, MO, USA). RPMI 1640 medium neurobasal medium, B27 supplement, fetal bovine serum (FBS), L-glutamine, IR-dye-tagged secondary antibodies, penicillin, and streptomycin and other cell culture reagents were purchased from Invitrogen (Gaithersburg, MD).
Mito-Apocynin Synthesis
The novel compound Mito-apocynin (Mito-Apo) was synthesized by modifying the previously described protocol for Mito-Q and long chain Mito-Apo-C11 synthesis (Ghosh et al. 2010; Dranka et al. 2014). Figure 1 shows the key steps involved in the synthesis. Briefly, acetylvanillic acid chloride was synthesized from first mixing acetylvanillic acid with thionyl chloride, and then the mixture was dissolved in dichloromethane and aminoethyltriphenylphosphonium bromide and pyridine. The acetylated Mito-Apo was purified on a silica gel column followed by removal of the acetyl protective group. The final product was purified and characterized by HPLC and LC-MS as described previously (Ghosh et al. 2010; Dranka et al. 2014).
Primary Neuronal Cultures and Treatments
Primary mesencephalic and primary striatal cultures were prepared from the ventral mesencephalon and striatum respectively from gestational 14-day-old mouse embryos as described previously (Ghosh et al. 2013; Ngwa et al. 2014). Briefly, mesencephalic tissues from E14 mouse embryos were dissected and maintained in ice-cold calcium-free Hank’s balanced salt solution and then dissociated in Hank’s balanced salt solution containing trypsin-0.25 % EDTA for 20 min at 37 °C. The dissociated cells were then plated at an equal density of 0.6 million cells per well on 12-mm coverslips precoated with 0.1 mg/ml poly-D-lysine. Cultures were maintained in neurobasal media fortified with B-27 supplement, 500 mM L-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin. The cells were maintained in a humidified CO2 incubator (5 % CO2 and 37 °C) for 24 h. One-half of the culture media was replaced every other day. Approximately 6- to 7-day-old cultures were used for experiments. Both primary mesencephalic and striatal neuronal cells were exposed to 10 μM MPP+ in the presence or absence of Mito-Apo (10 μM) for 24 h.
Primary Microglial and Astroglial Cultures and Treatments
Primary microglial and astroglial cultures were prepared from C57BL/6 postnatal day 1 (P1) mouse pups. Mouse brains were harvested, meninges removed, and then placed in Dulbecco’s modified Eagle’s medium/F-12 nutrient media (DMEM-F12,GIBCO Cat # 11,320) supplemented with 10 % heat-inactivated FBS, 50 U/mL penicillin, 50 μg/mL streptomycin, 2 mM L-glutamine, 100 μM non-essential amino acids, and 2 mM sodium pyruvate (Invitrogen). The brain tissue was then incubated in 0.25 % trypsin for 30 min with gentle agitation. The trypsin reaction was stopped by adding an equal volume of DMEM/F12 complete media, and the brain tissue was washed three times. The tissue was then triturated gently to prepare a single cell suspension and then passed through a 70-μm nylon mesh cell strainer to remove tissue debris and aggregates. Next, the cell suspension was made up in DMEM/F12 complete media and seeded into T-75 flasks. Cell cultures were placed in a humidified CO2 incubator at 37 °C where the mixed glial cells were grown to confluence in media that was changed after 4 to 5 days. Using differential adherence and magnetic separation, microglial cells were separated from confluent mixed glial cultures to >97 % purity, as described in our recent publication (Gordon et al. 2011). Both microglia and astrocytes were allowed to recover for at least 48 h after plating. Primary microglia and astrocytes were treated in DMEM/F12 complete media containing 2 % FBS.
Immunocytochemistry
The primary mesencephalic and glial cultures were fixed with 4 % paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 20 min and processed for immunocytochemical staining, as described previously (Ghosh et al. 2010). First, nonspecific sites were blocked with 2 % bovine serum albumin, 0.5 % Triton X-100 and 0.05 % Tween-20 in PBS for 45 min at RT. Cells were then incubated overnight at 4 °C with different primary antibodies such as TH (1:1600, mouse monoclonal; EMD Millipore, Billerica, MA) and GFAP (1:1000, mouse monoclonal; EMD Millipore). Appropriate secondary antibodies (Alexa Fluor 488 or 594 or 555; Invitrogen) were used followed by incubation with 10 μg/ml Hoechst 33342 (Invitrogen) for 5 min at RT to stain the nucleus. Coverslips containing stained cells were washed twice with PBS and mounted on poly-D-lysine-coated slides (Sigma, St Louis). Images of cells were captured with a SPOT digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI) attached to an inverted fluorescence microscope (model TE-2000 U; Nikon, Tokyo, Japan).
Quantification of TH-Positive Cell Numbers and Neuronal Processes
From each cover-slipped primary neuronal culture, the number of TH-positive dopaminergic neurons and the length of their neuronal processes were quantified using MetaMorph software, Version 5.0 (Molecular Devices) as described previously (Ghosh et al. 2013). For counting TH cells, pictures were first taken at 20× magnification, then thresholded. After that, neurons were counted using the Integrated Morphometry Analysis function. For measurement of neuronal processes, pictures were taken using a 60× oil immersion lens, and the lengths of the processes were marked by applying the region and length measurement function in the Integrated Morphometry analysis.
High-Affinity [3 H]Dopamine Uptake Assays
Cells in each well were washed twice with 1 ml of Krebs-Ringer buffer (16 mM NaH2PO4, 16 mM Na2HPO4, 119 mM NaCl, 4.7 mM KCl, 1.8 mM CaCl2, 1.2 mM MgSO4, 1.3 mM EDTA, and 5.6 mM glucose, pH 7.4). The cells were then incubated with 25 nM [3 H]dopamine in Krebs-Ringer buffer (0.4 ml/well) for 20 min at 37 °C. Nonspecific uptake of dopamine was determined in parallel wells incubated with tritiated dopamine and 10 μM mazindol, an inhibitor of neuronal dopamine uptake. Briefly, the cells were washed 3 times with ice-cold Krebs-Ringer buffer (1 ml/well) and lysed with 1 N NaOH (0.5 ml/well). After washing, lysates were mixed with 5 ml of scintillation fluid (Scinti-verse BD), and radioactivity was determined with a liquid scintillation counter (Tri-Carb 1600 TR; Packard, Meriden, CT). Specific dopamine uptake was calculated by subtracting the amount of radioactivity observed in the presence of mazindol from that observed in its absence.
Animals and Treatments
Eight- to 10-week-old male C57BL/6 mice weighing 24 to 28 g were housed in standard conditions: constant temperature (22 ± 1 °C), relative humidity (30 %), and a 12-h light cycle. Animal experiments were approved and supervised by the Institutional Animal Care and Use Committee (IACUC) at Iowa State University (Ames, IA). Sub-acute treatment paradigm: Mice received Mito-Apo (3 mg/kg/day) in 10 % ethanol by oral gavage beginning 1 day before the MPTP treatment, 5 days of co-treatment with MPTP (25 mg/kg/day, intraperitoneal injection), Mito-Apo treatment continued daily for another 6 days after ending MPTP and mice were sacrificed 24 h later. Control mice received 10 % ethanol in saline. Acute Paradigm: Mice received Mito-Apo (3 mg/kg/day) in 10 % ethanol by oral gavage beginning 1 day before the MPTP treatment and 3 days of co-treatment with MPTP (25 mg/kg/day, intraperitoneal injection). Mice were sacrificed 24 h after the last dose of MPTP. Control mice received 10 % ethanol in saline.
HPLC Analysis of Striatal Dopamine and its Metabolites
Samples were prepared and quantified as described previously (Ghosh et al. 2010). In brief, 7 days after MPTP injection, mice were sacrificed and striata were collected and stored at −80 °C. On the day of analysis, neurotransmitters from striatal tissues were extracted using an antioxidant extraction solution (0.1 M perchloric acid containing 0.05 % Na2EDTA and 0.1 % Na2S2O5) and isoproterenol (internal standard). Dopamine (DA), 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) were separated isocratically using a reversed-phase column with a flow rate of 0.6 mL/min. An HPLC system (ESA Inc., Bedford, MA) with a thermostatted automatic sampler (model 542; ESA Inc.) was used for these experiments. The electrochemical detection system consisted of a Coulochem model 5100A with a microanalysis cell (model 5014A, ESA Inc.) and a guard cell (model 5020, ESA Inc.). Data acquisition and analysis were performed using EZStart HPLC Software (ESA Inc.).
HPLC Detection of Mito-Apo in the Brain
Mice were administered 3 mg/kg Mito-Apo by oral gavage and then sacrificed at 1, 2, 4, or 24 h (n = 4 per time-point). Samples of various brain regions were dissected out and stored at -80 °C. Tissue homogenates were then made using an antioxidant buffer containing 0.2 M perchloric acid, 0.1 % Na2S2O5, and 0.05 % Na2EDTA. The standards prepared and used for quantification were 0.1 μg, 0.3 μg, 1.0 μg, 10 μg, and 30 μg of Mito-Apo. Samples were centrifuged and passed through a 0.2-μm filter before analysis on an Agilent Technologies 1200 Series HPLC system. Samples were injected at a volume of 20 μL. Separation was performed with a Kinetex C18 column, using a gradient elution (solvent A = 90 % water, 10 % acetonitrile and solvent B = 100 % acetonitrile) operated over seven minutes at a flow rate of 1.5 mL/min and a temperature of 40 °C. Mito-Apo was quantified by UV absorbance (collected at wavelengths of 262 nm and 292 nm) with an average retention time of 3.8 min. Standards were fit by linear regression and data were analyzed using GraphPad Prism 4.0 software (GraphPad Software Inc., San Diego, CA).
qRT-PCR
After treatment, total RNA was extracted from the SN of mouse brains using Absolutely RNA Miniprep Kit (Agilent Technologies, Santa Clara, CA) following the manufacturer’s protocol. Total RNA was treated with DNase I to remove DNA contamination and then reversibly transcribed into first-strand cDNA using the SuperScript III first-strand synthesis system (Invitrogen, Carlsbad, CA) as described in the kit instructions. SYBR-green quantitative PCR was performed with validated primers of TNF-α, IL-1β, iNOS, and IL-6 as well as control 18S primers (Qiagen, Valencia, CA) using RT2 SYBR® Green qPCR Master Mix (SABiosciences, Frederick, MD).
Western Blotting
Mice were sacrificed three days after MPTP treatment and the SN was dissected out. SN lysates containing equal amounts of protein were loaded in each lane and separated in a 10–15 % SDS-polyacrylamide gel, as described previously (Jin et al. 2011). Proteins were transferred to a nitrocellulose membrane and nonspecific binding sites were blocked with Licor Odyssey blocking buffer. The membranes were then incubated with different primary antibodies such as anti-IBA-1 (Abcam), anti-GFAP (EMD Millipore), anti-iNOS (Santa Cruz), anti-gp91phox (Abcam), anti-3NT (EMD Millipore) and anti-4HNE (R & D Systems). Next, membranes were incubated with one of the following secondary antibodies: Alexa Fluor 680 goat anti-mouse, Alexa Fluor 680 donkey anti-goat (Invitrogen) or IR dye 800 donkey anti-rabbit (Rockland). To confirm equal protein loading, blots were reprobed with a β-actin antibody (Sigma; 1:10,000 dilution). Western blot images were captured with a Licor Odyssey machine (Licor, NE), and the bands were quantified using NIH ImageJ software.
Immunohistochemistry (IHC)
Three days after the last MPTP treatment, mice were perfused with 4 % PFA and post-fixed with PFA and 30 % sucrose. Next, fixed brains were cut into 30-μm sections and were incubated overnight at 4 °C with different primary antibodies such as anti-IBA-1, anti-GFAP, anti-iNOS, anti-3NT, anti-gp91phox and anti-4HNE. Appropriate secondary antibodies (Alexa Fluor 488, 594 or 555 from Invitrogen) were used, followed by incubation with 10 μg/ml Hoechst 33342 for 5 min at RT to stain the nucleus. Images of sections were captured with a SPOT digital camera (Diagnostic Instruments) attached to an inverted fluorescence microscope (Nikon TE-2000 U).
DAB Immunostaining and Stereological Counting
DAB immunostaining was performed in striatal and SN sections, as described previously (Ghosh et al. 2009; Dranka et al. 2013). In brief, 30-μm sections were incubated overnight at 4 °C with one of the following primary antibodies: anti-TH antibody (EMD Millipore, rabbit anti-mouse, 1:1800), anti-IBA-1 (Abcam, goat anti-mouse, 1:1000) or anti-GFAP (EMD Millipore, mouse anti-mouse, 1:1000). Next, sections were incubated in biotinylated anti-rabbit, -goat or -mouse secondary antibody followed by incubation with avidin peroxidase (Vectastain ABC Elite kit, Vector laboratories). Immunolabeling was observed using diaminobenzidine (DAB), which yielded a brown stain. Total numbers of TH-positive neurons in SN were counted stereologically with Stereo Investigator software (MBF Bioscience, Williston, VT, USA) using an optical fractionator (Ghosh et al. 2010).
Behavioral Measurements
Four days after the last MPTP injection, open-field and rotarod experiments were conducted as described previously (Ghosh et al. 2010). Briefly, a computer-controlled open-field test (Versamax monitor, model RXYZCM-16, AccuScan, Columbus, OH) was used to measure the spontaneous activity of mice. The activity chamber was 40 × 40 × 30.5 cm, made of clear Plexiglas and covered with a ventilated Plexiglas lid. Data were collected and analyzed by a VersaMax Analyzer (AccuScan, model CDA-8, Columbus, OH). Before any treatment, mice were placed inside the infrared monitor for 5 min daily for 3 consecutive days to acclimatize them. Locomotor activities were monitored during 10-min test sessions. Locomotor coordination was assessed using the rotarod (AccuScan) preset to a constant 20-rpm speed. Mice were given a 5–7 min rest interval to eliminate stress and fatigue.
Data Analysis
Data analysis was performed using Prism 4.0 software. Raw data were first analyzed using one-way analysis of variance and then Tukey’s post-test was performed to compare all treatment groups. Differences with p < 0.05 were considered significant.
Results
Mito-Apo Protects Against MPP+-induced Neurotoxicity in Primary Mesencephalic Cultures
Primary mesencephalic and primary striatal cultures were isolated from the E14 embryos of C57BL/6 mice and processed as described in “Materials and Methods”. After six days in culture, primary mesencephalic neurons were exposed to 10 μM MPP+ in the presence or absence of 10 μM Mito-Apo. After a 24-h treatment, tyrosine hydroxylase (TH)-immunostaining assays for cell morphology, TH+ neuron counting and neurite length and [3 H] dopamine uptake assays for dopamine neuronal functions were then performed. For TH ICC studies, cultures were fixed and stained for TH, and images (Fig. 2a) were captured with a Nikon TE 2000 U microscope. Compared to control cultures, MPP+-treated TH-immunoreactive neurons underwent extensive degenerative changes, as evidenced by reduced neurites and shrunken cell bodies (Fig. 2a). However, these changes were largely reversed by Mito-Apo co-treatment. Quantification of TH cell counts from multiple images from the neuronal cultures revealed that MPP+ reduced the TH+ cell count by approximately 75 % (Fig. 2b) compared to untreated controls. Treating with 10 μM Mito-Apo significantly protected against the MPP+-induced loss of TH+ cells. Similarly, MPP+ induced an ~85 % loss in TH neurite length (Fig. 2c) compared to untreated controls (p < 0.01), whereas neurite length in Mito-Apo-cotreated cultures was significantly longer relative to MPP+-alone, remaining comparable to that of untreated controls. The functional integrity of dopaminergic neurons was also examined by dopamine reuptake capacity, which serves as a neurite functional viability indicator (Ghosh et al. 2013). Treating with MPP+ for 24 h dramatically diminished dopamine uptake compared to untreated controls, while Mito-Apo significantly improved dopamine uptake in MPP+-co-treated cultures (Fig. 2d). Together, these data suggest that Mito-Apo elicits neuroprotective effects in the MPP+-induced dopaminergic degenerative model.

Mito-Apo protected dopaminergic neurons against MPP+-induced toxicity in primary mesencephalic cultures. Mesencephalic tissues from E14 mouse embryos were cultured and grown on poly-D-lysine-coated coverslips. The neuronal cultures were treated with 10 μM MPP+ for 24 h in the presence or absence of 10 μM Mito-Apo. After treatment, primary neurons were fixed with 4 % PFA and incubated with anti-TH antibody and viewed under a Nikon TE2000 fluorescence microscope. a TH immunolabeling in primary mesencephalic cultures from substantia nigra. b Quantification of TH immunoreactive (i.r.) dopaminergic neurons. c Quantification of TH i.r. dopaminergic axons. d Dopamine uptake assay from primary striatal culture. Data are represented by the mean ± SEM of three independent experiments. ***, p < 0.001 vs. Control; @, p < 0.05 vs. MPP+; **, p < 0.01 vs MPP+
Mito-Apo Attenuates MPP+-induced Increases in the Expression of 3-Nitrotyrosine (NT) and 4-Hydroxynonenal (HNE) in Primary Mesencephalic Cultures
Next, we investigated whether Mito-Apo could also attenuate MPP+-induced oxidative and nitrative stress in primary mesencephalic cultures. We used 3-NT staining as a marker of nitric oxide-dependent oxidative stress and 4-HNE as a marker of membrane lipid peroxidation induced by hydroxyl radicals (•OH) (Stadler et al. 2008). Double-immunolabeling with antibodies directed against TH and 3-NT revealed that MPP+ significantly increased the immunoreactivity of 3-NT-modified proteins in primary dopaminergic neurons, while adding Mito-Apo significantly inhibited 3-NT immunoreactivity levels in MPP+-treated cultures (Fig. 3a). Similarly, we observed an increased 4-HNE immunoreactivity in dopaminergic neurons in the presence of MPP+ as evidenced by 4-HNE and TH double-immunolabeling, and co-treating with Mito-Apo effectively attenuated 4-HNE immunoreactivity, demonstrating the efficacy of Mito-Apo in inhibiting oxidative damage in dopaminergic neurons (Fig. 3b). Together, these results suggest that Mito-Apo mitigates MPP+-induced increases in the levels of 3-NT and 4-HNE in TH+ neurons, further supporting a neuroprotective role for Mito-Apo.

Mito-Apo attenuated MPP+-induced oxidative stress in primary mesencephalic cultures. Mesencephalic tissues from E14 mouse embryos were cultured and grown on poly-D-lysine-coated coverslips. The neuronal cultures were treated with 10 μM MPP+ for 24 h in the presence or absence of 10 μM Mito-Apo. Cells were fixed with 4 % PFA and incubated with different antibodies and viewed under a Nikon TE2000 fluorescence microscope. a Double-labeling of TH and 3-NT. b Double-labeling of TH and 4-HNE
Mito-Apo Mitigates MPP+-induced Glial Activation and Inducible Nitric Oxide Synthase (iNOS) Upregulation in Primary Microglia and Astrocyte Cultures
Activation of microglia and astrocytes plays an important role in the pathogenesis of PD, as well as in other neurodegenerative disorders (Dauer and Przedborski 2003; Gao et al. 2003b; Ghosh et al. 2007). It has been well-established that MPP+ causes glial cell proinflammatory activation, which is believed to exacerbate dopaminergic degeneration by augmenting oxidative stress (Block and Hong 2007). Ionized calcium-binding adaptor molecule 1 (IBA1) and glial fibrillary acidic protein (GFAP) are markers of microglia and astrocyte activation, respectively (Ghosh et al. 2012). The key proinflammatory enzyme iNOS is typically elevated in disease conditions (Kavanagh et al. 2014) and thereby serves as a marker for glial pro-inflammatory activation. To determine if Mito-Apo protects against MPP+-induced microglia activation and astrogliosis, we treated mouse primary microglia and astrocytes with 10 μM MPP+ in the presence or absence of 10 μM Mito-Apo for 24 h. In immunocytochemistry experiments, the expression of IBA-1 and GFAP were significantly increased in MPP+-treated primary microglia (Fig. 4a) and astrocytes (Fig. 4b), respectively. In contrast, these effects were greatly inhibited by co-treating with Mito-Apo. In colocalization studies, MPP+ significantly increased iNOS expression in both IBA-1- and GFAP-expressing cells, whereas Mito-Apo significantly restored iNOS expression back to control levels in MPP+-cotreated cultures (Fig. 4a, b). Together, these results suggest that Mito-Apo co-treatment mitigates MPP+-induced glial activation and iNOS upregulation in primary microglia and astrocytes, suggesting an anti-inflammatory effect for Mito-Apo.

Mito-Apo attenuated MPP+-induced glial activation and iNOS expression. Isolated primary microglia or astroglia from one-day-old pups were treated with 10 μM MPP+ for 24 h in the presence or absence of 10 μM Mito-Apo. Cells were fixed with 4 % PFA and incubated with different antibodies and imaged through a Nikon TE2000 fluorescence microscope. a Double-labeling of IBA-1 and iNOS in primary microglia cells. b Double-labeling of GFAP and iNOS in primary astroglia cells
Mito-Apo Improves Motor Behavior in MPTP-treated Mice
After establishing the efficacy of Mito-Apo in mitigating inflammatory reactions and protecting dopaminergic neurons against MPP+ toxicity in primary cultures of neurons, microglia and astrocytes, we investigated the neuroprotective effect of Mito-Apo in a preclinical animal model of PD. The MPTP mouse model is a reliable and effective rodent model which has been extensively used to test putative neuroprotective agents (Grunblatt et al. 2000; Meissner et al. 2004; Zhang et al. 2007; Prediger et al. 2011; Seidl and Potashkin 2011; Ghosh et al. 2012). We first evaluated whether Mito-Apo improves MPTP-induced motor deficits by testing the behavioral performance in the open-field test and on the rotarod, as described recently (Ghosh et al. 2010). Animals receiving Mito-Apo (3 mg/kg/day) orally were evaluated four days after MPTP treatment. Representative activity maps (Fig. 5a) of open-field movements of saline-treated control, MPTP and MPTP/Mito-Apo-cotreated mice revealed decreased movements of MPTP-treated mice in contrast to improved locomotion in MPTP/Mito-Apo-cotreated mice. MPTP decreased horizontal activity (Fig. 5b), vertical activity (Fig. 5c), stereotypy count (Fig. 5d), total movement time (Fig. 5e), rearing count (Fig. 5f) and the time spent on the 20-rpm rotarod (Fig. 5g), which are all consistent with our previous observations (Ghosh et al. 2010). Importantly, most of the key motor deficits observed in the MPTP mouse model of PD were restored significantly by Mito-Apo treatment (Fig. 5b-g), suggesting potential neurobehavioral improvement with Mito-Apo treatment for PD.

Mito-Apo rescued MPTP-induced behavioral and locomotor deficits. Sub-Acute MPTP-treatment paradigm as described in the methods section was used for this experiment. Control mice received saline. Mice were tested for locomotor activities four days after the last dose of MPTP. a Movement track of mice collected over 10 min. Open-field data showing b horizontal activity; c vertical activity; d stereotypy count; e total movement time; f rearing count and g time spent on rotarod (sec) at 20-rpm speed. Data represented by the mean ± SEM of six to eight mice per group. ***, p < 0.001 vs. the control group; **, p < 0.01 vs. the MPTP group; *, p < 0.05 vs. the MPTP group; @, p < 0.05 vs. the Control group; #, p < 0.01 vs. the Control group
Mito-Apo Inhibits Microglia and Astrocyte Activation in the SN in Vivo
Increasing evidence suggests that activated glial cells in the SN pars compacta (SNpc) play an important role in dopaminergic neurodegeneration in human PD patients and in the MPTP mouse model (Dauer and Przedborski 2003; Block and Hong 2007; Ghosh et al. 2007; Panicker et al. 2015). Thus, we evaluated whether the neuroprotective effect of Mito-Apo is due to attenuated gliosis in the SN. For determining neuroinflammatory responses, a three day MPTP model was used (Przedborski et al. 2004; Jackson-Lewis and Przedborski 2007; Ghosh et al. 2012). Three days post-MPTP treatment, mice were sacrificed and nigral tissue sections were processed for DAB immunostaining of IBA-1 and GFAP. Increased numbers of activated microglia (characterized by amoeboid shape with thickened processes) and astrocytes (characterized by bigger cell bodies with thickened processes) were noted in the SN of MPTP-treated mice compared with those in saline-treated control mice (Fig. 6a, b). However, Mito-Apo greatly decreased the number of IBA1-positive microglial cells (Fig. 6a) and GFAP-positive astroglial cells (Fig. 6b) in the MPTP-treated SN. To further corroborate these findings, Western blotting revealed significantly increased expression of IBA-1 and GFAP in MPTP-treated SN tissues compared with saline-treated control samples, whereas orally administered Mito-Apo attenuated the expression of IBA-1 (Fig. 6c, d) and GFAP (Fig. 6e, f) in MPTP-treated mice. These results, therefore, support an anti-inflammatory property for Mito-Apo in vivo.

Mito-Apo blocked activation of microglia and astrocytes in the SN of MPTP-treated mice. Acute MPTP-treatment paradigm as described in the methods section was used for this experiment. Control mice received 10 % ethanol in saline. The substantia nigra (SN) tissue sections were immunostained with antibodies against IBA-1 a and GFAP b Insets, four-fold higher magnification of A-B. Dotted white lines indicate the SNpc. Representative Western blots illustrating the expression of IBA-1 c and GFAP e in SN. d Bar graph showing mean IBA-1/β-actin ratios ± SEM from Western blotting in SN of 6 mice per group. f Bar graph showing mean GFAP/β-actin ratios ± SEM from Western blotting in SN of 6 mice per group. ***, p < 0.001 vs. the Control group; *, p < 0.05 vs. the MPTP group
Mito-Apo Attenuates Activation of NADPH Oxidase in Activated Microglia of MPTP-treated Mice
It has been demonstrated that NADPH oxidase-induced oxidative stress plays a pivotal role in the neurodegeneration of dopaminergic neurons in the MPTP mouse model of PD (Vejrazka et al. 2005). Apocynin is known to suppress NADPH oxidase (Anantharam et al. 2007; Ghosh et al. 2012; Dranka et al. 2013; Dranka et al. 2014). Thus, in the mouse SN, we examined the protein expression of gp91phox, a major component of NADPH oxidase, three days after MPTP treatment. Western blot analysis showed an increased gp91phox expression in MPTP-treated mice compared to only weak expression in saline-treated mice (Fig. 7a, b). However, Mito-Apo attenuated the MPTP-induced gp91phox expression in the SN (p < 0.05; Fig. 7a, b). Double-immunolabeling studies confirmed that gp91phox immunoreactivity colocalized with IBA-1-positive microglia (Fig. 7c). Additionally, robust gp91phox immunoreactivity was seen specifically in larger cells whose shorter ramifications were thickened in the SN of MPTP-treated mice (Fig. 7c insets). Consistently, Mito-Apo attenuated MPTP-induced gp91phox protein expression in IBA-1-positive microglia in the SN. These results suggest that Mito-Apo effectively dampens the MPTP-induced activation of microglial NADPH oxidase.

Mito-Apo attenuated activation of NADPH oxidase in the SN of MPTP-treated mice. Acute MPTP-treatment paradigm as described in the methods section was used for this experiment. a Representative Western blots illustrating the expression of gp91phox in SN. b Bar graph showing mean gp91phox/β-actin ratios ± SEM from Western blotting in SN of 6 mice per group. c Substantia nigra tissue sections were double-labeled for gp91phox and IBA-1. Images were captured at 20X and 60X (insets) magnification. The SNpc zone is outlined in white dots. Inset pictures demonstrate colocalization of IBA-1 and gp91phox. **, p < 0.01 vs. the Control group; *, p < 0.05 vs. the MPTP group
Mito-Apo Suppresses iNOS Expression in the SN of MPTP-treated Mice
Recently, we reported increased levels of iNOS in the SN of MPTP-treated mice (Ghosh et al. 2009; Ghosh et al. 2012). Therefore, we investigated whether Mito-Apo modulates dopamine neuronal survival by inhibiting MPTP-induced iNOS expression. Immunoblotting of SN tissues collected three days after MPTP treatment revealed a marked increase in the expression of iNOS in MPTP-treated mice relative to PBS-treated control mice, which was attenuated when co-treated with Mito-Apo (p < 0.05; Fig. 8a, b). Additionally, immunofluorescence of iNOS in SN sections shows that MPTP markedly increased nigral iNOS protein expression, and that iNOS colocalized with IBA-1-positive microglia and GFAP-positive astrocytes, respectively (Fig. 8c, d). However, consistent with its inhibitory effect on glial activation, Mito-Apo attenuated MPTP-induced expression of iNOS in IBA-1- and GFAP-expressing cells (Fig. 8c, d).

Mito-Apo decreased MPTP-induced nitric oxide damage in the SN of MPTP-treated mice. Acute MPTP-treatment paradigm as described in the methods section was used for this experiment. a Representative Western blots illustrating the expression of iNOS in SN. b Bar graph showing mean iNOS/β-actin ratios ± SEM from Western blotting in SN of 6 mice per group. c Double-labeling of GFAP and iNOS and d IBA-1 and iNOS in SN region of ventral midbrain. Images were captured at 20X and 60X (insets) magnification. Inset pictures demonstrate colocalization of iNOS and GFAP or IBA-1 ***, p < 0.001 vs. the Control group; *, p < 0.05 vs. the MPTP group
Mito-Apo Attenuates Nitrative Modification and Lipid Peroxidation in the SN in Vivo
Nitration of protein tyrosine residues is believed to be a well-known marker of oxidative stress in PD and its animal models (Dawson and Dawson 2003; Sherer et al. 2003; Sanders et al. 2014; Schapira et al. 2014). Under oxidative stress conditions, iNOS-derived oxidants selectively nitrate tyrosine residues, generating 3-NT (Ara et al. 1998). To observe iNOS-mediated oxidative damage to tyrosine residues, we checked the expression of 3-NT in the SN three days after MPTP treatment in the presence or absence of Mito-Apo. Western blot analysis showed a robust increase in 3-NT expression compared with that in PBS-treated control mice (Fig. 9a, b). However, Mito-Apo greatly attenuated the MPTP-induced 3-NT formation (p < 0.05; Fig 9b). Consistent with this result, IHC analysis demonstrated increased 3-NT expression in TH-positive dopaminergic neurons in the SN of MPTP-treated mice (yellow-colored cells show colocalization of TH and 3-NT) compared with PBS-treated control mice (Fig. 9c). However, this robust increase of 3-NT levels in MPTP-treated mice was dramatically inhibited by Mito-Apo, as very few TH-positive cells expressed nitrated protein (containing 3-NT) in their cytosol (Fig. 9c).

Mito-Apo decreased the formation of 3-nitrotyrosine in the SN of MPTP-treated mice. Acute MPTP-treatment paradigm as described in the methods section was used for this experiment. a Representative Western blots illustrating the expression of 3-NT in SN. b Bar graph showing mean 3-NT/β-actin ratios ± SEM from Western blotting in SN of 6 mice per group. c Double-labeling of TH and 3-NT in the SN. Images were captured at 20X and 60X (insets) magnifications. The SNpc zone is outlined in white dots. Inset pictures demonstrate colocalization of TH and 3-NT. ***, p < 0.001 vs. the Control group; *, p < 0.05 vs. the MPTP group
Studies from our lab and others have shown increased levels of 4-HNE in MPTP-treated mouse brains (Selley 1998; Fujita et al. 2009; Ghosh et al. 2012). In the present study, we also observed a robust increase in 4-HNE expression in the SN of MPTP-treated mice three days after MPTP administration compared to saline-treated mice (Fig. 10a, b). However, Mito-Apo significantly reduced the MPTP-induced expression of 4-HNE (p < 0.01; Fig. 10b). Consistent with the Western blotting data, IHC data show increased 4-HNE expression in the SN of MPTP-treated mice, with 70–80 % of TH-positive dopaminergic neurons expressing 4-HNE in their cytosol (Fig. 10c). In contrast, MPTP mice co-treated with Mito-Apo showed dramatically reduced 4-HNE expression in dopaminergic neurons of the SN (Fig. 10c). Together, these results demonstrate that Mito-Apo attenuates oxidative damage in nigral neurons in the MPTP mouse model of PD.

Mito-Apo inhibited MPTP-induced formation of 4-hydroxynonenol in the SN of MPTP-treated mice. Acute MPTP-treatment paradigm as described in the methods section was used for this experiment. a Representative Western blots illustrating the expression of 4-HNE in SN. b Bar graph showing mean 4-HNE/β-actin ratios ± SEM from Western blotting in SN of 6 mice per group. c Double-labeling of TH and 4-HNE in the SN. Images were captured at 20X and 60X (insets) magnifications. The SNpc zone is outlined in white dots. Inset pictures demonstrate colocalization of TH and 4-HNE. ***, p < 0.001 vs. the Control group; **, p < 0.01 vs. the MPTP group
Mito-Apo Inhibits the Expression of Pro-inflammatory Molecules in Vivo in the Midbrain of MPTP-treated Mice
Activated glial cells release pro-inflammatory cytokines along with iNOS, thereby damaging dopaminergic neurons. Our qRT-PCR analysis revealed a marked increase in mRNA expression of iNOS, TNF-α, IL-1β and IL-6 in the ventral midbrain three days after MPTP administration (Fig. 10a-d). However, Mito-Apo (3 mg/kg/day) strongly inhibited the MPTP-induced expression of iNOS, TNF-α and IL-1β in the ventral midbrain (Fig. 11a-c). Although Mito-Apo appeared to similarly attenuate the expression of IL-6, the effect was not significant (Fig. 11d). These results suggest that Mito-Apo suppresses the expression of major pro-inflammatory molecules in the ventral midbrain of MPTP-treated mice.

Mito-Apo attenuated the expression of proinflammatory molecules in the ventral midbrain of MPTP-treated mice. Acute MPTP-treatment paradigm as described in the methods section was used for this experiment. The mRNA expression of iNOS a, TNF-α b, IL-1β c and IL-6 d was analyzed by quantitative real-time PCR. Data represented by the mean ± SEM of six mice per group. **, p < 0.01 vs. Control; *, p < 0.05 vs. Control; @, p < 0.05 vs MPTP; #, p < 0.01 vs MPTP; ns = non-significant
Mito-Apo Improves Nigrostriatal Dopaminergic Neuron Survival and Striatal Neurotransmitter Levels in MPTP-treated Mice
To demonstrate that Mito-Apo protects against MPTP-induced dopaminergic neuronal degeneration in the SN, we performed TH-immunohistochemical studies (Ghosh et al. 2010; Ghosh et al. 2012; Ghosh et al. 2013). Sectioned brains collected seven days post-MPTP were immunostained with TH to detect dopaminergic neurons and fibers. DAB immunostaining revealed a significant loss of TH-positive cell bodies and terminals in the SN and striatum, respectively, in subacutely MPTP-treated mice as compared to saline-treated mice (Fig. 12a, b). Higher magnified (10X) pictures (Fig. 12b, lower panels) clearly demonstrate neuron loss in the SNpc, lateralis (SNl) and reticularis (SNr) of MPTP-treated mice. Oral administration of Mito-Apo (3 mg/kg/day) significantly mitigated MPTP-induced dopaminergic neuronal cell death in the SN and terminal loss in the striatum as determined by TH-immunostaining (Fig. 12a, b). Additionally, stereological counting of TH+ neurons in the SN further confirmed the neuroprotective property of Mito-Apo (Fig. 12c).

Mito-Apo prevented MPTP-induced neurotoxicity in the nigrostriatal axis. Sub-Acute MPTP-treatment paradigm as described in the methods section was used for this experiment. DAB-immunohistochemistry for TH in and striatum a and SN b, upper panel 2X magnification; lower panel 10X magnification. c Stereological counting of TH-positive neurons in the SN. Striatal dopamine d, DOPAC e and HVA f were measured by HPLC. Data represented by mean ± SEM of six to eight mice per group. ***, p < 0.001 vs. Control; @, p < 0.01 vs. MPTP; #, p < 0.05 vs MPTP
To further determine whether Mito-Apo protects against neurotransmitter depletion induced by MPTP, we measured striatal dopamine, DOPAC and HVA by HPLC (Fig. 12d-f). Our results revealed an ~80 % loss of striatal dopamine in MPTP-treated mice relative to saline-treated mice. In contrast, Mito-Apo (3 mg/kg/day) increased dopamine levels by ~50 % when compared to MPTP-alone (p < 0.001; Fig. 12d). MPTP also caused a ~ 76 % and ~70 % loss of DOPAC and HVA, respectively, in the striatum, and Mito-Apo treatment significantly attenuated MPTP-induced depletion of those metabolites (Fig. 12d-f). It is possible that neuroprotection afforded by Mito-Apo could due to it’s interference with the toxic metabolic conversion of MPTP to MPP+ by inhibiting MAO-B enzyme activity. To address this possibility, we measured the level of MPP+ in striatum 3 h after the final MPTP injection, with or without Mito-Apo treatment. We found that Mito-Apo had no effect on striatal levels of MPP+ (MPTP-treated mice, 87.38 ± 13.59 ppb; MPTP plus Mito-Apo treated mice, 83.12 ± 9.86 ppb). Taken together, our histological and neurochemical data clearly demonstrated that Mito-Apo imparts a remarkable neuroprotective effect in the MPTP model of PD.
Brain Bioavailability of Mito-Apo
The next logical step is to determine the amount of orally administered Mito-Apo that reached striatal and nigral tissues. Briefly, naïve C57BL/6 mice were oral gavaged 3 mg/kg Mito-Apo and then sacrificed at 1, 2, 4, or 24 h, and striatal and nigral regions were dissected and processed for HPLC detection as described in “Materials and Methods”. Striatal Mito-Apo levels were determined to be 72, 65, 43 and 19 ng/mg tissue at 1, 2, 4 and 24 h, respectively (Fig. 13). Similarly, Mito-Apo levels in the SN were determined to be 84, 48, 47 and 32 ng/mg tissue at 1, 2, 4 and 24 h, respectively (Fig. 13). These results suggest that Mito-Apo rapidly crosses the blood brain barrier to reach its target tissues, the striatum and SN, and persists in the CNS to produce its neuroprotective effect. We therefore can readily attribute the remarkable neuroprotective effect of Mito-Apo in the MPTP model of PD to its excellent bioavailability in the CNS.

Mito-Apo bioavailability in the brain. Mice were administered Mito-Apo (3 mg/kg) once orally and sacrificed at 1, 2, 4, or 24 h post-gavage. Control mice received saline. Striatal and nigral regions were dissected out and processed for HPLC-UV detection as described in “Materials and Methods”. a Linear regression of Mito-Apo standards at 292 nm. Quantification of chromatographs for detection of Mito-Apo in SN tissue b and striatum c. d, e Representative chromatographs of brain tissues analyzed for Mito-Apo detection at various time points in the SN and striatum, respectively. Data are represented as mean ± SEM of four mice per group. **, p < 0.01 vs. the control group
Discussion
Neuroinflammation, oxidative and nitrative stress induced by NADPH oxidase and mitochondria-derived superoxide are key pathophysiological events contributing to the degeneration of dopaminergic neurons in PD (Wu et al. 2003; Mosley et al. 2006; Przedborski 2007; Tansey and Goldberg 2010; Cristovao et al. 2012; Zhang et al. 2014). In this study, we synthesized a novel anti-oxidant, mito-apocynin (Mito-Apo), by conjugating triphenylphosphonium (TPP+) to apocynin to specifically target it to mitochondria. Mito-Apo attenuated the MPP+-induced loss of dopaminergic neurons and oxidative and nitrative stress in primary cultures. In animal studies, Mito-Apo blocked MPTP-induced glial activation, iNOS and gp91phox expression, and the formation of peroxynitrite (ONOO-) and 4-HNE, suggesting an anti-neuroinflammatory and antioxidant property. Importantly, Mito-Apo also protected against MPTP-induced motor deficits, striatal neurotransmitter depletion and nigrostriatal dopaminergic degeneration in animal studies, suggesting an in vivo neuroprotective property.
Many conventional antioxidant compounds (e.g., vitamin E, tempol, CoQ, creatine, minocycline and ubiquinone) have failed to attenuate ROS/RNS in preclinical models due to their inability to accumulate in the mitochondria (Murphy 1997; Smith et al. 1999). Here we provide evidence that the mitochondria-targeted antioxidant Mito-Apo attenuates MPP+-induced increases in the expression of IBA-1 and GFAP in primary microglia and astrocyte cultures (Fig. 4), and rescued TH+ neurons in primary mesencephalic cultures (Fig. 2), suggesting that Mito-Apo exhibits both anti-neuroinflammatory and neuroprotective properties. Extension of these studies to the MPTP animal model revealed that Mito-Apo also effectively attenuated MPTP-induced IBA-1 and GFAP expression and TH+ neuronal loss in the mouse SNpc (Figs. 6 & 12).
Studies employing the MPTP animal model of PD have also shown NADPH oxidase activation, increased iNOS expression and elevated pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α (Nagatsu et al. 2000; Wang et al. 2009; Lofrumento et al. 2011; Lim et al. 2015). Moreover, nitric oxide (•NO) generated by iNOS and derived from activated glial cells is believed to contribute to neurodegenerative diseases such as Alzheimer's disease (AD), PD, HIV-associated dementia, Huntington's disease (HD) and Amyotrophic lateral sclerosis (ALS) (Galea et al. 1992; Ghosh et al. 2009). Pro-inflammatory cytokines such as TNF-α, IL-6 and IL-1β secreted from activated microglia facilitate intracellular death-related signaling pathways or activation of iNOS expression in the MPTP mouse model (Teismann et al. 2003). Furthermore, the promoter region of iNOS and pro-inflammatory cytokines contain the DNA binding sites for NF-κB, a transcription factor that regulates glial cell-mediated inflammatory reactions (Hayden and Ghosh 2004). In vivo studies have shown increased expression of iNOS and pro-inflammatory cytokines in ventral midbrain regions of MPTP-treated mice (Ghosh et al. 2007; Ghosh et al. 2009). Here we show that Mito-Apo attenuated MPP+-induced increases in iNOS expression in primary microglia and astrocyte cultures (Fig. 3). Furthermore, Mito-Apo treatment suppressed MPTP-induced expression of iNOS and the pro-inflammatory cytokines TNFα, IL-6 and IL-1β in the mouse SNpc, suggesting an in vivo anti-inflammatory role (Fig. 11).
NADPH oxidase (NOX) and mitochondria-derived ROS are known to augment pro-inflammatory events in activated microglia, particularly in neurodegenerative disorders, such as PD, AD, ALS and multiple sclerosis (Ansari and Scheff 2011; Cristovao et al. 2012; Zhang et al. 2014). Recently, several studies have implicated Nox1-derived ROS in dopaminergic cell death induced by paraquat and 6-OHDA, and both of these toxins upregulated Nox1 (Choi et al. 2012; Cristovao et al. 2012; Hernandes et al. 2013). NADPH oxidase- and mitochondria-derived ROS both play equally important key roles in proinflammatory microglial activation (Dikalov 2011; Mead et al. 2012; Bordt and Polster 2014). In the microglia, superoxide (O2•) is primarily produced by the three isoforms Nox1, Nox2 and Nox4, and among these, Nox2 is the most responsive and upregulated isoform in activated microglia (Fischer et al. 2012; Maghzal et al. 2012; Cooney et al. 2013).
Recent studies from our lab and others have shown that apocynin is a non-specific NADPH oxidase inhibitor (Hayashi et al. 2005; Sovari et al. 2008; Impellizzeri et al. 2011; Ghosh et al. 2012; Dranka et al. 2014). Functional studies have revealed apocynin to attenuate superoxide formation and oxidative stress in vivo in the spinal cord (Bedard and Krause 2007). Additionally, apocynin administered at a dose of 300 mg/kg/day protected against oxidative damage in an ALS model (Harraz et al. 2008), although this finding has been questioned by Trumbull et al. (2012). In vitro studies in dopaminergic neuronal cell lines and primary cultures also demonstrated the protective role of apocynin in the MPP+ model of PD (Anantharam et al. 2007). Recently, we have shown that diapocynin (apocynin dimer) reduced neuroinflammation and protected dopaminergic neurons in the MPTP mouse model of PD (Ghosh et al. 2012) by preventing the assembly and activation of the NADPH oxidase complex. Diapocynin is 13-fold more lipophilic than the monomer apocynin (Luchtefeld et al. 2008). Later, we demonstrated that diapocynin successfully prevented motor deficits in the leucine-rich repeat kinase 2 (LRRK2R1441G) transgenic mouse (Dranka et al. 2013). Very recently, we showed that treatment with a long-chain Mito-Apo analog, Mito-Apo-C11, prevented hyposmia and reduced motor deficits in the progressive degenerative LRRK2R1441G transgenic mouse model (Dranka et al. 2014). Here, we demonstrate that MPTP-induced increases in Nox2 enzyme (gp91phox) expression colocalized with IBA1-positive cells in the SNpc and that Mito-Apo effectively attenuated this increase in Nox2 expression, suggesting its antioxidant role in vivo (Fig. 7). Therefore, Mito-Apo may suppress MPTP-induced microglial Nox2 upregulation by inhibiting mitochondria generated superoxide.
Reactive glial cells induce •NO and O2 •–-generated by NADPH oxidase, and in mitochondria, react to form a potent oxidant, ONOO−, which causes oxidative and nitrative modification of proteins through nitration of the proteins’ tyrosine residues in dopaminergic neurons (Ara et al. 1998; Dawson and Dawson 2003). Studies have shown iNOS expression and increased 3-NT levels in activated microglia in the SN of human PD brains (Hunot et al. 1996). Previous studies also have shown that iNOS facilitates dopaminergic neurodegeneration via MPTP-induced production of RNS in the MPTP mouse model of PD, resulting in enhanced 3-NT levels (Liberatore et al. 1999; Dehmer et al. 2000). In this study, the increased 3-NT expression we observed in primary mesencephalic culture and dopaminergic neurons in MPTP-treated mice was significantly reduced by Mito-Apo treatment in both models (Figs. 3 and 9). Besides peroxynitrite, 4-HNE, a well-known marker for oxidative insult caused mainly by the hydroxyl radical (•OH), was also formed during the course of disease. As an unsaturated aldehyde, more 4-HNE is generated due to decomposition of lipid peroxides in the SN of PD brains than in healthy controls (Yoritaka et al. 1996). Previous studies also demonstrated that 4-HNE blocked mitochondrial respiration and induced caspase-dependent apoptosis in neurons (Picklo et al. 1999). In our study, 4-HNE expression increased in both primary mesencephalic cultures and mice treated with MPP+ and MPTP, respectively (Figs. 3 and 10), but co-treatment with Mito-Apo significantly blocked the 4-HNE expression. These results further strengthen the anti-oxidant capability of Mito-Apo.
Therapy effective at halting the degeneration of dopaminergic neurons and their striatal terminals remains elusive. Administration of levodopa, the gold standard drug for PD does not affect disease progression. Other putative neuroprotective agents such as glial cell line-derived neurotrophic factor (GDNF), brain derived neurotrophic factor (BDNF) and other small compounds have been tested in different animal models of PD with no or little success in clinical trials (Chen and Le 2006; Wu et al. 2011; Ybot-Gorrin et al. 2011; Athauda and Foltynie 2015). Additionally, these compounds also caused adverse side effects. Our novel compound, Mito-Apo, has several advantages compared to other experimental drugs: 1) orally administered Mito-Apo does not show any toxic effect in mice; 2) Mito-Apo, being a lipophilic molecule, diffuses through the blood-brain barrier and into mitochondria where it inhibits superoxide production; 3) Mito-Apo can be taken orally, which is the least painful route; 4) orally administered Mito-Apo protects the nigrostriatal axis in the MPTP model of PD; 5) Mito-Apo is at least 100-fold more efficacious than apocynin or diapocynin in preclinical animal PD model; and 6) low-dose Mito-Apo is CNS bioavailable and reaches its intended target tissues, the substantia nigra and striatum.
In summary, we have demonstrated that Mito-Apo protected dopaminergic neurons both in primary culture and an animal model in the presence of a potent dopaminergic toxin. Mito-Apo reduced oxidative and nitrative stress, glial activation and inflammatory reactions. Mito-Apo is CNS bioavailable and reversed the behavioral deficits caused by MPTP. Moreover, Mito-Apo restored dopamine and its metabolite levels and preserved TH+ neurons in the SNpc of MPTP-treated mice. These previously undiscovered properties of this novel mitochondria-targeted antioxidant strongly support continued research into potential clinical applications for Mito-Apo as a viable neuroprotective drug for treating Parkinson’s disease.
References
Anantharam V, Kaul S, Song C, Kanthasamy A, Kanthasamy AG (2007) Pharmacological inhibition of neuronal NADPH oxidase protects against 1-methyl-4-phenylpyridinium (MPP+)-induced oxidative stress and apoptosis in mesencephalic dopaminergic neuronal cells. Neurotoxicology 28:988–997
Ansari MA, Scheff SW (2011) NADPH-oxidase activation and cognition in Alzheimer disease progression. Free Radic Biol Med 51:171–178
Ara J, Przedborski S, Naini AB, Jackson-Lewis V, Trifiletti RR, Horwitz J, Ischiropoulos H (1998) Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Proc Natl Acad Sci U S A 95:7659–7663
Athauda D, Foltynie T (2015) The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat Rev Neurol 11:25–40
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313
Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR (2015) Oxidative stress and Parkinson’s disease. Front Neuroanat 9:91
Block ML, Hong JS (2007) Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem Soc Trans 35:1127–1132
Bordt EA, Polster BM (2014) NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: a bipartisan affair? Free Radic Biol Med 76:34–46
Chen S, Le W (2006) Neuroprotective therapy in Parkinson disease. Am J Ther 13:445–457
Choi DH, Cristovao AC, Guhathakurta S, Lee J, Joh TH, Beal MF, Kim YS (2012) NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson’s disease. Antioxid Redox Signal 16:1033–1045
Cooney SJ, Bermudez-Sabogal SL, Byrnes KR (2013) Cellular and temporal expression of NADPH oxidase (NOX) isotypes after brain injury. J Neuroinflammation 10:155
Cristovao AC, Choi DH, Baltazar G, Beal MF, Kim YS (2009) The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxid Redox Signal 11:2105–2118
Cristovao AC, Guhathakurta S, Bok E, Je G, Yoo SD, Choi DH, Kim YS (2012) NADPH oxidase 1 mediates alpha-synucleinopathy in Parkinson’s disease. J Neurosci 32:14465–14477
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39:889–909
Dawson TM, Dawson VL (2003) Molecular pathways of neurodegeneration in Parkinson’s disease. Science (New York. NY 302:819–822
Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB (2000) Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J Neurochem 74:2213–2216
Dikalov S (2011) Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med 51:1289–1301
Dranka BP, Gifford A, Ghosh A, Zielonka J, Joseph J, Kanthasamy AG, Kalyanaraman B (2013) Diapocynin prevents early Parkinson’s disease symptoms in the leucine-rich repeat kinase 2 (LRRK2R(1)(4)(4)(1)G) transgenic mouse. Neurosci Lett 549:57–62
Dranka BP, Gifford A, McAllister D, Zielonka J, Joseph J, O’Hara CL, Stucky CL, Kanthasamy AG, Kalyanaraman B (2014) A novel mitochondrially-targeted apocynin derivative prevents hyposmia and loss of motor function in the leucine-rich repeat kinase 2 (LRRK2(R1441G)) transgenic mouse model of Parkinson’s disease. Neurosci Lett 583:159–164
Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, Mahad D, Bradl M, van Horssen J, Lassmann H (2012) NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain J Neurol 135:886–899
Fujita K, Seike T, Yutsudo N, Ohno M, Yamada H, Yamaguchi H, Sakumi K, Yamakawa Y, Kido MA, Takaki A, Katafuchi T, Tanaka Y, Nakabeppu Y, Noda M (2009) Hydrogen in drinking water reduces dopaminergic neuronal loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. PLoS One 4:e7247
Galea E, Feinstein DL, Reis DJ (1992) Induction of calcium-independent nitric oxide synthase activity in primary rat glial cultures. Proc Natl Acad Sci U S A 89:10945–10949
Gao HM, Liu B, Hong JS (2003a) Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 23:6181–6187
Gao HM, Liu B, Zhang W, Hong JS (2003b) Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease. FASEB J 17:1954–1956
Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, Ghosh S, Mosley RL, Gendelman HE, Pahan K (2007) Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A 104:18754–18759
Ghosh A, Roy A, Matras J, Brahmachari S, Gendelman HE, Pahan K (2009) Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci 29:13543–13556
Ghosh A, Chandran K, Kalivendi SV, Joseph J, Antholine WE, Hillard CJ, Kanthasamy A, Kanthasamy A, Kalyanaraman B (2010) Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic Biol Med 49:1674–1684
Ghosh A, Kanthasamy A, Joseph J, Anantharam V, Srivastava P, Dranka BP, Kalyanaraman B, Kanthasamy AG (2012) Anti-inflammatory and neuroprotective effects of an orally active apocynin derivative in pre-clinical models of Parkinson’s disease. J Neuroinflammation 9:241
Ghosh A, Saminathan H, Kanthasamy A, Anantharam V, Jin H, Sondarva G, Harischandra DS, Qian Z, Rana A, Kanthasamy AG (2013) The peptidyl-prolyl isomerase Pin1 up-regulation and proapoptotic function in dopaminergic neurons: relevance to the pathogenesis of Parkinson disease. J Biol chem 288:21955–21971
Goldman SM (2014) Environmental toxins and Parkinson’s disease. Annu Rev Pharmacol Toxicol 54:141–164
Gordon R, Hogan CE, Neal ML, Anantharam V, Kanthasamy AG, Kanthasamy A (2011) A simple magnetic separation method for high-yield isolation of pure primary microglia. J Neurosci Methods 194:287–296
Grunblatt E, Mandel S, Youdim MB (2000) Neuroprotective strategies in Parkinson’s disease using the models of 6-hydroxydopamine and MPTP. Ann N Y Acad Sci 899:262–273
Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schoneich C, Engelhardt JF (2008) SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest 118:659–670
Hayashi T, Juliet PA, Kano-Hayashi H, Tsunekawa T, Dingqunfang D, Sumi D, Matsui-Hirai H, Fukatsu A, Iguchi A (2005) NADPH oxidase inhibitor, apocynin, restores the impaired endothelial-dependent and -independent responses and scavenges superoxide anion in rats with type 2 diabetes complicated by NO dysfunction. Diabetes, Obes metab 7:334–343
Hayden MS, Ghosh S (2004) Signaling to NF-kappaB. Genes Dev 18:2195–2224
Hernandes MS, Santos GD, Cafe-Mendes CC, Lima LS, Scavone C, Munhoz CD, Britto LR (2013) Microglial cells are involved in the susceptibility of NADPH oxidase knockout mice to 6-hydroxy-dopamine-induced neurodegeneration. PLoS One 8:e75532
Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 8:382–397
Hirsch EC, Vyas S, Hunot S (2012) Neuroinflammation in Parkinson’s disease. Parkinsonism & related disorders 18. Supplement 1:S210–S212
Hunot S, Boissiere F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y, Hirsch EC (1996) Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 72:355–363
Hunter RL, Dragicevic N, Seifert K, Choi DY, Liu M, Kim H-C, Cass WA, Sullivan PG, Bing G (2007) Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem 100:1375–1386
Impellizzeri D, Esposito E, Mazzon E, Paterniti I, Di Paola R, Bramanti P, Cuzzocrea S (2011) Effect of apocynin, a NADPH oxidase inhibitor, on acute lung inflammation. Biochem Pharmacol 81:636–648
Jackson-Lewis V, Przedborski S (2007) Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc 2:141–151
Jin H, Kanthasamy A, Ghosh A, Yang Y, Anantharam V, Kanthasamy AG (2011) Alpha-synuclein negatively regulates protein kinase cdelta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J Neurosci 31:2035–2051
Jin H, Kanthasamy A, Ghosh A, Anantharam V, Kalyanaraman B, Kanthasamy AG (2014) Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: preclinical and clinical outcomes. Biochim Biophys Acta 1842:1282–1294
Kavanagh E, Rodhe J, Burguillos MA, Venero JL, Joseph B (2014) Regulation of caspase-3 processing by cIAP2 controls the switch between pro-inflammatory activation and cell death in microglia. Cell Death dis 5:e1565
Klingelhoefer L, Reichmann H (2015) Pathogenesis of Parkinson disease[mdash]the gut-brain axis and environmental factors. Nat Rev Neurol 11:625–636
Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S (1999) Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med 5:1403–1409
Lim HW, In PJ, More SV, Park JY, Kim BW, Jeon SB, Yosep Y, Park EJ, Yoon SH, Choi DK (2015) Anti-neuroinflammatory effects of DPTP, a novel synthetic clovamide derivative in in vitro and in vivo model of neuroinflammation. Brain Res Bull 112:25–34
Lofrumento DD, Saponaro C, Cianciulli A, De Nuccio F, Mitolo V, Nicolardi G, Panaro MA (2011) MPTP-induced neuroinflammation increases the expression of pro-inflammatory cytokines and their receptors in mouse brain. Neuroimmunomodulation 18:79–88
Luchtefeld R, Luo R, Stine K, Alt ML, Chernovitz PA, Smith RE (2008) Dose formulation and analysis of diapocynin. J Agric Food Chem 56:301–306
Maghzal GJ, Krause KH, Stocker R, Jaquet V (2012) Detection of reactive oxygen species derived from the family of NOX NADPH oxidases. Free Radic Biol Med 53:1903–1918
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1291
Mead EL, Mosley A, Eaton S, Dobson L, Heales SJ, Pocock JM (2012) Microglial neurotransmitter receptors trigger superoxide production in microglia; consequences for microglial-neuronal interactions. J Neurochem 121:287–301
Meissner W, Hill MP, Tison F, Gross CE, Bezard E (2004) Neuroprotective strategies for Parkinson’s disease: conceptual limits of animal models and clinical trials. Trends Pharmacol Sci 25:249–253
Mosley RL, Benner EJ, Kadiu I, Thomas M, Boska MD, Hasan K, Laurie C, Gendelman HE (2006) Neuroinflammation, oxidative stress and the pathogenesis of Parkinson’s disease. Clin Neurosci Res 6:261–281
Mullin S, Schapira AHV (2015) Pathogenic mechanisms of neurodegeneration in Parkinson disease. Neurol Clin 33:1–17
Murakami M, Hirano T (2012) The molecular mechanisms of chronic inflammation development. Front Immunol 3:323
Murphy MP (1997) Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol 15:326–330
Nagatsu T, Mogi M, Ichinose H, Togari A (2000) Cytokines in Parkinson’s disease. J Neural Transm 143-151
Ngwa HA, Kanthasamy A, Jin H, Anantharam V, Kanthasamy AG (2014) Vanadium exposure induces olfactory dysfunction in an animal model of metal neurotoxicity. Neurotoxicology 43:73–81
Panicker N, Saminathan H, Jin H, Neal M, Harischandra DS, Gordon R, Kanthasamy K, Lawana V, Sarkar S, Luo J, Anantharam V, Kanthasamy AG, Kanthasamy A (2015) Fyn kinase regulates microglial neuroinflammatory responses in cell culture and animal models of Parkinson’s disease. J Neurosci 35:10058–10077
Picklo MJ, Amarnath V, McIntyre JO, Graham DG, Montine TJ (1999) 4-hydroxy-2(E)-nonenal inhibits CNS mitochondrial respiration at multiple sites. J Neurochem 72:1617–1624
Prediger RD, Aguiar Jr AS, Moreira EL, Matheus FC, Castro AA, Walz R, De Bem AF, Latini A, Tasca CI, Farina M, Raisman-Vozari R (2011) The intranasal administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a new rodent model to test palliative and neuroprotective agents for Parkinson’s disease. Curr Pharm Des 17:489–507
Przedborski S (2007) Neuroinflammation and Parkinson’s disease. Handb Clin neurol 83:535–551
Przedborski S, Tieu K, Perier C, Vila M (2004) MPTP as a mitochondrial neurotoxic model of Parkinson’s disease. J Bioenerg Biomembr 36:375–379
Rocha N, de Miranda AS, Teixeira AL (2015) Insights into Neuroinflammation in Parkinson's Disease: From Biomarkers to Anti-Inflammatory Based Therapies. BioMed Res Int 2015:12.
Sanders LH, McCoy J, Hu X, Mastroberardino PG, Dickinson BC, Chang CJ, Chu CT, Van Houten B, Greenamyre JT (2014) Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson’s disease. Neurobiol Dis 70:214–223
Schapira AH, Olanow CW, Greenamyre JT, Bezard E (2014) Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: future therapeutic perspectives. Lancet 384:545–555
Seidl SE, Potashkin JA (2011) The promise of neuroprotective agents in Parkinson’s disease. Front Neurol 2:68
Selley ML (1998) (E)-4-hydroxy-2-nonenal may be involved in the pathogenesis of Parkinson’s disease. Free Radic Biol Med 25:169–174
Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT (2003) Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci 23:10756–10764
Smith RA, Porteous CM, Coulter CV, Murphy MP (1999) Selective targeting of an antioxidant to mitochondria. Eur J Biochem/FEBS 263:709–716
Sovari AA, Morita N, Karagueuzian HS (2008) Apocynin: a potent NADPH oxidase inhibitor for the management of atrial fibrillation. Redox Report: commun Free Radic Res 13:242–245
Stadler K, Bonini MG, Dallas S, Jiang J, Radi R, Mason RP, Kadiiska MB (2008) Involvement of inducible nitric oxide synthase in hydroxyl radical-mediated lipid peroxidation in streptozotocin-induced diabetes. Free Radic Biol Med 45:866–874
Strowig T, Henao-Mejia J, Elinav E, Flavell R (2012) Inflammasomes in health and disease. Nature 481:278–286
Tansey MG, Goldberg MS (2010) Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis 37:510–518
Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, Vila M, Jackson-Lewis V, Przedborski S (2003) Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc Natl Acad Sci U S A 100:5473–5478
Trumbull KA, McAllister D, Gandelman MM, Fung WY, Lew T, Brennan L, Lopez N, Morre J, Kalyanaraman B, Beckman JS (2012) Diapocynin and apocynin administration fails to significantly extend survival in G93A SOD1 ALS mice. Neurobiol Dis 45:137–144
Vejrazka M, Micek R, Stipek S (2005) Apocynin inhibits NADPH oxidase in phagocytes but stimulates ROS production in non-phagocytic cells. Biochim Biophys Acta 1722:143–147
Vila M, Przedborski S (2004) Genetic clues to the pathogenesis of Parkinson’s disease. Nat Med 10(Suppl):S58–S62
Wang WF, Wu SL, Liou YM, Wang AL, Pawlak CR, Ho YJ (2009) MPTP lesion causes neuroinflammation and deficits in object recognition in wistar rats. Behav Neurosci 123:1261–1270
Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S (2003) NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc Natl Acad Sci U S A 100:6145–6150
Wu Y, Le W, Jankovic J (2011) Preclinical biomarkers of Parkinson disease. Arch Neurol 68:22–30
Yan J, Fu Q, Cheng L, Zhai M, Wu W, Huang L, Du G (2014) Inflammatory response in Parkinson’s disease (review). Mol Med rep 10:2223–2233
Ybot-Gorrin I, Vivancos-Matellano F, Chacon-Pena JR, Alonso-Navarro H, Jimenez-Jimenez FJ (2011) Assessment of Parkinson disease: what do we need to show neuroprotection? Neurologist 17:S21–S29
Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y (1996) Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A 93:2696–2701
Zhang D, Anantharam V, Kanthasamy A, Kanthasamy AG (2007) Neuroprotective effect of protein kinase C delta inhibitor rottlerin in cell culture and animal models of Parkinson’s disease. J Pharmacol Exp Ther 322:913–922
Zhang FL, He Y, Zheng Y, Zhang WJ, Wang Q, Jia YJ, Song HL, An HT, Zhang HB, Qian YJ, Tong YL, Dong L, Wang XM (2014) Therapeutic effects of fucoidan in 6-hydroxydopamine-lesioned rat model of Parkinson’s disease: role of NADPH oxidase-1. CNS Neurosci Ther 20:1036–1044
Acknowledgments
This study was funded by NIH grant R01 NS039958 (to B.K. and A.K.), U.S. Army Medical Research and Materiel Command (grant no. W81XWH-11-1-0700) and by the Harry R. and Angeline E. Quadracci Chair Endowment (B.K.). The W. Eugene and Linda Lloyd Endowed Chair to A.G.K. is also acknowledged. A.G.K., V.A., and B.N. are grateful to the Iowa State Nanovaccine Initiative and B.N. acknowledges the Vlasta Klima Balloun Professorship. We thank Mr. Gary Zenitsky for his assistance in preparing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
A.G.K. and B.K. hold a patent entitled “Neuroprotective Compounds and Their Use” for the Mitoapocynin treatment of Parkinson’s disease. The other authors have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Ghosh, A., Langley, M.R., Harischandra, D.S. et al. Mitoapocynin Treatment Protects Against Neuroinflammation and Dopaminergic Neurodegeneration in a Preclinical Animal Model of Parkinson’s Disease. J Neuroimmune Pharmacol 11, 259–278 (2016). https://doi.org/10.1007/s11481-016-9650-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-016-9650-4