
Abstract
Purpose
Soils derived from different parent materials are known to support quite distinct indigenous community structures. Crop cultivation is also an important factor regulating bacterial community structure in soils. However, the extent to which the soil parent material imposes limits on the diversity of organisms present and the extent to which cropping practices are able to supplant the effects of soil parent materials are largely unknown.
Materials and methods
Five ancient paddy soils developed on three different parent materials were collected from the major rice production regions in southern China. The 16S rRNA genes were amplified from each soil DNA sample and then analyzed using Tag-pyrosequencing and terminal restriction fragment length polymorphism (T-RFLP) to characterize bacterial communities and their relative abundance. Total bacterial abundance was determined by 16S rRNA gene quantitative PCR (qPCR). Canonical correspondence analysis (CCA) was used to investigate the relationship between the bacterial distribution and soil properties.
Results and discussion
Cluster analysis of the T-RFLP and pyrosequencing data indicated that although the bacterial communities in the surface layers (0–20 cm) could be distinguished from those recovered from depths of 20–40 and 40–60 cm in all soils, the bacterial community structures in soils with similar parent materials clustered regardless of soil depth. Bacteria of the phylum Proteobacteria dominated in all soils with significantly higher proportions in the top soils. The relative abundances of Chloroflexi and Acidobacteria were closely associated with the soil parent material. This suggested that the changes in bacterial community compositions induced by rice cultivation did not mask those determined by soil parent material. CCA indicated that soil pH, available Mn, and total K and Fe contents explained most of the variance in bacterial distribution due to differences in parent materials. qPCR revealed that the bacterial abundance in top soils (0–20 cm) also varied with parent material.
Conclusions
The nature of the parent material is an important factor in determining soil bacterial community structure, and although long-term rice cultivation resulted in shifts in this structure, the community retained characteristics of the original populations.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Soil parent material is one of the major factors controlling soil formation (Jenny, 1941), with the nature of parent materials having a key role in regulating soil physicochemical properties such as texture, acidity, and base saturation (Anderson 1988; Imaya et al. 2005; Woodruff et al. 2009). Studies carried out in different regions have shown that soil characteristics vary between soils according to the type of parent materials (Imaya et al. 2005; Gökbulak and Özcan 2008; Appleton and Adlam 2012). For instance, Gökbulak and Özcan (2008) compared five forest soils developed from different parent materials in NE Turkey and concluded that soils formed on granite were coarse-grained in texture with lower moisture content at field capacity and lower pH values than other soils. Other studies carried out on brown forest soils in Japan showed that soils derived from volcanic ash were finely textured and contained markedly larger amounts of carbon and nitrogen compared to granite- and gabbro-formed soils (Imaya et al. 2005). Similar results have also been observed in Mediterranean woodland ecosystems in NE Spain and in Nigeria (Kooijman et al. 2005; Olowolafe 2002). Parent material is also known to determine the amounts and forms of soil chemical and mineral elements such as Ca, Fe, and K content (Appleton and Adlam 2012; Smith et al. 2011; Bini et al. 2011).
Given their influence over the physicochemical properties of soils, it is reasonable to hypothesize that soil parent materials may also influence the structure and dynamics of soil organisms. Although data on the effects of parent material on soil microbial communities are limited, it has been shown that changes in microbial community composition are highly correlated with variations in soil properties (Tripathi et al. 2012; Kuramae et al. 2012). In particular, soil pH has been identified as a key environmental factor shaping soil microbial communities across a range of ecosystems (Griffiths et al. 2011). Lauber et al. (2009) showed that differences in soil pH could explain a significant portion of the variability in bacterial community structure across a range of soils in North and South America. Similar studies in forest and non-forest soils along the central and southern Malay Peninsula and Northern Borneo also showed the importance of soil pH in structuring microbial communities (Tripathi et al. 2012). Works by Faoro et al. (2010) on bacterial diversity in the acid forest soils with a narrow pH range suggested that differences in the Ca2+/Mg2+ ratio and Al3+ and phosphorus levels had a significant influence on bacterial diversity. The studies on soils from six different land use patterns in The Netherlands indicated that besides pH, the C/N ratio and total phosphate content also had a significant impact on microbial community structures (Kuramae et al. 2012). These studies show the complexity of the interaction between microbial community structure and soil properties and suggest that there is unlikely to be one major driver of bacterial community structure in soils, physicochemical properties that act in combination to shape the bacterial community.
Soil properties play a key role in determining soil bacterial community structure and, meanwhile, reflect the nature of the parent material (Woodruff et al. 2009; Bini et al. 2011; Imaya et al. 2005). However, information on the importance of soil parent material in determining the diversity and relative abundance of bacterial communities is less well understood.
Anthropogenic activities such as soil management are also known to have a significant influence on soil microbial communities (Bissett et al. 2011; Acosta-Martínez et al. 2010). Continuous application of organic fertilizers has been shown to decrease markedly the proportion of β-Proteobacteria and Verrucomicrobia (Wu et al. 2011) and result in significant increases in the ratio of Gram-positive to Gram-negative bacteria and of bacteria to fungi (Marschner et al. 2003). When rice paddy fields were converted to vegetable production, the community structure was shown to change with a significant decrease in the relative abundance of Chloroflexi (Sheng et al. 2013). To date, many of these studies have been done at experimental sites in the same region and on similar parent materials. More information is needed on how the parent material and soil management interact in the establishment of microbial community structure and function in soils.
This paper explores the relationship between soil parent material and bacterial diversity in ancient paddy systems in Southern China using real-time PCR, 454 pyrosequencing, and T-RFLP analyses.
2 Materials and methods
2.1 Sites description and soil sampling
Five ancient paddy soils were selected from sites in southern China (Electronic supplementary material, Fig. S1). Soil LZ, formed on latosol parent material, was collected from Leizhou in Guangdong province (20°33′ N, 110°04′ E). Soils TY-B and YT were sampled from Baodongyu in Taoyuan in Hunan province (28°55′ N, 111°26′ E) and Yingtan in Jiangxi province (28°12′ N, 116°56′ E), respectively. Both of them were developed from quaternary red clays. Soils JX and TY-G were taken from Jiaxing in Zhejiang province (30°38′ N, 120°46′ E) and Gushi in Taoyuan in Hunan province (28°55′ N, 111°26′ E), respectively. These two soils were from alluvium deposits. According to local county records, all of the sampling sites had been continuously cropped with paddy rice for over a hundred years. Over recent decades, N, P, and K fertilizers were generally applied annually at rates of between 105–135 kg N ha–1, 45–52 kg P ha–1, and 78–117 kg K ha–1. Agronomic managements such as fertilization and water management were similar between sites and consistent with practices designed to deliver high grain yields.
Soil samples were collected between December 2010 and January 2011 when the fields had been drained and were in fallow. Each sampling site covered an area of about 1 ha, and at each site, three soil profiles were prepared. Each profile was 1 m in width and opened to 1 m in depth. The profile was sampled by taking three separate sub-samples at each of 0–20, 20–40-, and 40–60-cm depths. In total, 45 soil sub-samples were collected (five soils × three depths × three replicates). Each of the sub-samples was then divided into two portions. One portion was transferred to a sterile plastic bag and transported to the laboratory on ice and stored at –80 °C prior to molecular analyses. The other was air-dried for analysis of soil physical and chemical properties. Sub-samples were labeled as soil LZ1–TY-G1 (0–20 cm), LZ2–TY-G2 (20–40 cm), and LZ3–TY-G3 (40–60 cm).
2.2 Soil physical and chemical properties
Soil texture was analyzed using a fixed pipette method (Dane and Topp 2002). Soil organic carbon (SOC) was determined by K2Cr2O7 oxidation (Kalembas and Jenkinso 1973). Total nitrogen (TN) was measured with automatic flow injection after digestion in H2SO4, and total phosphorus (TP) and potassium (TK) were measured by inductively coupled plasma spectrometry (ICP) after fusion in NaOH. Total Mn (TMn) and Fe (TFe) were detected by ICP following digestion in HF-HCl-HNO3. Atomic absorption spectroscopy was employed to determine available K (AK) and Fe and Mn (AFe and AMn), whilst available P (AP) was measured following extraction with 0.5 M NaHCO3. Soil pH was determined at a soil to water ratio of 1:2.5 (Bao 2000). The physical and chemical properties of the soils are shown in Table 1.
2.3 DNA extraction and analysis of T-RFLP and pyrosequencing
DNA was extracted from 0.5 g (fresh weight) soil using the Fast DNA Spin Kit (MP Biomedicals, Carlsbad, CA, USA) following the manufacturer’s instructions. The extracted DNA was quantified using a Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). Each of the 45 soil sub-samples was extracted separately.
PCR for T-RFLP analysis was carried out with primer F299 labeled with 6-FAM (6-carboxyfluorescein) at the 5′ end and reverse primer R1307 for T-RFLP analysis (Wu et al. 2011). The 50-μl reaction mixture contained 1× PCR mix (TIANGEN, China), 20 pM of each primer, and 30 ng of DNA template. Amplification conditions were as follows: 95 °C for 2 min followed by 35 cycles of 95 °C for 30 s, 50 °C for 45 s, and 72 °C for 60 s. PCR amplification of each sample was replicated three times and then combined to provide a composite sample. Mixed PCR products were run on an agarose gel (1 % w/v), and fragments of expected length were excised and purified with Gel DNA Purification Kit (TIANGEN, China). Restriction digestion was conducted in a 20-μl reaction mixture containing 10 U of endonuclease MspI (Fermentas, Canada), and the precise lengths of the peaks from the amplified 16S rRNA gene products (T-RFLP) were determined using an ABI 3100 DNA sequencer (Applied Biosystems, USA).
For 454 pyrosequencing, barcoded (7 bp) 16S rRNA gene universal primers 16S-5F (5′-TGGAGAGTTTGATCCTGGCTCAG) and 16S-531R (5′-TACCGCGGCTGCTGGCAC) were used to amplify the V1–V3 region of about 500 bp. The PCR reactions were performed in a total volume of 50 μl containing 15 ng of template DNA, 1 U of Pyrobest DNA polymerase (TaKaRa, Japan), 1× Pyrobest Buffer, and 0.1 μM of each forward and reverse primer. Thermocycling was conducted in a Mastercycler pro gradient PCR Cycler (Eppendorf AG, Hamburg, German) as follows: 95 °C for 2 min followed by 25 cycles of 94 °C for 30 s, 58 °C for 30 s, 72 °C for 30 s, and a final elongation step at 72 °C for 5 min. Since the T-RFLP analyses showed to be reproducible (Fig. S1), the PCR products for each of the 15 soil samples were separately pooled and then purified using the QIA quick Gel Extraction Kit (Qiagen, Hilden, Germany). Equal amounts of the purified PCR products from 15 samples were sent to the Public Laboratory of BioEnergy and BioProcess, Qingdao Institute of BioEnergy and Bioprocess Technology, Chinese Academy of Sciences for pyrosequencing analysis on a Roche GS FLX 454 sequencer (Roche, New Jersey).
2.4 Comparison of bacterial community structures based on T-RFLP analysis
Bacterial community structures in each sub-sample (45 samples) were determined by T-RFLP. Data analysis of T-RFLP profiles was performed using PeakScan version 1.0. software (Applied Biosystems, Inc.), and only fragments with a signal above 1 % of the sum of all peak heights were included in the statistical analyses. The peak heights of T-RFs that differed in size by ≤2 bp in an individual profile were combined and considered as one fragment. Similarly, only T-RFs with a size of more than 50 bp were used for analysis. Cluster analysis of soils was done with an unweighted pair–group method with arithmetic averages (UPGMA algorithm)-applied Bray–Curtis distances between samples. The relationship between the bacterial distributions and soil properties was investigated using canonical correspondence analysis (CCA) and the CANOCO statistical package for Windows 4.5 (Biometris, Wageningen, The Netherlands). A Mantel test based on 999 random permutations was used to examine the significant correlations between the differences in soil chemical properties and bacterial community structures.
2.5 Analysis of bacterial diversity using phylogenetic analysis of pyrosequencing data
Barcodes were used to assign sequences to samples that were then quality-trimmed using MOTHUR, version 1.9.1. Low-quality sequences (<150 bp in length with an average quality score of < 25) were excluded from further analyses. The Ribosomal Database Project (RDP) pyrosequencing pipeline and multiclassifier function were used to align and assign taxonomic identities to the sequence data at 80 % confidence level (Claesson et al. 2009). The remaining sequences were assigned to operational taxonomic units (OTUs) using the UCLUST classifier at 97 % sequence similarity level. Dendrograms showing the similarity between the community structures in different samples were done using the “tree.shared” command in MOTHUR. Cluster analysis was also performed using an UPGMA algorithm based on Bray–Curtis distances between communities.
2.6 Quantitative PCR (qPCR)
Primers 1369 F/1492R (Suzuki et al. 2000) were used for Quantitative PCR (qPCR) amplification of the 16S rRNA gene. PCR assays were carried out in a volume of 10 μl containing 1× SYBR Premix Ex Taq TM (Takara, Japan), 150 nM forward and reverse primers, and 5 ng of template DNA. Thermal cycling conditions were as follows: an initial cycle of 95 °C for 30 s followed by 5 cycles of 95 °C for 5 s, 62 °C for 20 s, 72 °C for 20 s, and 30 cycles of 95 °C for 5 s, 57 °C for 20 s, 72 °C for 20 s. All qPCR amplifications were performed using an ABI Prism 7900 sequence detection system. A standard curve for the 16S rRNA gene was prepared using a tenfold dilution series (102–109 copies) of the plasmids containing the target gene fragment. The efficiency of the reactions was 105 %, and the R 2 value for the curves was 0.99. A single, sharp peak was observed for each of the melting curves.
3 Results
3.1 Variations in bacterial abundance
Sub-soils at 40–60-cm depth are rarely affected by rice cultivation or weathering and are used in this study to represent the situation of the parent materials. The real-time PCR data showed that the 16S rRNA gene copy numbers of the five sub-soils (40–60 cm) were between 1.47E+8 and 5.32E+8, with no significant differences between parent materials (Fig. 1). Although the 16S rRNA gene copy numbers in sub-soils (20–40 cm) were significantly higher than that in the 40–60-cm layer, the variation trends among soils were similar in these two layers. The top soils (0–20 cm), however, harbored the highest abundance of bacteria within the soil profile and varied according to parent materials. The alluvial top soils (JX1 and TY-G1) contained the highest 16S rRNA gene copy numbers (average 8.4 E+9 copies g−1 soil, respectively), followed by the quaternary red clays (TY-B1 and YT1). The latosol (LZ1) had the lowest bacterial abundance.

Copy number for the 16S rRNA gene in soils derived from a latosol (LZ), quaternary red clays (TY-B and YT) and alluvium deposits (JX and TY-G); 0–20, 20–40 and 40–60 cm indicate the sampling depth down the profile. Significant differences (p < 0.05) between the soils are shown using letters a, b, or c. Statistical analysis between soils for each layer was performed separately. Soils with the same letter at each depth are not significantly different at the p < 0.05 level
3.2 Bacterial community composition analysis by T-RFLP
The T-RF patterns showed that the structure of bacterial communities differed between parent materials (Fig. 2). The alluvial soils (JX and TY-G) shared a similar structure that was characterized by a major peak T-RF 189 bp that accounted for 32–39 % of the total T-RF population. Among soils, the proportion of T-RF 189 bp was significantly lower (15–26 %), and T-RF 163 bp was markedly higher in the quaternary red clays (TY-B and YT) than in the alluvial soils. T-RF 149 bp was only found in the quaternary red clays. The bacterial composition of the latosolic soil (LZ) was characterized by several equally distributed T-RFs (T-RF 128 bp, 163 bp, 169 bp, and 189 bp). The distribution of T-RFs down the profile also reflected the nature of the soil parent materials. The alluvial top soils (JX1 and TY-G1) exhibited similar T-RF patterns to those of their sub-soils (JX3 and TY-G3), except for the relative abundance of T-RF 163 bp that increased markedly in the top soil. Unlike alluvial soils, differences in community structure along the profile were more apparent in the quaternary red soils (TY-B and YT), especially in the proportion of T-RF 163 bp that was significantly higher at the top of the profile. The relative abundance of T-RF 217 bp and 275 bp in the latosol (LZ) increased significantly towards the top of the profile and was a major bacterial component of the top soil.

Relative abundance of 16S rRNA gene T-RFs following digestion with endonuclease MspI in soils derived from latosol (LZ), quaternary red clays (TY-B and YT) and alluvium deposits (JX and TY-G). Numbers 1, 2, and 3 following the capital letters (LZ–E) represent sampling depths of 0–20, 20–40, and 40–60 cm, respectively. The numbers in the legend represent the fragment lengths of the T-RFs
CCA analysis was used to investigate the relationships between soil bacterial composition (T-RFLP data) and soil physicochemical properties. The results showed that in the top soil, bacterial composition was significantly (p < 0.05) correlated with soil pH and available Mn (Fig. 3) with these two variables explaining 36.8 and 29.3 % of the variation between samples, respectively. In the sub-soils (20–40 cm), however, microbial compositions were significantly (p < 0.05) correlated with total Fe (TFe), available Mn (AMn), and available K (AK) contents. Together these three parameters accounted for over 80 % of the variance (36.2, 34.3, and 10.3 %, respectively). Significant environmental variables in the sub-soils (40–60 cm) included total Fe (TFe), total K (TK), and available Mn (AMn). Together these three variables explained 41 % (TK) and 42 % (TFe plus AMn) of the variance between the samples in the sub-soils (40–60 cm).

Ordination plots of the results of the CCA analyses used to investigate the relationship between bacterial populations and soil chemical properties. Soils were sampled at 0–20, 20–40, and 40–60 cm down the profile. Soil properties are represented on the plots as soil organic carbon (SOC), total nitrogen (TN), total potassium (TK), total phosphorus (TP), total iron (TFe), total manganese (TMn), available phosphorus (AP), available potassium (AK), available iron (AFe), available manganese (AMn). LZ latosol, TY-B and YT quaternary red clays, JX and TY-G alluvium deposits
3.3 Taxonomic and phylogenetic analysis by pyrosequencing
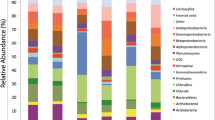
A total of 174,087 16S rRNA gene sequences (range 6,118 to 16,042 per sample) were obtained from the soil samples following 454 sequencing with an average read length of 442 bp (range 150 to 554 bp). Of these sequences, 113,011 (65 %) were assigned to 22 phyla using the RDP classifier with 80 % confidence level. Proteobacteria, Chloroflexi, Acidobacteria, Actinobacteria, and Firmicutes (relative abundance > 3.5 %) were the main phyla found in all soils (Fig. 4). Among the phyla identified, the distribution of Acidobacteria and Chloroflexi was closely related to the soil parent materials (Fig. 4). The relative abundance of Acidobacteria in the quaternary red soils (TY-B and YT) and the alluvial soils (JX and TY-G) averaged 17 and 8 %, respectively, with these proportions generally maintained throughout the profile. However, the proportion of Acidobacteria in the latosol soil (LZ) significantly decreased up the profile and was the lowest of the five top soils. The quaternary red soils (TY-B and YT) and alluvial soils (JX and TY-G) maintained relatively steady proportions of Chloroflexi throughout the soil profiles. In the quaternary red soils, the relative abundance of Chloroflexi was lower than in the alluvial soils. Although the Chloroflexi fraction in the latosol soil (LZ) was greater than that in the other top soils, the proportion of Chloroflexi was the lowest in the sub-soils (40–60 cm).

Relative abundance of major bacterial phyla in soils derived from latosol (LZ), quaternary red clays (TY-B and YT) and alluvium deposits (JX and TY-G). Numbers 1, 2, and 3 following the capital letters (LZ–TY-G) represent sampling depths of 0–20, 20–40, and 40–60 cm, respectively. The relative abundance is given as a percentage of the total classified 16S rRNA gene sequences in each of the dominant phyla. Taxonomic assignments of sequences are based on classification (80 % confidence level) to the phylum level using the RDP classifier tool
Figure 5 shows the relative abundances of different genera in the test soils. At 40–60-cm depth, soils derived from the same parent material show highly similar distributions (Fig. 5) with the quaternary red soils (TY-B3 and YT3) characterized by three major genera, GP1, Ktedonobacter, and Nitrospira. These taxa accounted for over 50 % of the identified sequences. The latosol (LZ3) showed a similar structure to the quaternary red soils, whilst those in the two alluvial soils (JX3 and TY-G3) were more evenly distributed with no dominant taxa. Long-term rice cultivation resulted in clear differentiation in the bacterial community structure of the surface soils (0–20 cm) compared to their corresponding sub-soils (40–60 cm). In general, the quaternary red clays formed top soils with similar bacterial communities to those of the corresponding sub-soils except that the relative abundance of Nitrospira decreased with depth whilst sequences of Verrucomicrobia subdivision 3 genera incertae sedis increased significantly with depth. In the alluvial soils, there were no major differences in the relative abundances of genera between the upper and lower soil layers. However, the bacterial structure of the latosolic top soil was markedly different between the top soil and the sub-soil (40–60-cm depth).

Relative abundance of major bacterial genera in soils derived from latosol (LZ), quaternary red clays (TY-B and YT), and alluvium deposits (JX and TY-G); 0–20, 20–40, and 40–60 cm indicate the sampling depth down the profile. The relative abundance is given as a percentage of each genus against the total 16S rRNA gene sequences. Only the genera with relative abundance > 0.1 % were shown. For clarity, the genus is abridged with suffix “genera incertae sedis” to the genus with suffix “*” in the legend text. For example, “OP11 genera incertae sedis” was shortened to “OP11*”. For easy retrieval, subdivision 3 genera incertae sedis, Nitrospira, Ktedonobacter, and Singulisphaera are labeled in the figure using their abbreviation Sub, Nit, Kte, and Sin, respectively
Cluster analysis showed that the bacterial community composition of the alluvial soils (JX and TY-G), the quaternary red clays (TY-B and YT), and the latosol (LZ) clustered, separately, and independently of soil depth (Fig. 6). Similar results were found with the cluster analysis of the T-RFLP data (Electronic supplementary material, Fig. S2).

Cluster analysis of the pyrosequencing data showing the relationships between soils derived from latosol (LZ), quaternary red clays (TY-B and YT), and alluvium deposits (JX and TY-G) at an OTU definition level of 0.03. Numbers 1, 2, and 3 following the capital letters (LZ–TY-G) represent 0–20, 20–40, and 40–60 cm layer of the soil profile, respectively
4 Discussion
4.1 Parent material effects on bacterial abundance
The introduction of 16S rRNA gene analyses has significantly advanced our understanding of microbial diversity in soils, yet our understanding of how that diversity is formed and structured remains largely unexplored. Developments in qPCR and more recently in high-throughput pyrosequencing have meant that we are now better able to determine not only what organisms are present but also how these communities are structured both spatially and temporally. Using qPCR, the work reported here has shown that differences in bacterial abundance, as determined by 16S rRNA gene copy number, in the sub-soils (40–60 cm) show no significant relationship with the soil parent material. However, in the top soils (0–20 cm), the bacterial abundances varied markedly with parent material. Below 40 cm, sub-soils are largely unaffected by rice cultivation and characterized by low nutrient conditions for bacterial growth (Frenzel et al. 1999). Consequently, the impact of soil parent materials on microbial structure may be constrained by nutrient availability (Rumpel and Kogel-Knabner 2011). In top soils managed under rice over hundreds years, the weathering of soil parent materials can increase the availability of mineral elements (Anderson 1988), coupling with the effects of fertilizer and the crop exudates, which can significantly increase the soil carbon content and the availability of N, P, and K (Treonis et al. 2010; Manlay et al. 2002; Wu et al. 2011; Shen et al. 2010). This more favorable nutrient environment helps sustain microbial growth and fosters the development of diversity, structure, and function (Fierer et al. 2003; Eilers et al. 2012).
It has been shown that variation in soil physicochemical parameters such as organic matter, pH, moisture, and temperature can have profound impacts on the abundance of the microorganisms (Wu et al. 2011; Fierer et al. 2003; Herold et al. 2014). However, differences in soil properties such as pH, organic carbon content, texture, and nutrient availability can also be determined by differences in parent material (Prakongkep et al. 2008; He et al. 2004; Shi et al. 2009), which can directly or indirectly impact on microbial form and function in soils (Wagai et al. 2011; Ulrich and Becker 2006). In the work reported here, the alluvial top soils supported the largest bacterial population, followed by the quaternary red soils and then the latosol (Fig. 1). Soils derived from quaternary red clays and latosols are developed under relatively intensive weathering and leaching conditions (Li 1983) and are generally characterized by high level of acidity and calcium and potassium deficiency (Jiang et al. 2010). This means that they are poorly fertile and usually unsuitable for agriculture without the application of fertilizer. Alluvial soils are near neutral (Table 1) and had higher organic matter and nitrogen contents with less nutrient leaching due to the high groundwater table (Xiang et al. 2005). Thus, the alluvial top soils had higher carbon content and greater nutrient availability for microorganisms than the other soils, which serves to explain the higher bacterial abundance in these top soils.
4.2 Succession of bacterial community structure along the soil profiles
Crop cultivation has been clearly linked to changes in the composition of microbial communities in cultivated horizon (Fierer et al. 2003; Acosta-Martínez et al. 2010). In this study, we detected that the bacterial community structures changed significantly with soil depth in all profiles. A number of other studies have also found that microbial communities in sub-surface are distinct from the surface communities (Fritze et al. 2000; Blume et al. 2002; Fierer et al. 2003). These changes in bacterial community structure have been shown to closely associate with reductions in the availability of oxygen and the quality and availability of soil organic carbon with depth (Potthoff et al. 2006; Fierer et al. 2003). The oligotrophic environment in the sub-soils restricts bacterial activity, and as a result, some surface-dwelling bacteria may be less likely to thrive in the deeper soil environment (Agnelli et al. 2004; Goberna et al. 2005; Will et al. 2010). The relative abundance of Proteobacteria, for example, was obviously greater in the top soils than the sub-soils (Fig. 4). Proteobacteria abundance was positively related to SOC content (Asuming-Brempong et al. 2008; Davinic et al. 2012). In contrast, the observed increase in the relative abundance of Nitrospira with depth (Fig. 4) parallels the results from other studies that have detected relatively large Nitrospira population in oligotrophic habitats (Keshri et al. 2013). The less extreme changes in temperature and moisture were also expected to be an important factor causing the differentiation of the bacterial communities within the studied profiles (Lavahum et al. 1996).
4.3 Relationship between parent material and bacterial community structure
Although long-term rice cultivation caused some shifts in the bacterial community structure in top soils, it was found that each soil supports a distinct bacterial community structure that reflects the characteristics of the soil parent material, independent of soil depth. Differences in community composition extended beyond the dominant Acidobacteria, which were on average 92 % more abundant in the quaternary red clay than in soils formed on the other two parent materials. Meanwhile, the alluvium soils harbored a significantly larger population of Chloroflexi (Fig. 4). Some recent studies have also shown that there are significant parent material controls over soil microbial community composition in forest ecosystems in the early stages of soil development (Wagai et al. 2011) and during biocrust formation in dryland soils (Steven et al. 2013). These controls are due to the impact of the parent material on the prevailing soil physicochemical properties. Here, the significant differences in multiple soil properties such as pH and available Mn and total K contents could help explain the differences in soil bacterial community structure between parent materials. Previous studies have shown that soil pH is a key factor in determining soil bacterial community structures (Griffiths et al. 2011; Lauber et al. 2009), especially the distribution of Acidobacteria (Nie et al. 2012; Tripathi et al. 2012), which can support that low pH soil TY-B and YT harbor higher proportions of Acidobacteria than other soils. Besides, several Acidobacteria sub-groups were also revealed to respond to high content of soil Mn (Navarrete et al. 2013). However, these edaphic factors are closely related to the chemistry and weathering extent of the parent material (Imaya et al. 2005; Heckman and Rasmussen 2011; Woodruff et al. 2009). Li (1983) demonstrated that latosolic soils developed under tropical conditions generally have high contents of iron and low amounts of potassium due to the long-term accumulation of iron and significant leaching of potassium during soil development.
Given the complex interplay between soil parent material, weathering, and soil mineral status and the importance of soil minerals in regulating soil chemical and physical properties such as nutrient availability, pH, and texture, it is perhaps not too surprising that soil mineral status influences the development of soil bacterial community structure. Much of what we currently know about the influence of soil conditions on bacterial community structure stems from work done on the effects of management on microbial diversity in the upper soil layers (0–20 cm), and more studies are needed to understand how microbial diversity changes with depth down the profile. The work reported in this paper suggests that parent material and soil formation, at least in part, already determine the diversity managed in the top soil.
5 Conclusions
The work reported here showed that the size of the bacterial population in the soils examined differed with parent material in the top soils but showed no clear relationship with parent material in the sub-soils. Although long-term rice cultivation induced some shifts in soil bacterial community in the top soils, soils formed on different parent materials support distinct structures regardless of soil depth. Soil properties such as pH, available Mn, and total K contents, which are largely determined by the nature of the soil parent material, were shown to be major factors determining bacterial community structure in these paddy soils.
References
Acosta-Martínez V, Dowd SE, Sun Y, Wester D, Allen V (2010) Pyrosequencing analysis for characterization of soil bacterial populations as affected by an integrated livestock-cotton production system. Appl Soil Ecol 45:13–25
Agnelli A, Ascher J, Corti G, Ceccherini MT, Nannipieri P, Pietramellara G (2004) Distribution of microbial communities in a forest soil profile investigated by microbial biomass, soil respiration and DGGE of total and extracellular DNA. Soil Biol Biochem 36:859–868
Anderson DW (1988) The effect of parent material and soil development on nutrient cycling in temperate ecosystems. Biogeochemistry 5:71–97
Appleton JD, Adlam KAM (2012) Geogenic control on soil chemistry in urban areas: a novel method for urban geochemical mapping using parent material classified data. Appl Geochem 27:161–170
Asuming-Brempong S, Gantner S, Adiku SGK, Archer G, Edusei V, Tiedje JM (2008) Changes in the biodiversity of microbial populations in tropical soils under different fallow treatments. Soil Biol Biochem 40:2811–2818
Bao SD (2000) Analysis of soil characteristics. Chinese Agricultural Press, Beijing
Bini C, Sartori G, Wahsha M, Fontana S (2011) Background levels of trace elements and soil geochemistry at regional level in NE Italy. J Geochem Explor 109:125–133
Bissett A, Richardson AE, Baker G, Thrall PH (2011) Long-term land use effects on soil microbial community structure and function. Appl Soil Ecol 51:66–78
Blume E, Bischoff M, Reichert J, Moorman T, Konopka A, Turco R (2002) Surface and subsurface microbial biomass, community structure and metabolic activity as a function of soil depth and season. Appl Soil Ecol 592:1–11
Claesson MJ, O’Sullivan O, Wang Q, Nikkilä J, Marchesi JR, Smidt H, de Vos WM, Ross RP, O’Toole PW (2009) Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structure in the human distal intestine. PLoS One 4(8):1–15
Dane JH, Topp GC (2002) Methods of soil analysis. In: Gee GW, Or D (eds) Physical methods. Soil Science Society of America, Madison, pp 255–278
Davinic M, Fultz LM, Acosta-Martinez V, Calderon FJ, Cox SB, Dowd SE, Allen VG, Zak JC, Moore-Kucera J (2012) Pyrosequencing and mid-infrared spectroscopy reveal distinct aggregate stratification of soil bacterial communities and organic matter composition. Soil Biol Biochem 46:63–72
Eilers KG, Debenport S, Anderson S, Fierer N (2012) Digging deeper to find unique microbial communities: the strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol Biochem 50:58–65
Faoro H, Alves AC, Souza EM, Rigo LU, Cruz LM, Al-Janabi SM, Monteiro RA, Baura VA, Pedrosa FO (2010) Influence of soil characteristics on the diversity of bacteria in the Southern Brazilian Atlantic forest. Appl Environ Microbiol 76:4744–4749
Fierer N, Schimel J, Holden P (2003) Variations in microbial community composition through two soil depth profiles. Soil Biol Biochem 35:167–176
Frenzel P, Bosse U, Janssen PH (1999) Rice roots and methanogenesis in a paddy soil: ferric iron as an alternative electron acceptor in the rooted soil. Soil Biol Biochem 31:421–430
Fritze H, Pietikainen J, Pennanen T (2000) Distribution of microbial biomass and phospholipid fatty acids in Podzol profiles under coniferous forest. Eur J Soil Sci 51:565–573
Goberna M, Insam H, Klammer S, Pascual JA, Sanchez J (2005) Microbial community structure at different depths in disturbed and undisturbed semiarid Mediterranean forest soils. Microb Ecol 50:315–326
Gökbulak F, Özcan M (2008) Hydro-physical properties of soils developed from different parent materials. Geoderma 145:376–380
Griffiths RI, Thomson BC, James P, Bell T, Bailey M, Whiteley AS (2011) The bacterial biogeography of British soils. Environ Microbiol 13:1642–1654
He M, Chen ZH, Iwasaki K, Sakurai K, Hu P, Wang Z, Dai L, Wang HY (2004) Pysico-chemical characteristics of the soils from the coastal plain in Shanghai, China. Soil Sci Plant Nutr 50:1237–1244
Heckman K, Rasmussen C (2011) Lithologic controls on regolith weathering and mass flux in forested ecosystems of the southwestern USA. Geoderma 164:99–111
Herold N, Schöning I, Gutknecht J, Alt F, Boch S, Müller J, Oelmann Y, Socher SA, Wilcke W, Wubet T, Schrumpf M (2014) Soil property and management effects on grassland microbial communities across a latitudinal gradient in Germany. App Soil Ecol 73:41–50
Imaya A, Ohta S, Tanaka T, Inagaki Y (2005) General chemical properties of brown forest soils developed from different parent materials in the submontane zone of the Kanto and Chubu districts, Japan. Soil Sci Plant Nutr 51:873–884
Jenny H (1941) Factors of soil formation: a system of quantitative pedology. McGraw-Hill, New York
Jiang J, Xu RK, Zhao AZ (2010) Comparison of the surface chemical properties of four soils derived from quaternary red earth as related to soil evolution. Catena 80:154–161
Kalembas SJ, Jenkinso DS (1973) Comparative study of titrimetric and gravimetric methods for determination of organic carbon in soil. J Sci Food Agric 24:1085–1090
Keshri J, Mody K, Jha B (2013) Bacterial community structure in a semi-arid haloalkaline soil using culture independent method. Geomicrobiol J 30(6):517–529
Kooijman AM, Jongejans J, Sevink J (2005) Parent material effects on Mediterranean woodland ecosystem in NE Spain. Catena 59:55–68
Kuramae EE, Yergeau E, Wong LC, Pijl AS, van Veen JA, Kowalchuk GA (2012) Soil characteristics more strongly influence soil bacterial communities than land-use type. FEMS Microbiol Ecol 79:12–24
Lauber CL, Hamady M, Knight R, Fierer N (2009) Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75:5111–5120
Lavahum MFE, Joergensen RG, Meyer B (1996) Activity and biomass of soil microorganisms at different depth. Biol Fert Soils 23:38–42
Li QQ (1983) Red earth of China. Science, Beijing
Manlay RJ, Chotte JL, Masse D, Laurent JY, Feller C (2002) Carbon, nitrogen and phosphorus allocation in agro-ecosystems of a West African savanna III. Plant and soil components under continuous cultivation. Agr Ecosyst Environ 88:249–269
Marschner P, Kandeler E, Marshchner B (2003) Structure and function of the soil microbial community in a long-term fertilizer experiment. Soil Biol Biochem 35:453–461
Navarrete AA, Kuramae EE, de Hollander M, Pijl AS, van Veen JA (2013) Acidobacterial community responses to agricultural management of soybean in Amazon forest soils. FEMS Microbiol 83:607–621
Nie M, Meng H, Li K, Wan JR, Quan ZX, Fang CM, Chen JK, Li B (2012) Comparison of bacterial and fungal communities between natural and planted pine forest in subtropical China. Curr Microbiol 64:34–42
Olowolafe EA (2002) Soil parent materials and soil properties in two separate catchment areas on the Jos Plateau Nigeria. GeoJournal 56:201–212
Potthoff M, Steenwerth KL, Jackson LE, Drenovsky RE, Scow KM, Joergensen RG (2006) Soil microbial community composition as affected by restoration practices in California grassland. Soil Biol Biochem 38:1851–1860
Prakongkep N, Suddhiprakam A, Kheoruenromne I, Smirk M, Gilkes RJ (2008) The geochemistry of Thai paddy soils. Geoderma 144:310–324
Rumpel C, Kogel-Knabner I (2011) Deep soil organic matter—a key but poorly understood component of terrestrial C cycle. Plant Soil 338:143–158
Shen JP, Zhang LM, Guo JF, Ray JL, He JZ (2010) Impact of long-term fertilization practices on the abundance and composition of soil bacterial communities in Northeast China. Appl Soil Ecol 46:119–124
Sheng R, Meng DL, Wu MN, Di HJ, Qin HL, Wei WX (2013) Effect of agricultural land use change on community composition of bacteria and ammonia oxidizers. J Soils Sediments 13:1246–1256
Shi XZ, Wang HJ, Yu DS, Weindorf David C, Cheng XF, Pan XZ, Sun WX, Chen JM (2009) Potential for soil carbon sequestration of eroded areas in subtropical China. Soil Till Res 105:322–327
Smith DB, Cannon WF, Woodruff LG (2011) A national-scale geochemical and mineralogical survey of soils of the conterminous United States. Appl Geochem 26:s250–s255
Steven B, Gallegos-Graves LV, Belnap J, Kuske CR (2013) Dryland soil microbial communities display spatial biogeographic patterns associated with soil depth and soil parent material. FEMS Microbiol Ecol 86:101–113
Suzuki MT, Taylor LT, DeLong EF (2000) Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′- nuclease assays. Appl Environ Microbiol 66:4605–4616
Treonis AM, Austin EE, Buyer JS, Maul JE, Spicer L, Zasada IA (2010) Effects of organic amendment and tillage on soil microorganisms and microfauna. Appl Soil Ecol 46:103–110
Tripathi BM, Kim M, Singh D, Lee-Cruz L, Lai-Hoe A, Ainuddin AN, Go R, Rahim RA, Husni MHA, Chun J, Adams JM (2012) Tropical soil bacterial communities in Malaysia: pH dominates in the Equatorial Tropics too. Microb Ecol 64:474–484
Ulrich A, Becker R (2006) Soil parent material is a key determinant of the bacterial community structure in arable soils. FEMS Microbiol Ecol 56:430–443
Wagai R, Kitayama K, Satomura T, Fujinuma R, Balser T (2011) Interactive influences of climate and parent material on soil microbial community structure in Bornean tropical forest ecosystems. Ecol Res 26:627–636
Will C, Thurmer A, Wollherr A, Nacke H, Herold N, Schrumpf M, Gutknecht J, Wubet T, Buscot F, Daniel R (2010) Horizon-specific bacterial community composition of german grassland soils, as revealed by pyrosequencing-based analysis of 16S rRNA genes. Appl Environ Microbiol 76:6751–6759
Woodruff LG, Cannon WF, Eberl DD, Smith DB, Kilburn JE, Horton JD, Garrett RG, Klassen RA (2009) Continental-scale patterns in soil geochemistry and mineralogy: results from two transects across the United State and Canada. Appl Geochem 24:1369–1381
Wu MN, Qin HL, Chen Z, Wu JS, Wei WX (2011) Effect of long-term fertilization on bacterial composition in rice paddy soil. Biol Fertil Soils 47:397–405
Xiang P, Huang H, Yan H, ZhouY ZH, Huang X (2005) Environmental cost of rice production in Dongting Lake area of Hunan Province. Chin J Appl Ecol 16:2187–2193
Acknowledgments
This work was supported by the Natural Science Foundation of China (41090282, 41330856) and the Chinese Academy (XDB15020200, 2012T1S0010).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Jizheng He
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Sample location map (JPEG 1248 kb)
Supplementary Fig. 2
Dendrogram showing the relationships between soils derived from latosol (LZ), quaternary red clays (TY-B and YT), and alluvium deposits (JX and TY-G) based on the results of T-RFLP analysis. Numbers 1, 2, and 3 following the capital letters (LZ–TY-G) represent sampling depths of 0–20, 20–40, and 40–60 cm, respectively (JPEG 631 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Sheng, R., Qin, H., O’Donnell, A.G. et al. Bacterial succession in paddy soils derived from different parent materials. J Soils Sediments 15, 982–992 (2015). https://doi.org/10.1007/s11368-014-1058-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-014-1058-2