
Abstract
Advanced age is associated with a decline in response to stress. This contributes to the establishment of chronic inflammation, one of the hallmarks of aging and age-related disease. Heat shock proteins (HSP) are determinants of life span, and their progressive malfunction leads to age-related pathology. To discuss the function of HSP on age-related chronic inflammation and illness. An updated review of literature and discussion of relevant work on the topic of HSP in normal aging and chronic inflammatory pathology was performed. HSP contribute to inflamm-aging. They also play a key role in age-associated pathology linked to chronic inflammation such as autoimmune disorders, neurological disease, cardiovascular disorder, and cancer. HSP may be targeted for control of their effects related to age and chronic inflammation. Research on HSP functions in age-linked chronic inflammatory disorders provides an opportunity to improve health span and delay age-related chronic disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aging is a complicated process not yet completely understood. Among many changes [1, 2], accumulation of random unrepaired molecular damage occurs over time. Caused by reactive oxygen species (ROS), as has been proposed by the free radical theory of aging [3], chronic oxidative stress eventually leads to tissue dysfunction, increasing frailty, and age-related disease. Heat shock proteins (HSP) are induced not only in response to thermal stress but also in response to a variety of stressors relevant to aging and life span [4]. When a cell encounters a stressor (e.g., infectious agents, radiation, intracellular stress, and stressing metabolic conditions) and molecular damage occurs, HSP contribute to its protection. This adaptive response, through maintenance of structural and functional integrity of client proteins (protein homeostasis), leads to anti-aging cellular effects [5].
HSP, however, have been involved in the pathogenesis of numerous diseases [6, 7]. Herein, the involvement of HSP in age-related chronic inflammatory disease is discussed. A special focus will be on the role of HSP, as components of the age milieu and their contribution to inflamm-aging, the chronic low grade of inflammation that also characterizes the aging process [8]. This approach is also relevant to understand HSP role in disease and their potential as therapeutic targets. The study of HSP functions in age-linked chronic inflammatory disorders provides an opportunity to extend life span and for design of forefront approaches to diagnose and treat age-related disease.
Age-related chronic inflammation
Chronic oxidative stress state and chronic low grade of inflammation are hallmarks of the free radical theory of aging [3] and inflamm-aging [8], respectively. Reference to the first concept was just brought up. In relation to the second point, first, it is important to refer to the immune system and its changes related to the aging process. Traditionally defined as a declining function of the immune system [9], immunosenescence refers to the effects of aging on adaptive and innate immunity [10,11,12,13]. Most recently, immunosenescence has been redefined: robust measures of immune parameters (biomarkers) are necessary to describe the state of immunosenescence in an individual [14]. To be in a state of good health, individuals must keep check of numerous components of immunity, including pro-inflammatory and anti-inflammatory mechanisms. It is clear that this capacity is lost in the elderly. An undisputed outcome of immunosenescence is higher incidence and mortality to infection, autoimmune diseases, cancer, and neurodegenerative diseases in the elderly population [9, 14].
In healthy subjects, advanced age is associated with a hyper inflammatory state [8]. Inflamm-aging is characterized by elevated circulating levels of pro-inflammatory mediators, such as the cytokines interleukin (IL)-6, IL-1β, tumor necrosis factor (TNF)-α, inflammatory mediator prostaglandin E2, and anti-inflammatory mediators, including IL-1 receptor antagonist, soluble TNF receptor, and acute phase proteins (including C-reactive protein and serum amyloid A) [8]. Many of these inflammatory mediators have been identified as predictors of all-cause mortality risk in longitudinal studies of several elderly cohorts (reviewed in [15, 16]) and are indicators of poor prognosis in age-related disorders [17,18,19,20,21]. Immunosenescence and inflamm-aging can be understood as highly intertwined processes. Immunosenescence is induced by inflamm-aging and vice versa [22]. Establishment of the senescence-associated secretory phenotype (SASP) is an example of this interaction. Reference to SASP is also important since it stems from complex microenvironmental interactions between different cellular and molecular players [10]. Unbalance in the interactions between factors of SASP leads to immunosenescence and inflamm-aging. SASP components include cytokines IL-6 and IL-8, growth factors, and proteases by senescent cells. These molecules are active modifiers of the microenvironmental milieu and promote senescence, chronic inflammation, and age-associated disease [23, 24].
A relatively understudied component of SASP are hormones [25]. In normal human liver cells, one function of the tumor suppressor p53 is to regulate expression of cytochrome P450 family 21 subfamily A polypeptide 2, corticosteroid-binding globulin, and sex hormone-binding globulin [26]. These proteins are relevant for the biological actions of steroid hormones and may have antitumor effects [26]. Due to the tumor suppressive role for sex hormone-related component of SASP [27], their role on immunosenescence needs to be established. The knowledge gained will provide understanding on the role of SASP and its components in modulating immunosenescence, inflamm-aging, and age-related pathology.
HSP and aging
Because of the aging process, cells undergo a decline in transcriptional pathways. Changes include a decreased ability of heat shock transcription factor (HSF1), master regulator of the heat shock response, to bind to HSP genes with stress [28]. Due in part to the concomitant decrease in HSP concentrations [29], the resulting impaired protein homeostasis of key clients contributes to the development of age-related pathology [4]. In the nematode Caenorhabditis elegans, HSP decreased cell death, age-related pathology, and enhanced life span. HSP suppression, by downregulation of HSF1, in turn shortened the life span [30, 31]. This response can be interpreted as a failed attempt of aging cells to maintain protein homeostasis.
Although the ability of HSF1 to bind to HSP genes decreases with age, there is evidence of age-related increased expression of HSP. For example, in Caenorhabditis elegans, the abundance of several human HSP homologs (e.g., HSP-16.48, HSP-43, HSP-17, and SIP-1) increased as nematodes aged [32]. Moreover, high transcript levels of HSP correlated with lower heat shock resistance, aberrant proteostasis, and shorter life span [32]. In Drosophila, aging led to the increase of HSPB8 and HSPA [33, 34]. Expression of these HSP predicted life span of adult flies during normal aging and following heat or oxidative stress [33]. Likewise, in Ames dwarf mice liver tissue, a decline of HSF1–DNA binding activity was accompanied by increase in expression of HSP, DNAJ, HSPA, and HSPC [35]. However, in Sprague–Dawley rats, a reduction of basal levels of HSPA was identified in post-mitotic tissues [36]. This decrease, leading to a reduction in basal levels of HSP, has been interpreted as a causal agent of tissue damage and promoter of age pathology. Similarly, HSPB1 increased in the brains of aged rhesus monkeys [37], possibly as a response to higher oxidative stress [37]. HSPB1, HSPA8, and HSPA rise significantly in late passage human senescent skin fibroblasts [38]. This elevation has been suggested as an adaptive response to cumulative intracellular stress through serial passaging [38]. Age-related surge in expression of HSP is therefore seen across the evolutionary scale. It has been postulated that the weakened heat shock response related to age could be traced back to HSF1–protein interactions and concomitant gain of HSP [35]. In this stage, there is a relevant role for the HSF1–DNA binding axis.
Changes in HSP levels in association with advanced age are a matter of controversy. A subject of discussion relates to the adaptation of cells to increased intracellular stress. Relevant to this point is to take into consideration the age-related decline in the ability of HSF1 to command the response to stress. Another intriguing variable to consider relates to the effect of repeated short periods of mild stress (hormesis). Known to stimulate the synthesis of HSP, hormesis improves cells functionality with no interfering with their replicative life span and promotes anti-aging mechanisms [38]. Although answering to these questions is not yet possible, some explanation for the observed discordances can be proposed. Many authors have suggested that discordances arise from the fact that HSP functions depend on the situation triggering their expression, as well as the compartment (intracellular vs. extracellular) in which HSP are present, and the context defined by a given physiological function [39, 40]. On this same regard, and especially relevant to the pro- versus anti-inflammatory role of HSP, other context-relevant factors are worth mentioning. Those include the concentration of HSP, the timing of exposure to HSP, and physio-pathological milieu at the target site [41]. Another explanation refers to the rate of aging of specific cell populations [42]. Post-mitotic cells and tissues age at a faster rate and are no longer able to synthesize HSP due to substantive damage accumulation [42]. This contributes to elimination of essential post-mitotic cells, and tissue deterioration [42]. On its side, accumulation of HSP in mitotic tissues, through blockage of apoptosis, could impair the elimination of damaged mitotic cells and contribute to age-related disease [42] (Table 1).
HSP and age-related chronic inflammation
To discuss the role of HSP on age-related inflammation, first is important to refer to their role in the inflammatory response. According to the danger theory, innate immune responses can be triggered by molecules released by stressed or damaged tissues, also referred to as danger-associated molecular pattern (DAMP) [43]. HSP were originally classified as DAMPs, but biological features of these proteins including lack of molecular patterns, downregulation of inflammatory signaling by some HSP receptors, and anti-inflammatory responses following immunization with HSP disqualify them as DAMPs [44, 45]. Several articles, in which the contaminants were effectively removed, namely, LPS, did not provide supportive evidence to a pro-inflammatory function for HSP [45]. These findings must call our attention to the role of HSP as DAMPs and their mechanistic involvement in inflammation in general. It seems that to dissect the reactivity of the immune system in conditions such as tissue damage, infection, or homeostasis, interactions to HSP in combination with DAMPs should be considered. Through CD14, and toll-like receptors (TLR), HSP regulate the expression of mediators of inflammation such as pro-inflammatory cytokines [46,47,48,49]. For example, in human monocytes, HSPD1 induced the secretion of IL-6, TNF-α, and granulocyte–macrophage colony-stimulating factor (GM-CSF) [50]. In another study, HSPA, through CD14 activation, stimulated expression of IL-1, IL-6, and TNF-α [51]. In general, evidence supports direct involvement of HSP in inflammation through signaling pathways involved in host response to pathogens, stressors, or physio-pathological processes.
An additional point worth discussion refers to the link between oxidative stress and inflammation. A relevant contribution to aging theory is the term oxi-inflamm-aging [52]. Among key arguments supporting this integrative theory of aging, researchers have identified a role for the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in aging immune cells [52]. Through its involvement on multiple aspects of innate and adaptive immune functions, NF-κB and members of its family of transcription factors serve as key regulators of the inflammatory response [53]. HSP regulate activation of NF-κB, particularly in immune cells. For example, in RAW 264.7 murine macrophages, NF-κB activation was attenuated by heat shock in a dose- and time-dependent manner [54]. Likewise, in human lymphoma cells, HSPA blocked nuclear translocation of NF-κB in response to inflammatory cytokines [55]. Similarly, in alveolar macrophages obtained from patients with active pulmonary tuberculosis, overexpression of HSPA decreased IκB-α phosphorylation, decreased activity the p65 subunit of NF-κB, and reduced TNF-α and IL-6 release [56]. Upregulated expression of HSP, via its regulatory effects on NF-κB, may suppress the effects of chronic inflammation and could be explored as an anti-aging alternative.
Immune cells, due to their capacity to produce oxidant and inflammatory compounds, lead to oxidative-inflammatory stress of the organism, which affects the rate of aging. HSP contribute to the balance between pro- and anti-inflammatory signals. Maintenance of this balance contributes to healthy age. If imbalance is maintained over time, a likely outcome is development and progression of disease. Having discussed these basic concepts, let us focus on specific examples related to the involvement of HSP on age-associated pathology linked to chronic inflammation.
Role of HSP in cardiovascular disease
Heat shock proteins have immunomodulatory roles, including their participation in resolution of inflammatory responses. They are also a regulatory component of age-related chronic inflammation. Because of their critically relevant involvement in these processes, functional or dysfunctional HSP have been linked to numerous age-related maladies [57]. Atherosclerosis is a chronic cardiovascular condition, characterized by progressive accumulation of inflammatory scar tissue within the walls of arteries. Among clinical manifestations of atherosclerosis, cardiovascular disease (CVD) remains the leading cause of mortality within the elderly and burdens the health system with high socioeconomic costs. Not only originated from a lipid imbalance, atheroma formation includes a complex set of factors. Among them, HSP and their immunomodulatory effects contribute with inflammatory processes relevant to the development of atherosclerotic plaques [58]. Extracellular HSP, activated in the heart in response to numerous physiological or pathological stresses, seem cardio-protective [59]. Numerous HSP [i.e., HSPC, HSPA, HSPD1, and αB-crystallin (HSPB5)] have been identified as cardiokines, a term used to define a number of proteins secreted from the heart with profound local and global effects [60]. Through their regulatory effects on expression of diverse cell types including cardiomyocytes, neurons, monocytes, macrophages, and endothelial cells, HSP exert regulatory influence on the inflammatory response, thought to be one of the chief mechanisms in the process of atherosclerosis [61].
Since atherosclerosis is primarily a chronic inflammatory disease, the involvement of HSP is of special relevance. HSPB1 seems to have a cardio-protective role as lower levels of this HSP in atherosclerotic coronary arteries were inversely correlated with increasing plaque progression and age [62,63,64,65]. Patients with high incidence of stenosis had lower HSPB1 compared to those free of atherosclerosis [62]. Moreover, high serum HSPB1 levels predicted for better cardiovascular outcomes [62]. In support of the potential of HSPB1 as a therapeutic target for atherosclerosis, in an experimental mouse model of atherosclerosis, exogenous administration of HSPB1 levels reduced lesion progression and promoted features of plaque stability [62]. HSPB1 may also serve as a marker of age. A prospective, nested, case-controlled study of healthy participants of the Women’s Health Study who subsequently developed myocardial infarction, ischemic stroke, or cardiovascular death was established [64]. Baseline HSPB1 plasma levels were inversely associated with age, but not with other established cardiovascular risk factors or with development of future CVD [64]. Limitations of the study included a female only population, relatively advanced age of participants, and a potential moderately advanced stage of coronary atherogenesis.
The aforementioned findings should encourage researchers to further study the potential for HSP as markers of the rate of aging, life span, and age-related pathology linked to chronic inflammation. As has been demonstrated for other HSP (e.g., HSPA) [42], this is an area of active investigation and promising prospects. Recent findings show that chronic whole-body heat treatment led to a robust expression of HSP, relieved atherosclerotic lesions, cardiometabolic abnormalities, and enhanced survival through the life span [66]. These findings were accompanied by replacement of SASP by an anti-inflammatory response related to the Sirtuin (SIRT1)-HSF1-HSP molecular axis [66]. Interventions restarting the anti-inflammatory response of HSP may therefore have a strong curative effect on age-related atherosclerotic lesions, via restoration of immunoinflammatory balance in blood vessels.
Not only HSPB1 displays potential as an anti-atherosclerosis factor. Extracellular HSPB6 was secreted from adult rat cardiomyocytes through exosomes [67], which are vesicular structures. Using this route of secretion, HSPB6 exerted a wide range of inflammatory or immunosuppressive effects [68] related to proliferation, migration, and tube formation on human umbilical vein endothelial cells. These effects were mediated by activation of the receptor for the angiogenic vascular endothelial growth factor (VEGF). Similarly, in vivo, relative to non-transgenic hearts, capillary density was significantly enhanced in HSPB6-overexpressing hearts [67]. These findings need careful consideration because angiogenesis of capillaries may destabilize atherosclerotic plaques, which leads to plaque rupture and acute coronary syndrome. Similarly, extracellular HSPB1 levels were elevated in coronary blood effluents of patients or mouse hearts undergoing global ischemia [69]. Neutralizing antibodies against HSPB1 suppressed myocardial nuclear factor NF-κB activation, reduced IL-6 production, and improved cardiac function in mouse hearts [69]. TLR2 knockout or TLR4 mutation abolished NF-κB phosphorylation and reduced production of pro-inflammatory mediators induced by extracellular HSPB1 in human coronary vascular endothelial cells [69]. Over the basis of these findings, it has been proposed that extracellular HSPB1 plays a role in mediating post ischemic myocardial inflammatory response and cardiac dysfunction. In this case, a potential therapeutic avenue to treat patients undergoing cardiac surgery with obligatory global ischemia could be based on target suppression of myocardial inflammatory responses through blockage of extracellular HSPB1.
HSP in neuroinflammation
Neuroinflammation is a known feature of different acute (ischemic stroke and traumatic brain injury) and chronic neurodegenerative diseases (Alzheimer’s (AD) and Parkinson’s (PD)). Inflammation within the brain or spinal cord may be initiated in response to a variety of factors, including infection, traumatic brain injury, toxic metabolites, or autoimmunity. Increase in the incidence of neurodegenerative diseases, due in part to the aging population [70], is a prevalent problem of public health. Neuroinflammation also provides a relevant scenario for discussion of the involvement of HSP in age-related pathology [71, 72]. In the elderly, inflamm-aging is one of the contributors to neurodegenerative diseases. Age-related chronic inflammation modulates central neuronal immune cell activity and reactivity [73]. Unsurprisingly, the term neuro-inflamm-aging alludes to this phenomenon [74]. HSP have gained recognition in terms to their role in the development of neuroinflammation based on their ability to regulate proteostasis, modulate neuronal survival, and because of their immunomodulatory properties [75].
Many age-related neurodegenerative disorders are characterized by accumulation of toxic protein aggregates in neurons. Proteins such as Aβ and tau in AD, α-synuclein in PD, or huntingtin in Huntington’s disease are landmarks of these diseases. These toxic protein aggregates are not cleared in the elderly as neurodegenerative diseases progress, contribute to immunosenescence on microglia and macrophages, and can initiate a chronic inflammatory response [74]. The accumulation of misfolded proteins increases constantly in the brain during aging and may conduce to the elevation of the basal HSP as has been reported in aging rodents [76,77,78,79]. This information, however, must be carefully interpreted because a downregulation of HSPB1 has also been described in brains of old mice [77]. Despite this controversy, it may be postulated that HSP signaling is involved in brain aging. As such, modulation of HSP levels may serve as a strategy to delay onset of central nervous system disease.
Acute brain injuries such as ischemic stroke [80] and ischemia/reperfusion injury [81] modify the expression of HSP. Likewise, altered expressions of HSP have been detected in the brains of patients with chronic inflammatory diseases such as AD and PD [82,83,84]. Primarily, through their proteostatic and immunomodulatory activities, HSP would be neuroprotective. The neuroprotective roles of HSP include effects on the pro-inflammatory environment and reduction of protein aggregation associated with neurological diseases. For example, in microglial cells, HSPB1 inhibited the activation of NF-κB and reduced expression of TNF-α [85]. Similarly, in astroglial cell cultures, treatment with lipopolysaccharide and interferon gamma was associated with an increased synthesis of HSPA [86]. These findings imply that HSP, after being taken up by brain cells, would confer resistance to development of inflammatory pathology through their influence of immune function.
Multiple sclerosis (MS) is a multifocal inflammatory disease with autoimmune etiology affecting the CNS. In this disease, the target is myelin and the myelin-producing cell, the oligodendrocyte. A pathologic hallmark of MS is presence of inflammatory demyelinating lesions [87]. Another salient feature of this disease is high levels of HSPB5 in pre-active lesions of stressed oligodendrocytes [88]. HSPB5, produced by oligodendrocytes, activates regulatory microglial responses during MS [89]. A comprehensive gene expression analysis of HSPB5-induced transcript changes showed that human microglia expressed immune-regulatory mediators, known to suppress inflammation and to promote immune tolerance and tissue repair [89]. Additional evidence in support of the protective role of anti-HSP immune responses in MS was suggested by a study showing HSP antibodies in sera of relapsing MS and not in progressive MS [90]. Similar findings were reported in juvenile RA. In this case, only the relapsing disease featured HSP T cell responses (e.g., juvenile chronic arthritis: T cell reactivity to human HSP60 in patients with a favorable course of arthritis) [91]. Over the basis of these findings, reactive MS lesions exhibit a beneficial anti-inflammatory response, which can be interpreted as an effort to restore tissue homeostasis.
Presented evidence in animal models demonstrates that HSP regulate the inflammatory response in the brain. Indeed, data suggests that HSP are a basic mechanism of defense when brain neurons are exposed to harmful insults [92]. If the pro- or anti-inflammatory effects of HSP will protect neurons from age-related neurodegeneration will depend on many factors inherent to the biology of these proteins. Relevant to neurodegenerative disease are variables such as secretion of HSP to the extracellular compartment, different cell surface receptors, their combination present on the surface of brain cells, beneficial versus harmful outcomes of the inflammatory process, and specific effects of intra- and extracellular HSP [93]. Other points to consider refer to whether HSP attenuate cerebral injury when applied post-insult, the limit of their protection, and how general effects are when different neurodegenerative diseases are taken into consideration. Additional research will shed light on the protective role of HSP in controlling excessive protein aggregation, neuroinflammation, and neuron survival during brain aging and in neurodegenerative disease.
HSP and autoimmunity
Overexpression of HSP in inflamed tissues and their presence in the extracellular space are features relevant to their immunogenicity [94]. In fact, exported HSP are highly immunogenic in nature, and the immune response to these proteins has been observed in various inflammatory and autoimmune diseases [95]. HSP mediate autoimmunity by their ability to stimulate various immunocytes, in particular regulatory T cells that act to suppress immune response [96, 97]. HSP also promote autoimmunity by driving peptide-specific immune responses [98]. Additionally, HSP can generate antigen-specific T cell responses [99]. In this last case, peptides including HSP are internalized, proceeded, and presented by antigen-presenting cells to the major histocompatibility complex.
Whether upregulation of HSP in chronically inflamed tissues represents a beneficial response to disease or plays a role in pathogenesis of autoimmune diseases remains unclear [100]. Multiple sclerosis is considered a standard model of an autoimmune disease with involvement of chronic inflammation, and HSP. Due to destruction of myelin, there is accumulation of antibodies to this protein along with accumulation of immune cells, and immune mediators [101]. The inflammatory and oxidative environment leads to overexpression of HSP [101]. On one hand, HSP, as has been observed for HSPA, through its binding to immunogenic peptides, promote local inflammation and autoimmunity. However, HSPA can prevent accumulation of protein aggregates [102], act as an anti-apoptotic factor [103], and halt neurodegeneration and autoimmunity. In this case, HSPA protects glial cells and neurons of the CNS against the effects of inflammation and subsequent neurodegeneration. Over the basis of these findings, HSP may be proposed as a tool to combat the development of MS. However, this approach should be carefully considered due to the role of HSP on autoimmune reactions, a characteristic of this disease.
Regardless of the exposed limitations relevant to the treatment of MS, capability of HSP to target antigen-presenting cells and elicit cross-presentation can be exploited as a strategy to downregulate inflammation and autoimmunity. Active immunization with full-length HSP suppressed inflammation [97, 104, 105]. This evidence let researchers to propose that HSP could be considered as components of vaccines to prevent or suppress autoimmune diseases [39, 106]. In support of this idea, immunization with HSPD1 was effective in preventing inflammation in pre-clinical models of autoimmune diseases such as arthritis [97, 107,108,109], type I diabetes [110], atherosclerosis [96, 97], and autoimmune-like psoriasis [106]. Auspicious results of active immunization using HSP have also been observed in rheumatoid arthritis patients [111]. Over the basis of these findings, research has been stimulated into the development of therapeutic HSP-based peptide vaccines for the restoration of immune tolerance in inflammatory diseases.
Another point relevant to this discussion relates to the ability of HSP to trigger immune responses leading to the production of autoantibodies. HSP are released from tissues in patients with inflammatory diseases or autoimmune disorders. This stimulates humoral responses and the production of anti-HSP autoantibodies and autoreactive cells against HSP [112]. In general, presence of autoantibodies to human antigens from organs and tissues is strongly influenced by diverse variables including age, gender, and disease [113]. For autoantibodies found elevated in the elderly, their presence seems to be an adaptive response to damaged tissue and high exposure to apoptotic cells rather than an autoimmune response [114]. Anti-HSP autoantibodies have been detected in the serum of healthy individuals [115, 116]. Regarding HSP, levels of HSPD1 and HSPA declined with age in the serum of elderly subjects, and no significant age-related change was observed for HSPD1 or HSP65 autoantibody levels [117]. Anti-HSPA antibodies, however, tended to increase with age [117]. To explain some of these discordances, it has been proposed that age-related increased immunogenicity to HSP arises from the reactivity of the immune system not only to autologous HSP but also to homologs from pathogen sources known to be persistently expressed in infections in aged patients. In addition, post-translation modifications resulting from the age process would make HSP immunogenic, thus stimulating autoantibody production. As a result, HSP immunogenicity will increase with advanced age [118].
Although autoantibodies against HSP have been identified in various autoimmune diseases [106], their physio-pathological role and value for predicting disease progression is a matter of debate. Autoantibodies against HSP, in part by reduction of expression of pro-inflammatory mediators and stimulation of anti-inflammatory mediators, were protective against progression of arthritis [119]. However, increased levels of autoantibodies to HSPD1, HSPA, and HSPC were not predictive of disease progression in rheumatoid arthritis patients [120]. Surprisingly, the authors of this study found that serum levels of autoantibodies against HSPA inversely correlated with serum levels of TNF-α [120]. Much like HSP, autoantibodies against HSP contribute to the regulation of immune responses. Critical questions needing answers in terms of the role of anti-HSP autoantibodies include elucidation of their participation as determinants of disease immunogenicity, or their ability to induce adaptive responses. Another point of discussion pertains to the upstream age-specific mediators that initiate the cascade of events that lead to immunosuppression in context of anti-HSP autoantibodies. It can be argued that disease-specific-associated autoantibodies against HSP may be an important point to consider for development of more effective individualized prognostication and allogeneic cell-based immunotherapy for age-related disease. Use of HSP as markers and application of HSP as vaccines must take into consideration their heterogeneity, immune escape roles of HSP, as well as their involvement in aspects related to chronic inflammation.
HSP and cancer
Genetic, environmental, and life style factors involved in the initiation, progression, and metastasis of cancer drive stress responses. Sources include those from intracellular (e.g., overexpression of oncoproteins, altered cellular metabolism, aneuploidy, and genomic instability) or external (e.g., microenvironment) origin. A good matter for discussion relates to the contributing roles of external sources of stress. The tumor microenvironment (TME) is defined by the interactions between cancer cells, local components of the tumor stroma, immunocytes, and exposure to extracellular metabolic conditions [24, 121,122,123]. The interactions occuring within the TME determine tumor growth, invasion, and metastasis [124,125,126], contribute to the natural selection of aggressive clones, and drive tumor establishment and progression.
The heat shock response contributes to cancer development [100]. As proof of that, intracellular HSP, though to originate from folding stress of key client proteins during malignant transformation, are abundantly expressed in most of cancer cells and promote their growth and survival [127,128,129]. Additionally, HSP have been detected in the serum of patients with several types of cancer [130,131,132,133,134,135,136]. This evidence, on one side, illustrates the high level of dependency of cancer cells on a large supply of HSP. In fact, cancer cells have been considered “addicted to chaperones,” as a stable increase in HSP allows for oncogenic protein accumulation and tumor progression [137]. On the other side, once more, the presented evidence underscores the relevance of HSP localization on cancer etiology. This point deserves more discussion, on light of evidence showing that HSP accumulate at the surface of tumor cells, where they can also affect the oncogenic process [138].
Through their modulatory actions, HSP contribute to the survival of cancer cells [139]. HSP regulate apoptotic cell death induced during normal processes such as cell division and cellular aging. HSP also determine the fate of tumor cells in response to cellular damage and oncogenic development. HSP are engaged in apoptosis and cellular senescence [127]. For example, relative to young individuals, mitotic tissues in the elderly have higher basal levels of HSPA [42]. Due to blockage of apoptosis, the consequent division of damaged cells can contribute to cancer. On the contrary, post-mitotic tissues in the elderly tissues experience a reduction of HSPA basal levels [42]. The effects in this case are also deleterious as essential post-mitotic cells are eliminated, which leads to tissue deterioration. The mechanisms leading to cell survival mediated by HSP relate to their known functional properties. For example, through its chaperoning property, HSPD1 directly binds and stabilizes survivin, an anti-apoptotic protein [140]. Through a similar mechanism, HSPD1 stabilizes the anti-apoptotic mediator clusterin [141]. Oncogenic roles of HSP are also related to their ability to form multiprotein complexes, which in turn override cell death mechanisms such as those involved in cell cycle checkpoints [140]. Of special relevance due to its involvement not only in apoptosis but also in the process of cellular senescence is the formation of a complex between HSP and tumor protein P53 [139, 140].
Independent of their anti-apoptotic role, HSP promote tumorigenesis by regulating cell signaling related to the generation of metabolites. Examples include effects on mitochondrial ATP [142], and production of ROS. Reactive oxygen species contribute to tumor progression through accumulation of cellular damage [143]. HSP not only drive tumor progression signaling but also stimulate tumor suppression [144], cancer cell migration, invasion, and metastasis [145]. The involvement of HSP as tumor survival factors remains a matter of active discussion, as it has been discovered that they can act as pro-apoptotic mediators. Through their ability to bind proteins involved in the apoptotic pathway [146], or by activation of apoptotic mediators [147], HSP may promote cell death in tumor cells. Thus, through their pro- or anti-apoptotic properties, HSP contribute to the survival of cancer cells and to tumor progression. In general, HSP are considered as oncoproteins. However, more research is needed to exactly establish their role in oncogenesis.
HSP and tumor-associated inflammation contribute to the manifestation of many hallmarks of cancer [148]. It is generally understood that chronic inflammation causes tissue injury, which in turn promotes tissue proliferation. Oxi-inflamm-aging, through its effects on cancer cells and other components of the TME such as stromal fibroblasts, immunocytes, and metabolites, causes damage to normal cells, leads to DNA damage and mutation, increases expression of HSP, and promotes tumor progression. On this regard, a role for NF-κB signaling has been identified as key in the modulation of the pro-inflammatory status by intracellular HSP in tumor cells. Through activation by direct interaction and modulation of serine-dependent phosphorylation of IκB kinase, cytosolic HSPD1 mediated enhanced NF-κB in cervical adenocarcinoma HeLa S3 cells [143]. Similarly, at a steady state, the B16 murine melanoma cell line constitutively released HSPD1 to the extracellular space. These cells had additionally increased expression of numerous pro- and anti-inflammatory cytokines, chemokines, and chemokine receptors through signaling mediated by TLR2 and activation of the transcription factor signal transducer and activator of transcription 3 (STAT3) [149]. Consistent with the involvement of HSP as a factor of tumor aggressiveness via persistent activation of mediators in inflammation, B16 cells, which are highly metastatic, had higher expression of TLR2 and had elevated baseline activation of STAT3, relative to low metastatic B16-F1 cells [149]. These findings provide evidence to the role of HSP in promoting a TME supportive of a pro-inflammatory milieu and tumor progression.
As a result of aging, HSP levels, antigenicity, and the normal tissue milieu are altered. These conditions will set the stage for the onset of chronic inflammation and tumor progression. This article referred to the roles of HSP in the regulation of intracellular signaling involved in inflammation. In addition, one should also consider HSP’s immune functions related to many aspects of malignant progression such as cell to cell communication [150, 151], antigenic presentation [152], and immunoescape [153]. Understanding HSP modes of action in oncogenic transformation, particularly in tumor inflammation, will enhance their therapeutic potential.
Conclusions
Through the common effort to address the role of aging as a major contributing factor for most chronic diseases, the field of geroscience has recently emerged. Defined as an interdisciplinary arena that aims to understand the relationship between aging and age-related diseases and disabilities [154], a priority of geroscience is addressing chronic diseases [155]. By increasing the understanding of aging biology, novel forms to improve health span and delay age-related chronic diseases will emerge. Among the main pillars of geroscience research, inflammation, responsiveness to stress, epigenetics, metabolism, macromolecular damage, proteostasis, and stem cells are proposed as drivers of the aging process [154, 155]. Advances in knowledge of the contributing role of HSP to chronic inflammation and age-related pathology are consistent with their involvement as mechanistic drivers of aging.
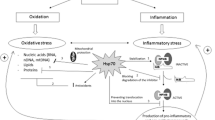
Because of their potential to favorably alter the onset or progression of multiple chronic diseases affecting the elderly, HSP are worth considering as therapeutic agents by geroscientists. As we have discussed, HSP are involved in many cellular processes related to the physiopathology of CVD, neurodegeneration, autoimmunity, and cancer (Fig. 1). Indeed, understanding of the fundamental mechanisms by which HSP contribute to aging has led to significant advancements in therapeutic interventions of these diseases. Life style interventions such as aerobic exercise and caloric restriction have proven successful in reducing age-related pathology, though increase in antioxidant effects, as well as counteracting the effects of age on the expression of HSP [36, 156, 157]. Likewise, pharmacological agents may help to reduce the effects of age on accumulation of defective and toxic proteins, with concomitant effects on the life span. Among them, plant medicinal compounds such as salicylate, resveratrol, curcumin, and celastrol activate HSF1, and its stress signaling cascade [158, 159] to promote salutary effects on aging, chronic inflammatory diseases, and neurodegeneration among other diseases [159, 160]. Similar effects have been observed when several small molecules, known to induce HSP, have been utilized in experimental models [161,162,163], and in a good number of clinical studies [164]. For instance, some small molecule compounds including those competitively inhibiting ATP hydrolysis of HSP90 and subsequent maturation of client proteins have been tested in phase I or II clinical trials for diverse types of cancer [128, 164, 165]. Versatility of interaction between some HSP, leading to formation of oligomeric species, has been exploited for development of small compounds with ability to target various age-related diseases in clinical trials for CVD [166, 167], neurodegeneration [75, 168, 169], and autoimmune diseases [95, 106]. In many of these studies, the therapeutic response was associated to production of anti-inflammatory mediators and decreased expression of inflammatory mediators [97, 170,171,172,173].

HSP in aging and chronic inflammatory diseases. Age leads to unbalance of cellular responses to stress mediated by HSP. This contributes to the establishment of chronic inflammation (inflamm-aging) and to age-associated pathology linked to chronic inflammation. Life style or pharmacological interventions aimed at restoring HSP functions provide an opportunity for improving outcomes in life span and various pathologies
HSP thus effectively induce immunoregulation and suppress disease in clinical trials. These effects are in part due to shift from a pro-inflammatory state to an anti-inflammatory state, which sets the stage for regulatory adaptive immune responses. Over the basis of these observations, it can be proposed that HSP have a crucial role in disease-protective immunoregulation [97]. A general principle also emerges from these studies: The reduced capacity to maintain HSP homeostasis with advanced age contributes to a decreasing resistance to chronic diseases. On the contrary, restoring HSP expression will likely contribute to effective HSP-mediated immunoregulation. As scientists [57] have suggested, fine adjustment of HSP-related therapy including refinement related to overcoming issues related to disease-specific expression of HSP vs. its ubiquitous expression in non-disease tissues, off target effects on client proteins, and selectivity of targeted downstream pathways are issues relevant to the observed toxicity of current HSP inhibitors in clinical trials. Answer to these questions will improve the success of therapy based on HSP as reflected by the concomitant beneficial outcomes in life span and various pathologies.
Data availability
All utilized data is publicly available.
Code availability
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- CVD:
-
Cardiovascular disease
- DAMP:
-
Danger-associated molecular pattern
- HSF1:
-
Heat shock transcription factor
- HSP:
-
Heat shock proteins
- HSPB5:
-
αB-crystallin
- IL:
-
Interleukin
- MS:
-
Multiple sclerosis
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- PD:
-
Parkinson’s disease
- ROS:
-
Reactive oxygen species
- SASP:
-
Senescence-associated secretory phenotype
- STAT3:
-
Signal transducer and activator of transcription 3
- TLR:
-
Toll-like receptors
- TME:
-
Tumor microenvironment
- TNF:
-
Tumor necrosis factor
References
Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120:437–47. https://doi.org/10.1016/j.cell.2005.01.027.
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039.
Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. https://doi.org/10.1093/geronj/11.3.298.
Murshid A, Eguchi T, Calderwood SK. Stress proteins in aging and life span. Int J Hyperthermia. 2013;29:442–7. https://doi.org/10.3109/02656736.2013.798873.
Rattan SI, Eskildsen-Helmond YE, Beedholm R. Molecular mechanisms of anti-aging hormetic effects of mild heat stress on human cells. Nonlinearity Biol Toxicol Med. 2004;2:105–16. https://doi.org/10.1080/15401420490464376.
Cappello F, Marino Gammazza A, Palumbo Piccionello A, Campanella C, Pace A, Conway de Macario E, Macario AJ. Hsp60 chaperonopathies and chaperonotherapy: targets and agents. Expert Opin Ther Targets. 2014;18:185–208. https://doi.org/10.1517/14728222.2014.856417.
Kakkar V, Meister-Broekema M, Minoia M, Carra S, Kampinga HH. Barcoding heat shock proteins to human diseases: looking beyond the heat shock response. Dis Model Mech. 2014;7:421–34. https://doi.org/10.1242/dmm.014563.
Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–54.
Stahl EC, Brown BN. Cell therapy strategies to combat immunosenescence. Organogenesis. 2015;11:159–72. https://doi.org/10.1080/15476278.2015.1120046.
Gomez CR, Boehmer ED, Kovacs EJ. The aging innate immune system. Curr Opin Immunol. 2005;17:457–62. https://doi.org/10.1016/j.coi.2005.07.013.
DeVeale B, Brummel T, Seroude L. Immunity and aging: the enemy within? Aging Cell. 2004;3:195–208.
Pawelec G. Hallmarks of human “immunosenescence”: adaptation or dysregulation? Immun Ageing. 2012;9:15. https://doi.org/10.1186/1742-4933-9-15.
Montgomery RR, Shaw AC. Paradoxical changes in innate immunity in aging: recent progress and new directions. J Leukoc Biol. 2015;98:937–43. https://doi.org/10.1189/jlb.5MR0315-104R.
G Pawelec Does the human immune system ever really become “senescent”? F1000Res 2017 6 https://doi.org/10.12688/f1000research.11297.1
Krabbe KS, Pedersen M, Bruunsgaard H. Inflammatory mediators in the elderly. Exp Gerontol. 2004;39:687–99.
Piber D, Olmstead R, Cho JH, Witarama T, Perez C, Dietz N, Seeman TE, Breen EC, Cole SW, Irwin MR. Inflammaging: age and systemic, cellular, and nuclear inflammatory biology in older adults. J Gerontol A Biol Sci Med Sci. 2019;74:1716–24. https://doi.org/10.1093/gerona/glz130.
Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation. 2003;107:391–7. https://doi.org/10.1161/01.cir.0000055014.62083.05.
Vasto S, Carruba G, Lio D, Colonna-Romano G, Di Bona D, Candore G, Caruso C. Inflammation, ageing and cancer. Mech Ageing Dev. 2009;130:40–5. https://doi.org/10.1016/j.mad.2008.06.003.
Pierce BL, Ballard-Barbash R, Bernstein L, Baumgartner RN, Neuhouser ML, Wener MH, Baumgartner KB, Gilliland FD, Sorensen BE, McTiernan A, Ulrich CM. Elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients. J Clin Oncol. 2009;27:3437–44. https://doi.org/10.1200/JCO.2008.18.9068.
Singh T, Newman AB. Inflammatory markers in population studies of aging. Ageing Res Rev. 2011;10:319–29. https://doi.org/10.1016/j.arr.2010.11.002.
Pansarasa O, Pistono C, Davin A, Bordoni M, Mimmi MC, Guaita A, Cereda C. Altered immune system in frailty: genetics and diet may influence inflammation. Ageing Res Rev. 2019;54:100935. https://doi.org/10.1016/j.arr.2019.100935.
Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, Witkowski JM, Franceschi C. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol. 2017;8:1960. https://doi.org/10.3389/fimmu.2017.01960.
Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d’Adda di Fagagna F, Bernard D, Hernando E, Gil J. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. https://doi.org/10.1016/j.cell.2008.03.038.
Elkhattouti A, Hassan M, Gomez CR. Stromal fibroblast in age-related cancer: role in tumorigenesis and potential as novel therapeutic target. Front Oncol. 2015;5:158. https://doi.org/10.3389/fonc.2015.00158.
Chesnokova V, Zhou C, Ben-Shlomo A, Zonis S, Tani Y, Ren SG, Melmed S. Growth hormone is a cellular senescence target in pituitary and nonpituitary cells. Proc Natl Acad Sci U S A. 2013;110:E3331-3339. https://doi.org/10.1073/pnas.1310589110.
Charni M, Molchadsky A, Goldstein I, Solomon H, Tal P, Goldfinger N, Yang P, Porat Z, Lozano G, Rotter V. Novel p53 target genes secreted by the liver are involved in non-cell-autonomous regulation. Cell Death Differ. 2016;23:509–20. https://doi.org/10.1038/cdd.2015.119.
Gomez CR, Nomellini V, Kovacs EJ (2018) Sex hormones and immunosenescence. In: Handbook of Immunosenescence. pp 1–58. https://doi.org/10.1007/978-3-319-64597-1_42-1
Barna J, Csermely P, Vellai T. Roles of heat shock factor 1 beyond the heat shock response. Cell Mol Life Sci. 2018;75:2897–916. https://doi.org/10.1007/s00018-018-2836-6.
Hou Y, Wei H, Luo Y, Liu G. Modulating expression of brain heat shock proteins by estrogen in ovariectomized mice model of aging. Exp Gerontol. 2010;45:323–30. https://doi.org/10.1016/j.exger.2009.10.006.
Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell. 2004;15:657–64. https://doi.org/10.1091/mbc.e03-07-0532.
Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A. 2009;106:14914–9. https://doi.org/10.1073/pnas.0902882106.
Maniere X, Krisko A, Pellay FX, Di Meglio JM, Hersen P, Matic I. High transcript levels of heat-shock genes are associated with shorter lifespan of Caenorhabditis elegans. Exp Gerontol. 2014;60:12–7. https://doi.org/10.1016/j.exger.2014.09.005.
Yang J, Tower J. Expression of hsp22 and hsp70 transgenes is partially predictive of drosophila survival under normal and stress conditions. J Gerontol A Biol Sci Med Sci. 2009;64:828–38. https://doi.org/10.1093/gerona/glp054.
King V, Tower J. Aging-specific expression of Drosophila hsp22. Dev Biol. 1999;207:107–18. https://doi.org/10.1006/dbio.1998.9147.
Jurivich DA, Manocha GD, Trivedi R, Lizakowski M, Rakoczy S, Brown-Borg H. Multifactorial attenuation of the murine heat shock response with age. J Gerontol A Biol Sci Med Sci. 2020;25:1846–52. https://doi.org/10.1093/gerona/glz204.
Colotti C, Cavallini G, Vitale RL, Donati A, Maltinti M, Del Ry S, Bergamini E, Giannessi D. Effects of aging and anti-aging caloric restrictions on carbonyl and heat shock protein levels and expression. Biogerontology. 2005;6:397–406. https://doi.org/10.1007/s10522-005-4906-z.
Schultz C, Dick EJ, Cox AB, Hubbard GB, Braak E, Braak H. Expression of stress proteins alpha B-crystallin, ubiquitin, and hsp27 in pallido-nigral spheroids of aged rhesus monkeys. Neurobiol Aging. 2001;22:677–82. https://doi.org/10.1016/s0197-4580(01)00229-9.
Fonager J, Beedholm R, Clark BF, Rattan SI. Mild stress-induced stimulation of heat-shock protein synthesis and improved functional ability of human fibroblasts undergoing aging in vitro. Exp Gerontol. 2002;37:1223–8. https://doi.org/10.1016/s0531-5565(02)00128-6.
Gomez CR (2019) Hsp60 in cancer immunity: biological basis, diagnostic potential and therapeutic opportunities. In: Heat Shock Protein 60 in Human Diseases and Disorders. Heat Shock Proteins. pp 117–134. https://doi.org/10.1007/978-3-030-23154-5_9
Cappello F, Conway de Macario E, Rappa F, Zummo G, Macario AJL. Immunohistochemistry of human Hsp60 in health and disease: from autoimmunity to cancer. Methods Mol Biol. 2018;1709:293–305. https://doi.org/10.1007/978-1-4939-7477-1_21.
Moudgil KD, Thompson SJ, Geraci F, De Paepe B, Shoenfeld Y. Heat-shock proteins in autoimmunity Autoimmune Dis. 2013;2013:621417. https://doi.org/10.1155/2013/621417.
Martinez de Toda I, De la Fuente M. The role of Hsp70 in oxi-inflamm-aging and its use as a potential biomarker of lifespan. Biogerontology. 2015; 16:709-721. https://doi.org/10.1007/s10522-015-9607-7
Matzinger P. An innate sense of danger. Semin Immunol. 1998;10:399–415. https://doi.org/10.1006/smim.1998.0143.
Broere F, van der Zee R, van Eden W. Heat shock proteins are no DAMPs, rather ‘DAMPERs.’ Nat Rev Immunol. 2011;11:565. https://doi.org/10.1038/nri2873-c1.
van Eden W, Spiering R, Broere F, van der Zee R. A case of mistaken identity: HSPs are no DAMPs but DAMPERs. Cell Stress Chaperones. 2012;17:281–92. https://doi.org/10.1007/s12192-011-0311-5.
Giuliano JS Jr, Lahni PM, Wong HR, Wheeler DS. Pediatric sepsis–part V: extracellular heat shock proteins: alarmins for the host immune system. Open Inflamm J. 2011;4:49–60. https://doi.org/10.2174/1875041901104010049.
Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, Wagner H. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. 2001;276:31332–9. https://doi.org/10.1074/jbc.M103217200.
Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–42. https://doi.org/10.1038/74697.
Coelho V, Broere F, Binder RJ, Shoenfeld Y, Moudgil KD. Heat-shock proteins: inflammatory versus regulatory attributes. Cell Stress Chaperones. 2008;13:119–25. https://doi.org/10.1007/s12192-008-0018-4.
Galdiero M, de l’Ero GC, Marcatili A. Cytokine and adhesion molecule expression in human monocytes and endothelial cells stimulated with bacterial heat shock proteins. Infect Immun. 1997;65:699–707.
Dubey A, Prajapati KS, Swamy M, Pachauri V. Heat shock proteins: a therapeutic target worth to consider. Vet World. 2015;8:46–51. https://doi.org/10.14202/vetworld.2015.46-51.
De la Fuente M, Miquel J. An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr Pharm Des. 2009;15:3003–26. https://doi.org/10.2174/138161209789058110.
Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017; 2. https://doi.org/10.1038/sigtrans.2017.23
Schell MT, Spitzer AL, Johnson JA, Lee D, Harris HW. Heat shock inhibits NF-kB activation in a dose- and time-dependent manner. J Surg Res. 2005;129:90–3. https://doi.org/10.1016/j.jss.2005.05.025.
Guzhova IV, Darieva ZA, Melo AR, Margulis BA. Major stress protein Hsp70 interacts with NF-kB regulatory complex in human T-lymphoma cells. Cell Stress Chaperones. 1997;2:132–9. https://doi.org/10.1379/1466-1268(1997)002%3c0132:msphiw%3e2.3.co;2.
Wang CH, Chou PC, Chung FT, Lin HC, Huang KH, Kuo HP. Heat shock protein70 is implicated in modulating NF-kappaB activation in alveolar macrophages of patients with active pulmonary tuberculosis. Sci Rep. 2017;7:1214. https://doi.org/10.1038/s41598-017-01405-z.
Charmpilas N, Kyriakakis E, Tavernarakis N. Small heat shock proteins in ageing and age-related diseases. Cell Stress Chaperones. 2017;22:481–92. https://doi.org/10.1007/s12192-016-0761-x.
Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol. 2009;27:165–97. https://doi.org/10.1146/annurev.immunol.021908.132620.
Willis MS, Patterson C. Hold me tight: role of the heat shock protein family of chaperones in cardiac disease. Circulation. 2010;122:1740–51. https://doi.org/10.1161/CIRCULATIONAHA.110.942250.
Doroudgar S, Glembotski CC. The cardiokine story unfolds: ischemic stress-induced protein secretion in the heart. Trends Mol Med. 2011;17:207–14. https://doi.org/10.1016/j.molmed.2010.12.003.
Wu YS, Zhu B, Luo AL, Yang L, Yang C. The role of cardiokines in heart diseases: beneficial or detrimental? Biomed Res Int. 2018;2018:8207058. https://doi.org/10.1155/2018/8207058.
Seibert TA, Hibbert B, Chen YX, Rayner K, Simard T, Hu T, Cuerrier CM, Zhao X, de Belleroche J, Chow BJ, Hawken S, Wilson KR, O’Brien ER. Serum heat shock protein 27 levels represent a potential therapeutic target for atherosclerosis: observations from a human cohort and treatment of female mice. J Am Coll Cardiol. 2013;62:1446–54. https://doi.org/10.1016/j.jacc.2013.05.041.
Batulan Z, Pulakazhi Venu VK, Li Y, Koumbadinga G, Alvarez-Olmedo DG, Shi C, O’Brien ER. Extracellular release and signaling by heat shock protein 27: role in modifying vascular inflammation. Front Immunol. 2016;7:285. https://doi.org/10.3389/fimmu.2016.00285.
Kardys I, Rifai N, Meilhac O, Michel JB, Martin-Ventura JL, Buring JE, Libby P, Ridker PM. Plasma concentration of heat shock protein 27 and risk of cardiovascular disease: a prospective, nested case-control study. Clin Chem. 2008;54:139–46. https://doi.org/10.1373/clinchem.2007.094961.
Lepedda AJ, Cigliano A, Cherchi GM, Spirito R, Maggioni M, Carta F, Turrini F, Edelstein C, Scanu AM, Formato M. A proteomic approach to differentiate histologically classified stable and unstable plaques from human carotid arteries. Atherosclerosis. 2009;203:112–8. https://doi.org/10.1016/j.atherosclerosis.2008.07.001.
Bruxel MA, Tavares AM, Zavarize Neto LD, de Souza Borges V, Trevisan Schroeder H, Martins Bock P, Lavina Rodrigues MI, Belló-Klein A, Jr Homem de Bittencourt PI. Chronic whole-body heat treatment relieves atherosclerotic lesions, cardiovascular and metabolic abnormalities, and enhances survival time restoring the anti-inflammatory and anti-senescent heat shock response in mice. Biochimie. 2019;156:33–46. https://doi.org/10.1016/j.biochi.2018.09.011.
Zhang X, Wang X, Zhu H, Kranias EG, Tang Y, Peng T, Chang J, Fan GC. Hsp20 functions as a novel cardiokine in promoting angiogenesis via activation of VEGFR2. PLoS ONE. 2012;7:e32765. https://doi.org/10.1371/journal.pone.0032765.
De Maio A, Vazquez D. Extracellular heat shock proteins: a new location, a new function. Shock. 2013;40:239–46. https://doi.org/10.1097/SHK.0b013e3182a185ab.
Jin C, Cleveland JC, Ao L, Li J, Zeng Q, Fullerton DA, Meng X. Human myocardium releases heat shock protein 27 (HSP27) after global ischemia: the proinflammatory effect of extracellular HSP27 through toll-like receptor (TLR)-2 and TLR4. Mol Med. 2014;20:280–9. https://doi.org/10.2119/molmed.2014.00058.
Cova I, Markova A, Campini I, Grande G, Mariani C, Pomati S. Worldwide trends in the prevalence of dementia. J Neurol Sci. 2017;379:259–60. https://doi.org/10.1016/j.jns.2017.06.030.
Kim JY, Yenari MA. The immune modulating properties of the heat shock proteins after brain injury. Anat Cell Biol. 2013;46:1–7. https://doi.org/10.5115/acb.2013.46.1.1.
Banjara M, Ghosh C. Sterile neuroinflammation and strategies for therapeutic intervention. Int J Inflam. 2017;2017:8385961. https://doi.org/10.1155/2017/8385961.
Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW. Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain. 2016;139:653–61. https://doi.org/10.1093/brain/awv395.
Costantini E, D’Angelo C, Reale M. The role of immunosenescence in neurodegenerative diseases. Mediators Inflamm. 2018;2018:6039171. https://doi.org/10.1155/2018/6039171.
Dukay B, Csoboz B, Toth ME. Heat-shock proteins in neuroinflammation. Front Pharmacol. 2019;10:920. https://doi.org/10.3389/fphar.2019.00920.
Crum TS, Gleixner AM, Posimo JM, Mason DM, Broeren MT, Heinemann SD, Wipf P, Brodsky JL, Leak RK. Heat shock protein responses to aging and proteotoxicity in the olfactory bulb. J Neurochem. 2015;133:780–94. https://doi.org/10.1111/jnc.13041.
Carnemolla A, Labbadia JP, Lazell H, Neueder A, Moussaoui S, Bates GP. Contesting the dogma of an age-related heat shock response impairment: implications for cardiac-specific age-related disorders. Hum Mol Genet. 2014;23:3641–56. https://doi.org/10.1093/hmg/ddu073.
Gupte AA, Morris JK, Zhang H, Bomhoff GL, Geiger PC, Stanford JA. Age-related changes in HSP25 expression in basal ganglia and cortex of F344/BN rats. Neurosci Lett. 2010;472:90–3. https://doi.org/10.1016/j.neulet.2010.01.049.
Gleixner AM, Pulugulla SH, Pant DB, Posimo JM, Crum TS, Leak RK. Impact of aging on heat shock protein expression in the substantia nigra and striatum of the female rat. Cell Tissue Res. 2014;357:43–54. https://doi.org/10.1007/s00441-014-1852-6.
Sharp FR, Zhan X, Liu DZ. Heat shock proteins in the brain: role of Hsp70, Hsp 27, and HO-1 (Hsp32) and their therapeutic potential. Transl Stroke Res. 2013;4:685–92. https://doi.org/10.1007/s12975-013-0271-4.
Bartelt-Kirbach B, Slowik A, Beyer C, Golenhofen N. Upregulation and phosphorylation of HspB1/Hsp25 and HspB5/alphaB-crystallin after transient middle cerebral artery occlusion in rats. Cell Stress Chaperones. 2017;22:653–63. https://doi.org/10.1007/s12192-017-0794-9.
Turturici G, Sconzo G, Geraci F. Hsp70 and its molecular role in nervous system diseases. Biochem Res Int. 2011;2011:618127. https://doi.org/10.1155/2011/618127.
Marino Gammazza A, Bavisotto CC, Barone R, de Macario EC, Macario AJ. Alzheimer’s disease and molecular chaperones: current knowledge and the future of chaperonotherapy. Curr Pharm Des. 2016;22:4040–9. https://doi.org/10.2174/1381612822666160518141437.
Lyon MS, Milligan C. Extracellular heat shock proteins in neurodegenerative diseases: new perspectives. Neurosci Lett. 2019;711:134462. https://doi.org/10.1016/j.neulet.2019.134462.
Liu L, An D, Xu J, Shao B, Li X, Shi J. Ac2-26 induces IKKbeta degradation through chaperone-mediated autophagy via HSPB1 in NCM-treated microglia. Front Mol Neurosci. 2018;11:76. https://doi.org/10.3389/fnmol.2018.00076.
Calabrese V, Copani A, Testa D, Ravagna A, Spadaro F, Tendi E, Nicoletti VG, Giuffrida Stella AM. Nitric oxide synthase induction in astroglial cell cultures: effect on heat shock protein 70 synthesis and oxidant/antioxidant balance. J Neurosci Res. 2000;60:613–22. https://doi.org/10.1002/(SICI)1097-4547(20000601)60:5%3c613::AID-JNR6%3e3.0.CO;2-8.
Moll NM, Rietsch AM, Thomas S, Ransohoff AJ, Lee JC, Fox R, Chang A, Ransohoff RM, Fisher E. Multiple sclerosis normal-appearing white matter: pathology-imaging correlations. Ann Neurol. 2011;70:764–73. https://doi.org/10.1002/ana.22521.
van Noort JM, Bsibsi M, Gerritsen WH, van der Valk P, Bajramovic JJ, Steinman L, Amor S. Alphab-crystallin is a target for adaptive immune responses and a trigger of innate responses in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol. 2010;69:694–703. https://doi.org/10.1097/NEN.0b013e3181e4939c.
Bsibsi M, Holtman IR, Gerritsen WH, Eggen BJ, Boddeke E, van der Valk P, van Noort JM, Amor S. Alpha-B-crystallin induces an immune-regulatory and antiviral microglial response in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol. 2013;72:970–9. https://doi.org/10.1097/NEN.0b013e3182a776bf.
Quintana FJ, Farez MF, Viglietta V, Iglesias AH, Merbl Y, Izquierdo G, Lucas M, Basso AS, Khoury SJ, Lucchinetti CF, Cohen IR, Weiner HL. Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis. Proc Natl Acad Sci U S A. 2008;105:18889–94. https://doi.org/10.1073/pnas.0806310105.
de Graeff-Meeder ER, van Eden W, Rijkers GT, Prakken BJ, Kuis W, Voorhorst-Ogink MM, van der Zee R, Schuurman HJ, Helders PJ, Zegers BJ. Juvenile chronic arthritis: T cell reactivity to human HSP60 in patients with a favorable course of arthritis. J Clin Invest. 1995;95:934–40. https://doi.org/10.1172/JCI117801.
Calabrese V, Bates TE, Stella AM. NO synthase and NO-dependent signal pathways in brain aging and neurodegenerative disorders: the role of oxidant/antioxidant balance. Neurochem Res. 2000;25:1315–41. https://doi.org/10.1023/a:1007604414773.
Calderwood SK, Gong J, Murshid A. Extracellular HSPs: the complicated roles of extracellular HSPs in immunity. Front Immunol. 2016;7:159. https://doi.org/10.3389/fimmu.2016.00159.
van Eden W, Jansen MAA, Ludwig I, van Kooten P, van der Zee R, Broere F. The enigma of heat shock proteins in immune tolerance. Front Immunol. 2017;8:1599. https://doi.org/10.3389/fimmu.2017.01599.
van Eden W. Immune tolerance therapies for autoimmune diseases based on heat shock protein T-cell epitopes. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2018; 373. https://doi.org/10.1098/rstb.2016.0531
Landstein D, Ulmansky R, Naparstek Y. HSP60: a double edge sword in autoimmunity. Oncotarget. 2015;6:32299–300. https://doi.org/10.18632/oncotarget.5869.
van Eden W, van der Zee R, Prakken B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev. 2005;5:318–30. https://doi.org/10.1038/nri1593.
Zonneveld-Huijssoon E, Albani S, Prakken BJ, van Wijk F. Heat shock protein bystander antigens for peptide immunotherapy in autoimmune disease. Clin Exp Immunol. 2013;171:20–9. https://doi.org/10.1111/j.1365-2249.2012.04627.x.
Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev. 2002;2:185–94. https://doi.org/10.1038/nri749.
Edkins AL, Price JT, Pockley AG, Blatch GL. Heat shock proteins as modulators and therapeutic targets of chronic disease: an integrated perspective. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2018; 373. https://doi.org/10.1098/rstb.2016.0521
Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14:183–93. https://doi.org/10.1016/S1474-4422(14)70256-X.
Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol. 2013;14:630–42. https://doi.org/10.1038/nrm3658.
Saile B, Eisenbach C, Dudas J, El-Armouche H, Ramadori G. Interferon-gamma acts proapoptotic on hepatic stellate cells (HSC) and abrogates the antiapoptotic effect of interferon-alpha by an HSP70-dependant pathway. Eur J Cell Biol. 2004;83:469–76. https://doi.org/10.1078/0171-9335-00409.
Wieten L, Berlo SE, Ten Brink CB, van Kooten PJ, Singh M, van der Zee R, Glant TT, Broere F, van Eden W. IL-10 is critically involved in mycobacterial HSP70 induced suppression of proteoglycan-induced arthritis. PLoS ONE. 2009;4:e4186. https://doi.org/10.1371/journal.pone.0004186.
van Herwijnen MJ, Wieten L, van der Zee R, van Kooten PJ, Wagenaar-Hilbers JP, Hoek A, den Braber I, Anderton SM, Singh M, Meiring HD, van Els CA, van Eden W, Broere F. Regulatory T cells that recognize a ubiquitous stress-inducible self-antigen are long-lived suppressors of autoimmune arthritis. Proc Natl Acad Sci U S A. 2012;109:14134–9. https://doi.org/10.1073/pnas.1206803109.
Tukaj S, Kaminski M. Heat shock proteins in the therapy of autoimmune diseases: too simple to be true? Cell Stress Chaperones. 2019;24:475–9. https://doi.org/10.1007/s12192-019-01000-3.
Stocki P, Dickinson AM. The immunosuppressive activity of heat shock protein 70. Autoimmune Dis. 2012;2012:617213. https://doi.org/10.1155/2012/617213.
Borges TJ, Wieten L, van Herwijnen MJ, Broere F, van der Zee R, Bonorino C, van Eden W. The anti-inflammatory mechanisms of Hsp70. Front Immunol. 2012;3:95. https://doi.org/10.3389/fimmu.2012.00095.
Lorenzo N, Altruda F, Silengo L, Del Carmen DM. APL-1, an altered peptide ligand derived from heat-shock protein, alone or combined with methotrexate attenuates murine collagen-induced arthritis. Clin Exp Med. 2017;17:209–16. https://doi.org/10.1007/s10238-016-0412-7.
van Halteren AG, Mosselman B, Roep BO, van Eden W, Cooke A, Kraal G, Wauben MH. T cell reactivity to heat shock protein 60 in diabetes-susceptible and genetically protected nonobese diabetic mice is associated with a protective cytokine profile. J Immunol. 2000;165:5544–51. https://doi.org/10.4049/jimmunol.165.10.5544.
Barbera A, Lorenzo N, van Kooten P, van Roon J, de Jager W, Prada D, Gomez J, Padron G, van Eden W, Broere F, Del Carmen DM. APL1, an altered peptide ligand derived from human heat-shock protein 60, increases the frequency of Tregs and its suppressive capacity against antigen responding effector CD4 + T cells from rheumatoid arthritis patients. Cell Stress Chaperones. 2016;21:735–44. https://doi.org/10.1007/s12192-016-0698-0.
Mandal K, Jahangiri M, Xu Q. Autoimmunity to heat shock proteins in atherosclerosis. Autoimmun Rev. 2004;3:31–7. https://doi.org/10.1016/S1568-9972(03)00088-0.
Nagele EP, Han M, Acharya NK, DeMarshall C, Kosciuk MC, Nagele RG. Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS ONE. 2013;8:e60726. https://doi.org/10.1371/journal.pone.0060726.
Manoussakis MN, Tzioufas AG, Silis MP, Pange PJ, Goudevenos J, Moutsopoulos HM. High prevalence of anti-cardiolipin and other autoantibodies in a healthy elderly population. Clin Exp Immunol. 1987;69:557–65.
Pockley AG, Shepherd J, Corton JM. Detection of heat shock protein 70 (Hsp70) and anti-Hsp70 antibodies in the serum of normal individuals. Immunol Invest. 1998;27:367–77. https://doi.org/10.3109/08820139809022710.
Pockley AG, Bulmer J, Hanks BM, Wright BH. Identification of human heat shock protein 60 (Hsp60) and anti-Hsp60 antibodies in the peripheral circulation of normal individuals. Cell Stress Chaperones. 1999;4:29–35. https://doi.org/10.1054/csac.1998.0121.
Rea IM, McNerlan S, Pockley AG. Serum heat shock protein and anti-heat shock protein antibody levels in aging. Exp Gerontol. 2001;36:341–52. https://doi.org/10.1016/s0531-5565(00)00215-1.
Cappello F, Conway de Macario E, Marino Gammazza A, Bonaventura G, Carini F, Czarnecka AM, Farina F, Zummo G, Macario AJ. Hsp60 and human aging: Les liaisons dangereuses. Front Biosci (Landmark Ed). 2013;18:626–37. https://doi.org/10.2741/4126.
Ulmansky R, Landstein D, Moallem E, Loeb V, Levin A, Meyuhas R, Katzavian G, Yair S, Naparstek Y. A humanized monoclonal antibody against heat shock protein 60 suppresses murine arthritis and colitis and skews the cytokine balance toward an anti-inflammatory response. J Immunol. 2015;194:5103–9. https://doi.org/10.4049/jimmunol.1500023.
Mantej J, Polasik K, Piotrowska E, Tukaj S. Autoantibodies to heat shock proteins 60, 70, and 90 in patients with rheumatoid arthritis. Cell Stress Chaperones. 2019;24:283–7. https://doi.org/10.1007/s12192-018-0951-9.
Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell. 2017;169:570–86. https://doi.org/10.1016/j.cell.2017.04.004.
Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol. 2018;15:366–81. https://doi.org/10.1038/s41571-018-0007-1.
Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124–34. https://doi.org/10.1038/nm.4409.
Schiavoni G, Gabriele L, Mattei F. The tumor microenvironment: a pitch for multiple players. Front Oncol. 2013;3:90. https://doi.org/10.3389/fonc.2013.00090.
Patel H, Nilendu P, Jahagirdar D, Pal JK, Sharma NK. Modulating secreted components of tumor microenvironment: a masterstroke in tumor therapeutics. Cancer Biol Ther. 2018;19:3–12. https://doi.org/10.1080/15384047.2017.1394538.
Arendt LM, Rudnick JA, Keller PJ, Kuperwasser C. Stroma in breast development and disease. Semin Cell Dev Biol. 2010;21:11–8. https://doi.org/10.1016/j.semcdb.2009.10.003.
Wu J, Liu T, Rios Z, Mei Q, Lin X, Cao S. Heat shock proteins and cancer. Trends Pharmacol Sci. 2017;38:226–56. https://doi.org/10.1016/j.tips.2016.11.009.
Chatterjee S, Burns TF. Targeting heat shock proteins in cancer: a promising therapeutic approach. International journal of molecular sciences. 2017; 18. https://doi.org/10.3390/ijms18091978
Beyene DA, Naab TJ, Kanarek NF, Apprey V, Esnakula A, Khan FA, Blackman MR, Brown CA, Hudson TS. Differential expression of Annexin 2, SPINK1, and Hsp60 predict progression of prostate cancer through bifurcated WHO Gleason score categories in African American men. Prostate. 2018;78:801–11. https://doi.org/10.1002/pros.23537.
Fanelli MA, Cuello Carrion FD, Dekker J, Schoemaker J, Ciocca DR. Serological detection of heat shock protein hsp27 in normal and breast cancer patients. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 1998;7:791–5.
Banerjee S, Lin CF, Skinner KA, Schiffhauer LM, Peacock J, Hicks DG, Redmond EM, Morrow D, Huston A, Shayne M, Langstein HN, Miller-Graziano CL, Strickland J, O’Donoghue L, De AK. Heat shock protein 27 differentiates tolerogenic macrophages that may support human breast cancer progression. Can Res. 2011;71:318–27. https://doi.org/10.1158/0008-5472.CAN-10-1778.
Feng JT, Liu YK, Song HY, Dai Z, Qin LX, Almofti MR, Fang CY, Lu HJ, Yang PY, Tang ZY. Heat-shock protein 27: a potential biomarker for hepatocellular carcinoma identified by serum proteome analysis. Proteomics. 2005;5:4581–8. https://doi.org/10.1002/pmic.200401309.
Huang Q, Ye J, Huang Q, Chen W, Wang L, Lin W, Lin J, Lin X. Heat shock protein 27 is over-expressed in tumor tissues and increased in sera of patients with gastric adenocarcinoma. Clin Chem Lab Med. 2010;48:263–9. https://doi.org/10.1515/CCLM.2010.043.
Thuringer D, Berthenet K, Cronier L, Solary E, Garrido C. Primary tumor- and metastasis-derived colon cancer cells differently modulate connexin expression and function in human capillary endothelial cells. Oncotarget. 2015;6:28800–15. https://doi.org/10.18632/oncotarget.4894.
Melle C, Ernst G, Escher N, Hartmann D, Schimmel B, Bleul A, Thieme H, Kaufmann R, Felix K, Friess HM, Settmacher U, Hommann M, Richter KK, Daffner W, Taubig H, Manger T, Claussen U, von Eggeling F. Protein profiling of microdissected pancreas carcinoma and identification of HSP27 as a potential serum marker. Clin Chem. 2007;53:629–35. https://doi.org/10.1373/clinchem.2006.079194.
Liao WC, Wu MS, Wang HP, Tien YW, Lin JT. Serum heat shock protein 27 is increased in chronic pancreatitis and pancreatic carcinoma. Pancreas. 2009;38:422–6. https://doi.org/10.1097/MPA.0b013e318198281d.
Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci. 2007;1113:202–16. https://doi.org/10.1196/annals.1391.012.
Gross C, Koelch W, DeMaio A, Arispe N, Multhoff G. Cell surface-bound heat shock protein 70 (Hsp70) mediates perforin-independent apoptosis by specific binding and uptake of granzyme B. J Biol Chem. 2003;278:41173–81. https://doi.org/10.1074/jbc.M302644200.
Beere HM, Green DR. Stress management–heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol. 2001;11:6–10. https://doi.org/10.1016/s0962-8924(00)01874-2.
Ghosh JC, Dohi T, Kang BH, Altieri DC. Hsp60 regulation of tumor cell apoptosis. J Biol Chem. 2008;283:5188–94. https://doi.org/10.1074/jbc.M705904200.
Chaiwatanasirikul KA, Sala A. The tumour-suppressive function of CLU is explained by its localisation and interaction with HSP60. Cell Death Dis. 2011;2:e219. https://doi.org/10.1038/cddis.2011.99.
Zhou C, Sun H, Zheng C, Gao J, Fu Q, Hu N, Shao X, Zhou Y, Xiong J, Nie K, Zhou H, Shen L, Fang H, Lyu J. Oncogenic HSP60 regulates mitochondrial oxidative phosphorylation to support Erk1/2 activation during pancreatic cancer cell growth. Cell Death Dis. 2018;9:161. https://doi.org/10.1038/s41419-017-0196-z.
Chun JN, Choi B, Lee KW, Lee DJ, Kang DH, Lee JY, Song IS, Kim HI, Lee SH, Kim HS, Lee NK, Lee SY, Lee KJ, Kim J, Kang SW. Cytosolic Hsp60 is involved in the NF-kappaB-dependent survival of cancer cells via IKK regulation. PLoS ONE. 2010;5:e9422. https://doi.org/10.1371/journal.pone.0009422.
Ruan W, Wang Y, Ma Y, Xing X, Lin J, Cui J, Lai M. HSP60, a protein downregulated by IGFBP7 in colorectal carcinoma. Journal of experimental & clinical cancer research : CR. 2010;29:41. https://doi.org/10.1186/1756-9966-29-41.
Tsai YP, Yang MH, Huang CH, Chang SY, Chen PM, Liu CJ, Teng SC, Wu KJ. Interaction between HSP60 and beta-catenin promotes metastasis. Carcinogenesis. 2009;30:1049–57. https://doi.org/10.1093/carcin/bgp087.
Kennedy D, Jager R, Mosser DD, Samali A. Regulation of apoptosis by heat shock proteins. IUBMB Life. 2014;66:327–38. https://doi.org/10.1002/iub.1274.
Xanthoudakis S, Roy S, Rasper D, Hennessey T, Aubin Y, Cassady R, Tawa P, Ruel R, Rosen A, Nicholson DW. Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO J. 1999;18:2049–56. https://doi.org/10.1093/emboj/18.8.2049.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Yang HZ, Cui B, Liu HZ, Mi S, Yan J, Yan HM, Hua F, Lin H, Cai WF, Xie WJ, Lv XX, Wang XX, Xin BM, Zhan QM, Hu ZW. Blocking TLR2 activity attenuates pulmonary metastases of tumor. PLoS ONE. 2009;4:e6520. https://doi.org/10.1371/journal.pone.0006520.
Quintana FJ, Cohen IR. The HSP60 immune system network. Trends Immunol. 2011;32:89–95. https://doi.org/10.1016/j.it.2010.11.001.
Gammazza AM, Caruso C, David S, Barone R, Rappa F, Campanella C, Conway de Macario E, Cappello F, Macario AJL. HSP60 is a ubiquitous player in the physiological and pathogenic interactions between the chaperoning and the immune systems. Curr Immunol Revs. 2017;13:44–55. https://doi.org/10.2174/1573395513666170412170540.
Pockley AG, Henderson B. Extracellular cell stress (heat shock) proteins-immune responses and disease: an overview. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2018; 373. https://doi.org/10.1098/rstb.2016.0522
Campanella C, Rappa F, Sciume C, Marino Gammazza A, Barone R, Bucchieri F, David S, Curcuru G, Caruso Bavisotto C, Pitruzzella A, Geraci G, Modica G, Farina F, Zummo G, Fais S, Conway de Macario E, Macario AJ, Cappello F. Heat shock protein 60 levels in tissue and circulating exosomes in human large bowel cancer before and after ablative surgery. Cancer. 2015;121:3230–9. https://doi.org/10.1002/cncr.29499.
Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, Rando TA, Richardson A, Schadt EE, Wyss-Coray T, Sierra F. Geroscience: linking aging to chronic disease. Cell. 2014;159:709–13. https://doi.org/10.1016/j.cell.2014.10.039.
Sierra F. The emergence of geroscience as an interdisciplinary approach to the enhancement of health span and life span. Cold Spring Harb Perspect Med. 2016;6:a025163. https://doi.org/10.1101/cshperspect.a025163.
Rinaldi B, Corbi G, Boccuti S, Filippelli W, Rengo G, Leosco D, Rossi F, Filippelli A, Ferrara N. Exercise training affects age-induced changes in SOD and heat shock protein expression in rat heart. Exp Gerontol. 2006;41:764–70. https://doi.org/10.1016/j.exger.2006.05.008.
Matai L, Chandra Sarkar G, Chamoli M, Malik Y, Shekhar Kumar S, Rautela U, Ranjan Jana N, Chakraborty K, Mukhopadhyay A. Dietary restriction improves proteostasis and increases life span through endoplasmic reticulum hormesis. Proc Natl Acad Sci U S A. 2019;116:17383–92. https://doi.org/10.1073/pnas.1900055116.
Jurivich DA, Sistonen RA, Kroes R, Morimoto I. Effect of sodium salicylate on the human heat shock response. Science (New York, NY). 1992;255:1243–5. https://doi.org/10.1126/science.1546322.
Cascão R, Fonseca JE, Moita LF. Celastrol: a spectrum of treatment opportunities in chronic diseases. Front Med (Lausanne). 2017;4:69. https://doi.org/10.3389/fmed.2017.00069.eCollection2017.
Trivedi R, Jurivich DA. A molecular perspective on age-dependent changes to the heat shock axis. Exp Gerontol. 2020;137:110969. https://doi.org/10.1016/j.exger.2020.110969.
Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–95. https://doi.org/10.1038/nm1298.
Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet. 2001;10:1307–15. https://doi.org/10.1093/hmg/10.12.1307.
Gomez-Pastor R, Burchfiel ET, Thiele DJ. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat Rev Mol Cell Biol. 2018;19:4–19. https://doi.org/10.1038/nrm.2017.73.
Wang L, Xu X, Jiang Z, You Q. Modulation of protein fate decision by small molecules: targeting molecular chaperone machinery. Acta Pharm Sin B. 2020;10:1904–25. https://doi.org/10.1016/j.apsb.2020.01.018.
Saini J, Sharma PK. Clinical, prognostic and therapeutic significance of heat shock proteins in cancer. Curr Drug Targets. 2018;19:1478–90. https://doi.org/10.2174/1389450118666170823121248.
Song YJ, Zhong CB, Wang XB. Heat shock protein 70: a promising therapeutic target for myocardial ischemia-reperfusion injury. J Cell Physiol. 2019;234:1190–207. https://doi.org/10.1002/jcp.27110.
Shao A, Zhou Y, Yao Y, Zhang W, Zhang J, Deng Y. The role and therapeutic potential of heat shock proteins in haemorrhagic stroke. J Cell Mol Med. 2019;23:5846–58. https://doi.org/10.1111/jcmm.14479.
Zhu Z, Reiser G. The small heat shock proteins, especially HspB4 and HspB5 are promising protectants in neurodegenerative diseases. Neurochem Int. 2018;115:69–79. https://doi.org/10.1016/j.neuint.2018.02.006.
Shorter J. Designer protein disaggregases to counter neurodegenerative disease. Curr Opin Genet Dev. 2017;44:1–8. https://doi.org/10.1016/j.gde.2017.01.008.
Prakken BJ, Samodal R, Le TD, Giannoni F, Yung GP, Scavulli J, Amox D, Roord S, de Kleer I, Bonnin D, Lanza P, Berry C, Massa M, Billetta R, Albani S. Epitope-specific immunotherapy induces immune deviation of proinflammatory T cells in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2004;101:4228–33. https://doi.org/10.1073/pnas.0400061101.
Koffeman EC, Genovese M, Amox D, Keogh E, Santana E, Matteson EL, Kavanaugh A, Molitor JA, Schiff MH, Posever JO, Bathon JM, Kivitz AJ, Samodal R, Belardi F, Dennehey C, van den Broek T, van Wijk F, Zhang X, Zieseniss P, Le T, Prakken BA, Cutter GC, Albani S. Epitope-specific immunotherapy of rheumatoid arthritis: clinical responsiveness occurs with immune deviation and relies on the expression of a cluster of molecules associated with T cell tolerance in a double-blind, placebo-controlled, pilot phase II trial. Arthritis Rheum. 2009;60:3207–16. https://doi.org/10.1002/art.24916.
Huurman VA, van der Meide PE, Duinkerken G, Willemen S, Cohen IR, Elias D, Roep BO. Immunological efficacy of heat shock protein 60 peptide DiaPep277 therapy in clinical type I diabetes. Clin Exp Immunol. 2008;152:488–97. https://doi.org/10.1111/j.1365-2249.2008.03656.x.
Broadley SA, Vanags D, Williams B, Johnson B, Feeney D, Griffiths L, Shakib S, Brown G, Coulthard A, Mullins P, Kneebone C. Results of a phase IIa clinical trial of an anti-inflammatory molecule, chaperonin 10, in multiple sclerosis. Mult Scler. 2009;15:329–36. https://doi.org/10.1177/1352458508099141.
Acknowledgements
The author thanks Drs. Venkat Mannam, Ingrid Espinoza, Amit Reddy, and Ms. Isabella Gomez-Espinoza for valuable discussions and critical reading of the manuscript.
Funding
This study was supported in part by Grant DOD PC094680 and PCF Creativity Award.
Author information
Authors and Affiliations
Contributions
CRG had the idea for the article, performed the literature search and data analysis, and drafted the work.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The author declares no competing interests.
Disclaimer
Dr. Gomez contributed to this article as an employee of the University of Mississippi Medical Center. The views expressed are his own and do not necessarily represent the views of the National Institutes of Health or the US Government.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Gomez, C.R. Role of heat shock proteins in aging and chronic inflammatory diseases. GeroScience 43, 2515–2532 (2021). https://doi.org/10.1007/s11357-021-00394-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-021-00394-2