
Abstract
The capacity for human monocytes to differentiate into antigen-presenting dendritic cells (DC) can be influenced by a number of immune modulating signals. Monocytes express intracellular cannabinoid type 1 (CB1) and 2 (CB2) receptors and we demonstrate that exposure to Δ9-tetrahydrocannabinol (THC) inhibits the forskolin-induced generation of cyclic adenosine monophosphate in a CB2-specific manner. In order to examine the potential impact of cannabinoids on the generation of monocyte-derived DC, monocytes were cultured in vitro with differentiation medium alone [containing granulocyte/macrophage-colony stimulating factor (GM-CSF) and Interleukin-4 (IL-4)] or in combination with THC. The presence of THC (0.25–1.0 μg/ml) altered key features of DC differentiation, producing a concentration-dependent decrease in surface expression of CD11c, HLA-DR and costimulatory molecules (CD40 and CD86), less effective antigen uptake, and signs of functional skewing with decreased production of IL-12 but normal levels of IL-10. When examined in a mixed leukocyte reaction, DC that had been generated in the presence of THC were poor T cell activators as evidenced by their inability to generate effector/memory T cells or to stimulate robust IFN-γ responses. Some of these effects were partially restored by exposure to exogenous IL-7 and bacterial superantigen (S. aureus Cowans strain). These studies demonstrate that human monocytes express functional cannabinoid receptors and suggest that exposure to THC can alter their differentiation into functional antigen presenting cells; an effect that may be counter-balanced by the presence of other immunoregulatory factors. The impact of cannabinoids on adaptive immune responses in individuals with frequent drug exposure remains to be determined.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The expression of cannabinoid receptors by human leukocytes suggests that both endogenous ligands and inhaled marijuana smoke might exert immunoregulatory properties that are distinct from their effects on the brain (Klein and Cabral 2006; Klein et al. 2005). Furthermore, while brain cells exclusively express cannabinoid receptor type 1 (CB1), leukocytes express both CB1 and CB2, with CB2 reported as the predominant subtype (Bouaboula et al. 1993, Munro et al. 1993; Nong et al. 2002). Both CB1 and CB2 are transmembrane G-protein coupled receptors that inhibit the generation of cyclic adenosine monophosphate (cAMP) and can signal through a variety of pathways including PI3-kinase, MAP kinase, NF-κΒ, AP-1, and NF-AT (Basu and Dittel 2011; Bosier et al. 2010). The resulting effects on host immunity have primarily been studied in animal models and suggest a coordinated down-regulation of cellular responses that can occur through altered trafficking, selective apoptosis, or functional skewing of antigen presenting cells and T cells away from T helper type 1 (Th1) or Th17 response patterns (Klein et al. 2000; Zhu et al. 2000; Nagarkatti et al. 2009; Rieder et al. 2010; Karmaus et al. 2013; Kong et al. 2014). Similar results have been observed when purified human T cells are stimulated in vitro in the presence of Δ9-tetrahydrocannabinol (THC) (Yuan et al. 2002). However, the extent to which the effects are observed in humans in vivo is unclear. Daily administration of marijuana or oral THC to research subjects in a prospective and randomized study had no obvious effect on T cell proliferation or cytokine production when blood cells were subsequently isolated and stimulated in vitro (Bredt et al. 2002). Sipe et al. (2005) examined the distribution and function of a common polymorphism in the human CB2 gene associated with the replacement of a glutamine by an arginine at amino acid position 63. Functionally, lymphocytes from subjects with either of these genotypes proliferated normally when stimulated with anti-CD3 antibody. However, when stimulated in the presence of an endocannabinoid, lymphocytes expressing the glutamine residue at position 63 were markedly inhibited while those expressing the arginine were only modestly suppressed. The arginine substitution also correlated with the prevalence of autoimmune disease in the subjects tested. Collectively, this body of work suggests that cannabinoids are biologically active immune regulators in humans.
Expanding upon this hypothesis, we examined the expression of cannabinoid receptors by human monocytes and the impact of THC on their differentiation into monocyte-derived dendritic cells (DC). Exposing monocytes to THC blocked many of the features normally associated with their differentiation into functional DC and impaired their capacity for T cell activation. Furthermore, the T cell activation that did occur was associated with a change in T cell phenotype and cytokine secretion. However, the impact of THC was partially overcome when DC and T cells were exposed to a combination of activation signals and exogenous cytokines. Our findings suggest that cannabinoids are capable of altering the differentiation and activation of cells involved in human cell-mediated immunity.
Materials and Methods
Primary Cells and Cell Lines
Human peripheral blood was obtained from healthy volunteers according to a protocol approved by the UCLA Institutional Review Board. Mononuclear cells (PBMC) were isolated by ficoll density gradient centrifugation. Chinese hamster ovary (CHO) cells were transfected with a plasmid encoding for human CB2 and transfectants (CHO-CB2) selected by growth in Kaighn’s F-12 medium containing 0.2 mg/ml G418 (Calbiochem, San Diego, CA).
Reagents and Antibodies
THC and SR144528 (selective CB2 antagonist) were provided by the National Institute on Drug Abuse (NIDA, Bethesda, MD). JWH-015 (selective CB2 agonist) was obtained from Enzo Life Sciences (Farmingdale, NY). All cannabinoids were solubilized in ethanol and diluted serially in DMSO and then culture medium prior to use (final ethanol concentration ≤ 0.01% and DMSO ≤ 0.125%). Interleukin (IL)-10, and IL-12 ELISA kits, fluorescent-labeled monoclonal antibodies (mAbs) directed against CD11c, CD13, CD14, CD40, CD45RA, CD86, and HLA-DR, and mAbs used for cell depletion and purification were all from BD-Biosciences (San Jose, CA). Monoclonal anti-CB2 and fluorescent-labeled anti-CB1 antibodies and recombinant human IL-4, IL-7, IL-12 and IL-15 were from R&D Systems (Minneapolis, MN). APC-labeled goat anti-mouse F(ab’)2 mAb, fluorescent-labeled anti-CD25 antibody and fluorescein-labeled dextran (FITC-dextran, MW 40,000) were purchased from Invitrogen (Carlsbad, CA). Granulocyte/macrophage-colony stimulating factor (GM-CSF) was obtained from Berlex Laboratories, Inc. (Richmond, CA).
Preparation of Monocytes, DC and T cells
Human monocytes were prepared from PBMC by immunomagnetic depletion (Miltenyi Biotec, Auburn, CA) and T cells were purified using a combination of mAb (anti-CD14, anti-CD16, anti-CD19) and anti-mouse Ig-conjugated immunomagnetic beads (Dynal, Lake Success, NY). DC were differentiated from monocyte precursors by culturing adherent PBMC in X-VIVO 15 medium (Lonza; Walkersville, MD) supplemented with GM-CSF (800 U/ml) and IL-4 (100 to 500 U/ml) according to a standard protocol (Kiertscher and Roth 1996). The effects of THC on this differentiation process were assessed by adding THC (0.25 to 1.0 μg/ml) or diluent alone (containing ethanol/DMSO) 10 min before the addition of GM-CSF and IL-4. Dendritic cells were recovered from the flasks on day 7 and the expression of cell surface markers characterized by fluorescence-activated cell sorting (FACS) using a FACS Calibur® cytometer with CellQuest® analysis software (Becton Dickinson, San Jose, CA). For mixed leukocyte reactions (MLR) and cytokine assays, DC were further purified by depleting T cells, natural killer cells and B cells using lineage-specific mAb (anti-CD3, anti-CD19, anti-CD56) and immunomagnetic beads (Dynal).
Analysis of CB1 and CB2 Expression
For CB1 and CB2 mRNA expression, total cellular RNA was isolated using Rneasy mini-kits (Qiagen, Valencia, CA) and RT-PCR was carried out as detailed below or through a commercial vendor employing a quantitative RT2 Profiler™ PCR Array (Qiagen). Total RNA was reverse-transcribed using the cDNA Cycle® kit from Invitrogen (Carlsbad, CA) and 2 μl of the reverse transcription (RT) product used in a 20 μl RT-PCR reaction containing 0.4 mM dNTM mix, 2 mM MgCl2, 2.5 U of Taq DNA polymerase, PCR buffers and 0.5 μM each of the forward and reverse primers for either CB1 (5’-caccttccgcaccatcaccac-3’; 5’-gtctcccgcagtcatcttctcttg-3’), CB2 (5’-catggaggaatgctgggtgac-3’; 5’-gaggaaggcgatgaacaggag-3’) or β-actin (5’-tgatggtgggcatgggtcag-3’; 5’-gtgttggcgtacaggtcttt-3’), all from Invitrogen. RT-PCR cycling conditions for CB1 and CB2 included an initial 5 min denaturation @ 94 °C followed by 35 cycles consisting of 45 s @ 94 °C, 45 s @ 64 °C, and 1 min @ 72 °C, with a final extension for 7 min @72 °C. Cycling conditions for β-actin were similar except for the use of only 30 cycles and an annealing temperature of 62 °C. RT-PCR products were resolved on 2% agarose gels and imaged with a UV transilluminator and a Polaroid photodocumentation camera. Expression of β-actin was used to control for loading and signal intensities measured by densitometry using NIH Imager software (NIH, Bethesda, MD). Using this approach, serial two-fold dilutions of total RT product from CHO-CB2 cells demonstrated a linear relationship between dilution factor and signal intensity over an 8-fold range.
Cell surface and intracellular expression of CB1 and CB2 receptors were determined by FACS analysis as previously described (Castaneda et al. 2013). Briefly, cell surface CB2 was detected with unlabeled mouse mAb directed against human CB2, followed by APC-labeled goat anti-mouse F(ab’)2. Isotype-matched mAb against an irrelevant antigen (mouse NK1.1) was used as a control. Cell surface CB1 was measured by anti-CB1-PE, with anti-mouse NK1.1-PE serving as a negative isotype control. For the detection of intracellular CB1 and CB2 receptor, cells were fixed with 1 % paraformaldehyde/PBS (Sigma Aldrich, St. Louis, MO) and treated with permeabilizing solution (BD Biosciences) prior to staining with mAb.
Forskolin-Induced cAMP Assay
Functional coupling of cannabinoid receptors to G-protein activity was assessed by measuring forskolin-induced cAMP levels in CHO-CB2 cells and fresh human monocytes. CHO-CB2 cells were cultured overnight at 5 × 105 cells/well in a 6-well plate. The next day, DMSO was added (50 μM) and cells cultured for an additional 18 h. THC (0.5 μg/ml), JWH-015 (0.025 μM), the combination of SR144528 (1 μM) and THC (0.5 μg/ml), or diluent alone, were then added to the wells and incubated for 15 min prior to an 18 h stimulation with 50 μM forskolin (Sigma-Aldrich). For studies with monocytes, 5 × 106 PBMC were placed into each well of a 24-well plate and monocytes allowed to adhere for 2 h in X-VIVO-15 medium. Non-adherent cells were then removed, wells rinsed, and the remaining monolayer treated in a manner identical to that described for CHO-CB2 cells. For both cell types, supernatants were harvested at the end of the forskolin stimulation and stored at −80 °C until assayed in duplicate for cAMP using a standard competitive enzyme immunoassay kit (Cayman Chemicals; Ann Arbor, MI). The final reaction product was read in a plate reader at 405 nm (Spectra/SLT Lab Instruments; Salzburg, Austria) and the amount of cAMP determined by regression analysis.
DC Endocytosis and Cytokine-Secretion Assays
DC that had been generated in the presence or absence of THC (0.5 μg/ml) for 7 days were cultured with FITC-dextran (1 mg/ml) for 60 min at either 4 °C or 37 °C and the assay terminated by adding 3 ml of ice-cold PBS containing 0.1% azide. Cell pellets were extensively washed and the DC immediately analyzed for accumulation of intracellular fluorescence by FACS. The degree of endocytosis was determined by comparing the intracellular uptake at 37 °C to that at 4 °C (i.e., difference in mean fluorescence intensity). In order to evaluate their capacity for cytokine production, control and THC-treated DC were also cultured for an additional 48 h at 0.5 × 106 cells/ml in X-VIVO 15 medium supplemented with GM-CSF (800 U/ml), IL-4 (500 U/ml) and 20 μg/ml of heat-killed, formalin-fixed Staphylococcus aureus Cowan (SAC, Calbiochem) as a cytokine-inducing agent. Supernatants were harvested and replicate samples assayed for the concentration of IL-10 and IL-12 by cytokine-specific ELISA. Results from duplicate wells were analyzed using a microplate reader and automated regression software (Spectra/SLT).
MLR and Cytokine Assays
DC and THC-DC were evaluated for their capacity to activate T cells in a standard MLR assay (Kiertscher and Roth 1996). Allogeneic CD45RA+ T cells were isolated by negative selection with specific antibody (anti-CD14, anti-CD16, anti-CD19, anti-CD45RO) and immunomagnetic beads, then labeled using the Vybrant CDSE/CFSE Cell Tracer Kit (Invitrogen-Molecular Probes, Eugene, OR) according to the manufacturer’s protocol. DC were cultured with 2 × 105 T cells at 1:50 DC:T cell ratios in X-VIVO 15 medium in 96 well round-bottom plates at 37 °C in a humidified CO2 incubator. For some experiments, DC and THC-DC were matured by culture with 20 μg/ml SAC for 18–24 h prior to co-culture with the T cells. In other experiments, the co-cultures were supplemented with 2 ng/ml of either IL-7, IL-12 or IL-15. On day 5 of co-culture, the T cells were collected and analyzed by FACS for proliferation (by CFSE dilution) and cell surface marker expression (by addition of marker-specific fluorescent antibodies). Cell-free supernatants were collected from the wells and assessed for cytokines by custom multiplex analysis (Aushon BioSystems, Billerica, MA). Each cytokine was measured in duplicate and represented as the average value ± SD.
Statistical Analysis
Data from individual experiments are represented as the mean ± SD for the indicated number of replicates. Pooled data from multiple experiments are represented as mean values or as a percentage of control, ± SE. Comparisons involving multiple groups were assessed by one-way ANOVA for the presence of an overall treatment effect at a level of p < 0.05. If an overall difference was identified, then a post-hoc analysis was performed to compare individual groups of interest using either a paired or unpaired t-test as appropriate for the relationship between experimental groups. Statistically significant differences were determined by the presence of p ≤ 0.05 for single comparisons or with a Bonferroni correction for multiple comparisons.
Results
Human Monocytes Express Functional Cannabinoid Receptors
As an initial step in understanding the potential interaction between cannabinoids and human monocyte-derived DC, monocytes were evaluated for the expression of the CB1 and CB2 receptor subtypes by RT-PCR (Fig. 1a) and flow cytometry (Fig. 1b). RT-PCR studies were carried out on monocytes that had been purified to >90% purity by either negative depletion or fluorescent cell sorting. mRNA encoding for both CB1 and CB2 were detected, although expression of CB2 predominated whether analyzed by standard RT-PCR (Fig. 1b, representative experiment) or by an automated quantitative RT-PCR using cells from 4 different donors (average CB2:CB1 ratio = 4.0; range =0.15 to 10.34).

Monocytes express high levels of CB2 mRNA and intracellular CB1 and CB2 protein. a: Peripheral blood monocytes were purified from PBMC by negative depletion using immunomagentic beads and the relative expression of CB1 and CB2 mRNA determined by semi-quantitative RT-PCR using densitometry and normalized for the expression of β-actin. Representative results from one of two determinations. b: mAbs specific for the N-terminal domains of CB1 and CB2 were used to identify expression of the corresponding proteins on the cell surface or at intracellular sites following permeabilization. Anti-CB1 was a fluorochrome-conjugated mAb while anti-CB2 was an unlabeled mAb detected by secondary stain with fluorochrome-conjugated goat-anti-mouse IgG antibody. For the intracellular analysis, cells were fixed with paraformaldehyde and permeabilized prior to mAb staining. Results are gated for CD13+ or CD14+ peripheral blood monocytes by flow cytometry and expressed as histograms with relative fluorescence intensity (RFI) on the X-axis. Black line (no fill) = unstained cells; Gray line (no fill) = isotype control mAb for CB1 (top panels) or isotype control mAb with secondary detection by GAM for CB2 (bottom panels); Black solid fill = staining by anti-CB1 (top panels) or anti-CB2 mAb with secondary detection by GAM (bottom panels). Representative results from one of 4 determinations
Despite the presence of mRNA, standard flow cytometry failed to detect CB1 or CB2 receptor protein on the cell surface of monocytes even though antibodies were directed against their N-terminal epitopes. However, when cells were fixed and permeabilized, specific staining for both CB1 and CB2 was detected, consistent with the presence of intracellular protein (Fig. 1b). Intracellular background staining with isotype control mAb was minimal for CB1 but dimly-positive for CB2, likely reflecting the need for APC-labeled goat anti-mouse F(ab’)2 as a secondary detection reagent. Due to these differences in fluorescent labels and staining protocols, the relative fluorescent intensity for CB1 and CB2 cannot be directly compared as measures of receptor concentration.
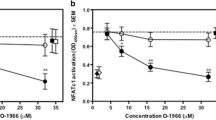
The presence of functional CB2 receptor complexes was then assessed by measuring the impact of different cannabinoids on forskolin-induced generation of cAMP (Fig. 2a). Using CHO-CB2 cells as a model, we confirmed that treatment with THC (0.5 μg/ml = 1.59 μM) significantly inhibited the generation of cAMP, as did JWH-015 (0.025 μM; selective CB2 agonist) at p < 0.01. Furthermore, the inhibition of cAMP by THC was blocked by pretreatment with SR144528, a selective CB2 receptor antagonist (p < 0.01). The same assays were repeated using purified human monocytes (Fig. 2b). Again, an overall CB2 agonist treatment effect was present. Pretreatment with either THC or JWH-015 inhibited the forskolin-induced generation of cAMP (68.0 + 4.2% and 58.3 + 5.7% of control levels, respectively) and the effects of THC were blocked by SR144528 (p < 0.01 for all comparisons). While monocytes express both CB1 and CB2, the predominance of CB2 mRNA and the response of these cells to CB2-selective agents suggest that CB2 acts as the dominant cannabinoid signaling pathway.

CB2 receptors on monocytes are coupled to G i proteins and activated by THC. CHO cells expressing human CB2 (CHO-CB2) (a) and adherent human monocytes (b) were pre-treated for 15 min with either diluent alone (control), THC (0.5 μg/ml), JWH-015 (0.025 μM), or the combination of SR144528 (1 μM) and THC (0.5 μg/ml), followed by an 18 h stimulation with forskolin (50 μM). Accumulation of cAMP in the culture supernatant was determined by a competitive enzyme immunoassay and presented as mean values ± SD of replicate measurements. JWH-015 is a selective CB2 agonist and SR144528 a selective CB2 antagonist. *p <0.01 compared to control. † p < 0.01 compared to THC treatment alone. Representative results from one of 4 experiments
Exposure to THC Alters the Phenotype of Monocyte-Derived DC
The differentiation of human monocytes into DC is associated with characteristic changes in cell surface proteins involved in antigen presentation (Kiertscher and Roth 1996). To evaluate the effects of THC on this aspect of differentiation, adherent PBMC were cultured for 7 days with GM-CSF and IL-4 and examined for the expression of typical monocyte and DC markers by flow cytometry (Fig. 3). Exposure to THC (0.25 to 1.0 μg/ml) did not prevent the normal down-regulation of CD14, but did inhibit the upregulation of other cell surface markers characteristic of antigen presenting cells including CD11c, HLA-DR, CD40 and CD86. The effects were concentration-dependent, with 0.5 μg/ml THC inhibiting expression of all of these markers by 40–60%. Interestingly, the response profiles were not uniform for every protein. THC produced a uniform decrease in the expression of CD11c and CD40 on all of the cells but resulted in two distinct subsets with respect to the expression of HLA-DR and CD86 – one population that did not express these markers and one that expressed relatively normal levels (Fig. 3). In the latter case, the relative proportions of these two subsets depended upon the concentration of THC, with higher levels of THC resulting in fewer marker-positive cells.

THC inhibited the expression of HLA-DR and other DC surface markers in a concentration-dependent manner. a: Adherent PBMC were cultured for 7 days with GM-CSF and IL-4 to induce their differentiation into DC in the presence of diluent alone or increasing concentrations of THC as indicated. Cell surface HLA-DR expression was measured by flow cytometry (percent of control mean fluorescence intensity). Representative experiment, n = 5. b-c: Adherent PBMC were cultured for 7 days with GM-CSF and IL-4 to induce their differentiation into DC in the presence of diluent alone (b) or THC at 0.5 μg/ml (c) added at the beginning of culture. Cell surface expression of CD14, CD11c, HLA-DR, CD86 and CD40 were measured by flow cytometry. Numbers represent mean fluorescent intensity for the entire population and the dashed line indicates the highest fluorescence intensity value for unstained cells. Data are representative from one of 9 experiments
Cannabinoids have been reported to promote the apoptosis of mouse bone marrow-derived DC under certain conditions (Do et al. 2004). In order to assure that the phenotypic changes observed in our studies were not the result of poor viability, DC that had been differentiated in the presence of either THC (0.25 to 1.0 μg/ml) or JWH-015 (0.25 to 0.75 μM) were stained with propidium iodide and Annexin-V-FITC. There was no significant impact of either cannabinoid on the number of recovered cells or on the frequency of apoptotic or dead cells (data not shown).
Dendritic Cells Generated in the Presence of THC are Functionally Altered
In addition to their high level expression of major histocompatibility complex and costimulatory molecules, monocyte-derived DC are usually characterized by their capacity for antigen uptake, as well as their secretion of cytokines that promote cell mediated immunity. Receptor-mediated endocytosis was measured by the uptake of FITC-dextran and was dramatically suppressed in cells that had been exposed to THC (Fig. 4a). The production of IL-10 and IL-12 was also assessed by stimulating cells with SAC and measuring cytokines released into the culture media at 48 h following stimulation. Interestingly, while the production of IL-12 was significantly suppressed (p < 0.01), the secretion of IL-10, which can bias T cell activation toward T helper type 2 (Th2) and/or T regulatory (Treg) phenotypes, was not altered (Fig. 4b). This differential effect on cytokine production is consistent with an immunoregulatory effect rather than a global suppression of DC function.

Exposure to THC impairs the capacity for receptor-mediated endocytosis and alters the relative production of IL-10 and IL-12 by activated DC. a: Purified DC that had been generated in the presence or absence of THC (0.5 μg/ml) for 7 days were cultured for 1 h with FITC-dextran (1 mg/ml) at either 4 °C or 37 °C and then analyzed by FACS for uptake. Numbers represent mean fluorescence intensity. b: Purified DC were cultured for an additional 48 h (0.5 × 106 cells/ml) with 20 μg/ml SAC as a cytokine-inducing agent. Release of IL-10 and IL-12 was measured by cytokine-specific ELISA and represented as mean values ± SD for replicate measurements. *p < 0.01 compared to control. N = 3 experiments
THC-DC are Poor T cell Stimulators and Fail to Induce Effector T cells
The capacity for DC to stimulate the activation and proliferation of antigen-specific T cells represents a final integrated measure of their function. DC that had been generated in the presence or absence of THC (THC-DC) were recovered from the 7 day culture of adherent PBMC, purified by negative depletion, and cultured with allogeneic T cells in a MLR assay (Fig. 5). In order to avoid direct effects on responder T cells, no further THC was added during the 5 days of DC:T cell co-culture. Proliferation was monitored by labeling cells with CFSE, which also allowed the phenotype of responder cells to be tracked with each cell division over time. While control DC were potent stimulators of both CD4+ and CD8+ responder T cells, the proliferative response to THC-DC was severely blunted (Fig. 5a). THC-treated DC induced T cell proliferation that ranged from only 17 to 32% of control values (p < 0.01, n = 6 experiments). Furthermore, while control DC promoted the conversion from naïve CD45RA+ T cells to activated CD45RO+ clones expressing the high affinity IL-2 receptor (CD25), this functional transformation into effector/memory T cells was almost completely absent when T cells were stimulated with THC-DC (Fig. 5b). There was a similar impact on the generation of effector cell cytokines as measured by the release of IFN-γ and TNF-α into the culture supernatant (Fig. 5c, p < 0.01).

Exposure to THC impairs the ability for DC to activate allogeneic T cells in a MLR assay. DC or THC-exposed DC were cultured with 2 × 105 CFSE-labeled allogeneic CD45RA+ T cells at a 1:50 DC:T cell ratio in a standard MLR assay. Following 5 days of culture, T cells were collected and stained with fluorescently-labeled monoclonal antibodies specific for CD8, CD45RA, and CD25. a: The percentage of T cells and T cell subsets that proliferated was determined by measuring CFSE dilution and cell surface marker expression (top panels). The percentage of T cells corresponding to each cell division number is displayed in the bottom panels. b: The proliferating cells were further analyzed for the development of an effector memory phenotype (upregulation of CD25, downregulation of CD45RA). The percentages indicate the percent of total T cells within the specified regions, with the numbers in parentheses indicating the MFI of those cells. c. The levels of IFN-γ and TNF-α present in the co-cultures were determined by multiplex analysis of the day 5 supernatants. *p < 0.01 comparing control DC and THC-DC. Representative results from one of 6 experiments (a-b), and one of two experiments (c)
DC Activators and Supplemental Cytokines Partially Restore the Function of THC-DC
A number of factors can help restore function to impaired antigen presenting cells or enhance their capacity to stimulate T cell responses. Given our findings with THC-DC, we hypothesized that a combination of DC activation and cytokine replacement might be effective for this purpose. In initial experiments, DC and THC-DC were exposed to heat-killed and fixed SAC for 18–24 h prior to co-culture with T cells. The goal was to replicate bacterial activation signals that might occur during an immune challenge in vivo. In other experiments, the co-cultures were supplemented with IL-7, IL-12 or IL-15 to replace key cytokines known to be involved in the proliferation and differentiation of effector/memory T cells. As demonstrated in Fig. 6, pre-treating control DC with SAC enhanced their capacity to stimulate T cell proliferation and maturation. In addition, exposing THC-DC to SAC restored some of their capacity to generate mature (CD45RO+/CD25+) responder T cells. This effect correlated with the upregulation of HLA-DR, CD80 and CD86 on THC-DC (data not shown). In addition, supplementing the co-cultures with IL-7 helped SAC-stimulated DC to further promote the expansion and phenotypic maturation of effector T cells, a synergistic effect that was not observed with either IL-12 or IL-15. When assessed in a limited number of experiments, IL-7 also increased the production of IFN-γ and TNF-α, consistent with a restoration of their effector/memory function (data not shown).

Addition of SAC or IL-7 can partially compensate for the diminished T cell stimulatory capacity of THC-exposed DC. DC or THC-exposed DC were cultured with 2 × 105 CFSE-labeled allogeneic CD45RA+ T cells at a 1:50 DC:T cell ratio as in Fig. 5. As indicated, some of the DC and THC-exposed DC were further matured by an 18–24 h exposure to 20 μg/ml SAC prior to co-culture with the T cells. Additionally, some of the DC-T cell co-cultures were supplemented with 2 ng/ml rhIL-7. The CFSE dilution and CD45RA staining profiles are shown for the various experimental groups, with the percentages indicating the percent of total T cells within the CFSE-diluted/CD45RA-dim regions. Representative results one of 6 experiments
Discussion
The human CB2 receptor was first cloned from a human myeloid cell line (Munro et al. 1993) and has been reported as the predominant cannabinoid receptor subtype expressed by immune cells (Galiègue et al. 1995). Consistent with this, we found that expression of CB2 mRNA predominated over CB1 when fresh human monocytes were purified and assayed by semi-quantitative RT-PCR techniques. However, neither cannabinoid receptor could be detected on the extracellular surface of monocytes when stained with mAbs known to be specific for their N-terminal sequences. We recently reported that CB2 may exist as an intracellular protein in immune cells (Castaneda et al. 2013) and others have suggested that CB1 may also function as an intracellular receptor (Brailoiu et al. 2011). Consistent with these observations, the addition of an initial fixation and permeabilization step resulted in positive staining by both anti-CB1 and anti-CB2 mAbs, but not by their respective isotype controls. Functional receptor protein was confirmed by assaying the capacity for cannabinoids to inhibit forskolin-induced changes in cAMP. Addition of THC, a pan-agonist with equal affinity for CB1 and CB2, blocked forskolin-induced cAMP in both transduced CHO-CB2 cells and in fresh human monocytes. In addition, this effect was recapitulated by exposure to JWH-015, a selective CB2 agonist, and the effects of THC were completely blocked by SR144528, a selective CB2 antagonist. These findings confirm reports that CB2 predominates as the functional cannabinoid receptor pathway in human monocytes and add the caveat that receptor expression occurs at an intracellular location rather than on the cell surface.
Monocytes act as myeloid precursors that can differentiate along a number of functionally-distinct pathways depending upon their interaction with cytokines, growth factors, infectious signals and other regulatory mediators (Sica and Mantovani 2012). When driven to differentiate into monocyte-derived DC under the influence of GM-CSF and IL-4 (Kiertscher and Roth 1996; Roth et al. 2000), their function can also be modulated by a variety of factors (Alonso et al. 2011). Concurrent exposure to IL-6 and macrophage-colony stimulating factor can divert differentiation toward macrophages instead of DC (Chomarat et al. 2000). Transforming growth factor (TGF)-β and IL-23 promote the development of DC that promote Th17 biased responses (Rajkovic et al. 2011). IL-10 promotes tolerogenic and Th2-promoting features (Steinbrink et al. 1997), while a variety of toll-like receptor ligands and immunostimulatory cytokines will promote DC that stimulate effector/memory T cells (Banchereau et al. 2000; Lanzavecchia and Sallusto 2000). In this setting, we hypothesized that exposure to THC during the process of DC differentiation would provide valuable insight regarding its immunoregulatory properties. Further, given the immunosuppressive effects that cannabinoids have on antigen-specific T cell responses in animals in vivo (Klein et al. 2000; Zhu et al. 2000) and on human T cell activation in vitro (Yuan et al. 2002; Sipe et al. 2005), we hypothesized that cannabinoids might render DC tolerogenic or otherwise skew their stimulatory activity.
Only a few studies have examined the interaction of cannabinoids with DC and in most cases the focus has been on murine models or on the effects of cannabinoids on differentiated DC (Do et al. 2004; Lu et al. 2006; Karmaus et al. 2013). Do et al. (2004) suggested that THC can impair immune responses by inducing DC apoptosis. However, they studied mouse bone marrow-derived DC and apoptosis occurred primarily when THC concentrations exceeded 5 μM. In our studies, immunoregulatory effects on human monocyte-derived DC were observed at lower THC concentrations (0.8–3.2 μM), more akin to peak levels that occur in the blood of marijuana smokers (Kosel et al. 2002), and had no effect on cell recovery or surface staining by Annexin-V. Instead of apoptosis, we observed broad-ranging effects of THC on the expression of MHC class II and costimulatory molecules, and the capacity for antigen uptake and IL-12 production. Furthermore, DC that had been exposed to THC during their in vitro differentiation (THC-DC) were impaired in their capacity to activate T cells – including both CD4+ and CD8+ responders. T cell proliferation and the acquisition of a memory/effector phenotype were both impaired, as was the release of Th1 cytokines. These effects of THC on the capacity for monocyte-derived DC to stimulate T cells are almost identical to the direct effects of THC on T cell activation (Yuan et al. 2002; Robinson et al. 2013), suggesting a coordinated immunoregulatory effect. It is interesting that other immunosuppressive factors, including IL-10 and TGF-β, share this capacity to act in a coordinated manner on both DC and T cells (Steinbrink et al. 1997; Rajkovic et al. 2011). As is the case with IL-10−/− knockout mice (Davidson et al. 1996), CB1−/−/CB2−/− double-knockout mice exhibit elevated levels of activated T cells and respond to antigen challenges by producing a higher number of activated effector cells and stronger IFN-γ responses (Karmaus et al. 2011). Collectively, these findings suggest an intrinsic role for endocannabinoid signaling as a homeostatic regulator of T cell activation.
There are a number of critical features that develop during the transition from monocytes into DC that enable them to activate antigen specific T cells (Banchereau et al. 2000; Lanzavecchia and Sallusto 2000). Among these are high levels of antigen expression in the context of cell-surface MHC, the upregulation of adhesion and costimulatory molecules, and the elaboration of immunostimulatory cytokines. Our studies suggest that cannabinoid receptor activation impacts on all of these. Exposure to THC during the differentiation of monocyte-derived DC impaired antigen uptake and prevented the normal upregulation of MHC class II. These findings are consistent with earlier reports by McCoy et al. (1999), where THC was found to impair the presentation of whole hen egg lysozyme, which required uptake and processing, but not the presentation of its immunodominant peptides, which bound directly to existing cell surface MHC. Dendritic cells that present antigen in the absence of adequate costimulatory molecules cannot fully activate T cells and may contribute to the development of T cell anergy (Banchereau et al. 2000; Lanzavecchia and Sallusto 2000). The inhibitory effects of THC on the expression of CD40, CD86 and other costimulatory molecules likely contributed to the failure of THC-DC to stimulate T cell proliferation. Finally, the relative production of IL-10 and IL-12 by DC plays a central role in their capacity to activate either Th1 (requiring IL-12) or Th2 (dependent upon IL-10) responses. In our studies, THC-DC produced only limited amounts of IL-12 but normal levels of IL-10. Lu et al. (2006b) reported a similar suppressive effect of THC on the expression of MHC and costimulatory molecules and on production of IL-12 by mouse bone marrow-derived DC that had been infected with Legionella pneumophila.
While these findings add to other compelling evidence that cannabinoids can exert important immunosuppressive effects, clinical evidence that marijuana smoking significantly impairs immune function in humans is limited. One explanation may be that inhaled THC never produces sufficient systemic levels, or that exposures may not be sustained for a sufficient period of time. to mediate immunosuppressive effects (Kosel et al. 2002; Desrosiers et al. 2014). Another explanation may be that the effects are short-lived or counterbalanced by the presence of other immune regulatory factors. The study of purified cells in vitro culture does not adequately replicate the complex environment that occurs during an immune challenge in vivo. In this study we hypothesized that the processes of DC activation and cytokine exposure that occur in response to an infectious challenge might modulate the impact of THC. Exposing DC and THC-DC to heat-killed and fixed SAC for 18–24 h enhanced their capacity for T cell activation; an effect that was more pronounced with THC-DC than with control DC. Adding IL-12 and IL-15 to the DC:T cell co-culture also enhanced T cell activation and proliferation, but these effects occurred equally with control and THC-DC. Furthermore, these cytokines promoted T cell proliferation and cytokine production even in the absence of stimulation by DC (data not shown). However, the addition of IL-7 to DC:T cell co-cultures had a dramatic effect on T cell proliferation, maturation and cytokine production that was restricted in part to co-cultures containing THC-DC. These studies suggest that the immunoregulatory effects of THC might be counterbalanced by the presence of a combination of DC activating signals and the production of cytokines by other cell types present in the local immune environment.
In summary, our experiments demonstrate that human monocytes express functional cannabinoid receptors, even if they are not detectable on the cell surface, and that exposure to THC alters their capacity to differentiate into immunostimulatory DC with prominent effects on antigen uptake and presentation, expression of costimulatory molecules, and production of IL-12. The end result is the generation of DC that fail to stimulate T cell proliferation or promote maturation into functional effector/memory T cells. While the effects are relatively potent when studied in isolation in vitro, there may be a number of immunoregulatory factors that could counteract or moderate the impact of cannabinoid exposure in vivo. The functional role that marijuana smoking has on host immunity and the response to immune challenges in vivo remains to be clarified.
References
Alonso MN, Wong MT, Zhang AL, Winer D, Suhoski MM, Tolentino LL, Gaitan J, Davidson MG, Kung TH, Galel DM, Nadeau KC, Kim J, Utz PJ, Söderström K, Engleman EG (2011) TH1, TH2, and TH17 cells instruct monocytes to differentiate into specialized dendritic cell subsets. Blood 118(12):3311–3320. doi:10.1182/blood-2011-03-341065
Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K (2000) Immunobiology of dendritic cells. Annu Rev Immunol 18:767–811. doi:10.1146/annurev.immunol.18.1.767
Basu S, Dittel BN (2011) Unraveling the complexities of cannabinoid receptor 2 (CB2) immune regulation in health and disease. Immunol Res 51(1):26–38. doi:10.1007/s12026-011-8210-5
Bosier B, Muccioli GG, Hermans E, Lambert DM (2010) Functionally selective cannabinoid receptor signalling: therapeutic implications and opportunities. Biochem Pharmacol 80(1):1–12. doi:10.1016/j.bcp.2010.02.013
Bouaboula M, Rinaldi M, Carayon P, Carillon C, Delpech B, Shire D, LeFur G, Casellas P (1993) Cannabinoid-receptor expression in human leukocytes. Eur J Biochem 214(1):173–180. doi:10.1111/j.1432-1033.1993.tb17910.x
Brailoiu GC, Oprea TI, Zhao P, Abood ME, Brailoiu E (2011) Intracellular cannabinoid type 1 (CB1) receptors are activated by anandamide. J Biol Chem 286(33):29166–29174. doi:10.1074/jbc.M110.217463
Bredt BM, Higuera-Alhino D, Shade SB, Hebert SJ, McCune JM, Abrams DI (2002) Short-term effects of cannabinoids on immune phenotype and function in HIV-l-infected patients. J Clin Pharmacol 42(11 Suppl):82S–89S. doi:10.1177/0091270002238798
Castaneda JT, Harui A, Kiertscher SM, Roth JD, Roth MD (2013) Differential expression of intracellular and extracellular CB(2) cannabinoid receptor protein by human peripheral blood leukocytes. J Neuroimmune Pharmacol 8(1):323–332. doi:10.1007/s11481-012-9430-8
Chomarat P, Banchereau J, Davoust J, Palucka AK (2000) IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol 1(6):510–514. doi:10.1038/82763
Davidson NJ, Leach MW, Fort MM, Thompson-Snipes L, Kühn R, Müller W, Berg DJ, Rennick DM (1996) T helper cell 1-type CD4+ T cells, but not B cells, mediate colitis in interleukin 10-deficient mice. J Exp Med 184(1):241–251
Desrosiers NA, Himes SK, Scheidweiler KB, Concheiro-Guisan M, Gorelick DA, Huestis MA (2014) Phase I and II cannabinoid disposition in blood and plasma of occasional and frequent smokers following controlled smoked cannabis. Clin Chem 60(4):631–643. doi:10.1373/clinchem.2013.216507
Do Y, McKallip RJ, Nagarkatti M, Nagarkatti PS (2004) Activation through cannabinoid receptors 1 and 2 on dendritic cells triggers NF-kappaB-dependent apoptosis: novel role for endogenous and exogenous cannabinoids in immunoregulation. J Immunol 173(4):2373–2382. doi:10.4049/jimmunol.173.4.2373
Galiègue S, Mary S, Marchand J, Dussossoy D, Carrière D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P (1995) Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem 232(1):54–61. doi:10.1111/j.1432-1033.1995.tb20780.x
Karmaus PW, Chen W, Crawford RB, Harkema JR, Kaplan BL, Kaminski NE (2011) Deletion of cannabinoid receptors 1 and 2 exacerbates APC function to increase inflammation and cellular immunity during influenza infection. J Leukoc Biol 90(5):983–995. doi:10.1189/jlb.0511219
Karmaus PWF, Chen W, Crawford R, Kaplan BLF, Kaminski NE (2013) Δ9-Tetrahydrocannabinol impairs the inflammatory response to influenza infection: role of antigen-presenting cells and the cannabinoid receptors 1 and 2. Toxicol Sci 131(2):419–433. doi:10.1093/toxsci/kfs315
Kiertscher SM, Roth MD (1996) Human CD14+ leukocytes acquire the phenotype and function of antigen-presenting dendritic cells when cultured in GM-CSF and IL-4. J Leukoc Biol 59(2):208–218
Klein TW (2005) Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol 5(5):400–411. doi:10.1038/nri1602
Klein TW, Cabral GA (2006) Cannabinoid-induced immune suppression and modulation of antigen-presenting cells. J Neuroimmune Pharmacol 1(1):50–64. doi:10.1007/s11481-005-9007-x
Klein TW, Newton CA, Nakachi N, Friedman H (2000) Delta9-tetrahydrocannabinol treatment suppresses immunity and early IFN-gamma, IL-12, and IL-12 receptor beta2 responses to Legionella pneumophila infection. J Immunol 164(12):6461–6466. doi:10.4049/jimmunol.164.12.6461
Kong W, Li H, Tuma RF, Ganea D (2014) Selective CB2 receptor activation ameliorates EAE by reducing Th17 differentiation and immune cell accumulation in the CNS. Cellular Immunol 287(1):1–17. doi:10.1016/j.cellimm.2013.11.002
Kosel BW, Aweeka FT, Benowitz NL, Shade SB, Hilton JF, Lizak PS, Abrams DI (2002) The effects of cannabinoids on the pharmacokinetics of indinavir and nelfinavir. AIDS 16(4):543–550
Lanzavecchia A, Sallusto F (2000) Dynamics of T lymphocyte responses:iIntermediates, effectors, and memory cells. Science 290(5489):92–97. doi:10.1126/science.290.5489.9
Lu T, Newton C, Perkins I, Friedman H, Klein TW (2006a) Cannabinoid treatment suppresses the T-helper cell-polarizing function of mouse dendritic cells stimulated with Legionella pneumophila infection. J Pharmacol Exp Ther 319(1):269–76. doi:10.1124/jpet.106.108381
Lu T, Newton C, Perkins I, Friedman H, Klein TW (2006b) Role of cannabinoid receptors in Delta-9-tetrahydrocannabinol suppression of IL-12p40 in mouse bone marrow-derived dendritic cells infected with Legionella pneumophila. Eur J Pharmacol 532(1–2):170–177. doi:10.1016/j.ejphar.2005.12.040
McCoy KL, Matveyeva M, Carlisle SJ, Cabral GA (1999) Cannabinoid inhibition of the processing of intact lysozyme by macrophages: evidence for CB2 receptor participation. J Pharmacol Exp Ther 289(3):1620–1625
Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365(6441):61–5. doi:10.1038/365061a0
Nagarkatti P, Pandey R, Rieder SA, Hegde VL, Nagarkatti M (2009) Cannabinoids as novel anti-inflammatory drugs. Future Med Chem 1(7):1333–1349. doi:10.4155/fmc.09.93
Nong L, Newton C, Cheng Q, Friedman H, Roth MD, Klein TW (2002) Altered cannabiniod receptor mRNA expression in peripheral blood mononuclear cells from marijuana smokers. J Neuroimmunol 127(1–2):169–176. doi:10.1016/S0165-5728(02)00113-3
Rajkovic I, Dragicevic A, Vasilijic S, Bozic B, Dzopalic T, Tomic S, Majstorovic I, Vucevic D, Djokic J, Balint B, Colic M (2011) Differences in T-helper polarizing capability between human monocyte-derived dendritic cells and monocyte-derived Langerhans’-like cells. Immunology 132(2):217–25. doi:10.1111/j.1365-2567.2010.03356.x
Rieder SA, Chauhan A, Singh U, Nagarkatti M, Nagarkatti P (2010) Cannabinoid-induced apoptosis in immune cells as a pathway to immunosuppression. Immunobiology 215(8):598–605. doi:10.1016/j.imbio.2009.04.001
Robinson RH, Meissler JJ, Breslow-Deckman JM, Gaughan J, Adler MW, Eisenstein TK (2013) Cannabinoids inhibit T-cells via cannabinoid receptor 2 in an in vitro assay for graft rejection, the mixed lymphocyte reaction. J Neuroimmune Pharmacol 8(5):1239–1250. doi:10.1007/s11481-013-9485-1
Roth MD, Gitlitz BJ, Kiertscher SM, Park AN, Mendenhall M, Moldawer N, Figlin RA (2000) Granulocyte/macrophage-colony stimulating factor and interleukin-4 enhance the number and antigen-presenting activity of circulating CD14+ and CD83+ cells in cancer patients. Cancer Res 60(7):1934–1941
Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122(3):787–795. doi:10.1172/JCI59643
Sipe JC, Arbour N, Gerber A, Beutler E (2005) Reduced endocannabinoid immune modulation by a common cannabinoid 2 (CB2) receptor gene polymorphism: possible risk for autoimmune disorders. J Leukoc Biol 78(1):231–238. doi:10.1189/jlb.0205111
Steinbrink K, Wolfl M, Jonuleit H, Knop J, Enk AH (1997) Induction of tolerance by IL-10-treated dendritic cells. J Immunol 159(10):4772–4780
Yuan M, Kiertscher SM, Cheng Q, Zoumalan R, Tashkin DP, Roth MD (2002) Δ9-Tetrahydrocannabinol regulates Th1/Th2 cytokine balance in activated human T-cells. J Neuroimmunol 133(1–2):124–131. doi:10.1016/S0165-5728(02)00370-3
Zhu LX, Sharma A, Stolina S, Gardner B, Roth MD, Tashkin DP, Dubinett SM (2000) Δ-9 tetrahydrocannabinol inhibits antitumor immunity by a CB2 receptor-mediated, cytokine-dependent pathway. J Immunol 165(1):373–80. doi:10.4049/jimmunol.165.1.373
Acknowledgments
Research reported in this publication was supported by the National Institute on Drug Abuse, National Institutes of Health, under award numbers 5-R01-DA003018 and 1-R01-DA037102. JT Castaneda was supported by a Ruth L. Kirschstein National Research Service Award (NRSA) Individual Predoctoral Fellowship to Promote Diversity in Health-Related Research from the National Institute on Drug Abuse, National Institutes of Health, under award numbers 1 F31 DA036293. Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research (CFAR) Flow Cytometry Core Facility that is supported by National Institutes of Health awards CA-16042 and AI-28697, and by the JCCC, the UCLA AIDS Institute, and the David Geffen School of Medicine at UCLA.
Conflicts of Interest
The authors declare that they have no conflict of interest.
Statement of Human Rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Roth, M.D., Castaneda, J.T. & Kiertscher, S.M. Exposure to Δ9-Tetrahydrocannabinol Impairs the Differentiation of Human Monocyte-derived Dendritic Cells and their Capacity for T cell Activation. J Neuroimmune Pharmacol 10, 333–343 (2015). https://doi.org/10.1007/s11481-015-9587-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-015-9587-z