
Abstract
In vitro models of angiogenesis are valuable tools for understanding the underlying mechanisms of pathological conditions and for the preclinical evaluation of therapies. Our laboratory developed the rat mesentery culture model as a new tool for investigating mechanistic cell–cell interactions at specific locations across intact blood and lymphatic microvascular networks ex vivo. The objective of this study was to report a method for evaluating the effect of aging on human stem cell differentiation into pericytes during angiogenesis in cultured microvascular networks. DiI labeled exogenous stem cells were seeded onto harvested adult Wistar rat mesenteric tissues and cultured in alpha-MEM + 1% serum for up to 5 days according to four experimental groups: (1) adult human adipose–derived stem cells (hASCs), (2) aged hASCs, (3) adult human bone marrow-derived stem cells (hBMSCs), and (4) aged hBMSCs. Angiogenesis per experimental group was supported by observation of increased vessel density and capillary sprouting. For each tissue per experimental group, a subset of cells was observed in typical pericyte location wrapped along blood vessels. Stem cell differentiation into pericytes was supported by the adoption of elongated pericyte morphology along endothelial cells and positive NG2 labeling. The percentage of cells in pericyte locations was not significantly different across the experimental groups, suggesting that aged mesenchymal stem cells are able to retain their differentiation capacity. Our results showcase an application of the rat mesentery culture model for aging research and the evaluation of stem cell fate within intact microvascular networks.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stem cell therapy has emerged as a promising method to treat a wide range of age-related diseases including peripheral artery disease (Hao et al. 2014) and myocardial infarction (Fuchs et al. 2001), where the underlying condition is impairment of the angiogenic process defined as the formation of new blood vessels. Impaired angiogenesis has been linked to aging through altered endothelial cell dynamics, including decreased capillary sprouting (Heiss et al. 2005; Hoetzer et al. 2007), cellular senescence (Minamino et al. 2004; Erusalimsky 2009), and diminished responses to growth factor signaling (Moriya and Minamino 2017). A potential therapeutic target to combat impaired angiogenesis are vascular pericytes, specialized support cells that function to promote angiogenesis and stabilize newly formed blood vessels through the regulation of endothelial cells (Gerhardt and Betsholtz 2003; Ozerdem 2006; Stapor et al. 2013; Kelly-Goss et al. 2014; Hodges et al. 2018). One of the proposed solutions to restore pericyte coverage and promote angiogenesis for the treatment of age-related diseases has been the use of differentiated stem cells (Mendel et al. 2013; Cronk et al. 2015; Kramerov and Ljubimov 2016).
Tissue-resident stem cell populations present a sustainable source for new pericytes to treat pathological angiogenesis. Mesenchymal stem cells (MSCs) are resident stem cells present in numerous tissue sources including the bone marrow, adipose, and blood (Izadpanah et al. 2006; Hou et al. 2016). These multipotent cells have been used in recent years to promote angiogenesis by cytokine signaling, direct cell incorporation, and differentiation into various cell types including pericytes (Rehman et al. 2004; Kondo et al. 2009; Kachgal and Putnam 2011; Mendel et al. 2013). For example, Rajantie et al. showed that a subpopulation of bone marrow-derived cells that participated in angiogenesis had the distinct morphology of vascular pericytes, expressed the pericyte marker NG2, and were found in close spatial association with endothelial cells along blood vessels (Rajantie et al. 2004). Mendel et al. differentiated adipose-derived stem cells (ASCs) into pericytes and intravitreally injected the cells to enhance retinal microvascular stabilization in a murine model of retinopathy. ASC derived pericytes incorporated into the host vasculature and adopted both pericyte morphology and marker expression (Mendel et al. 2013). While MSCs derived from bone marrow and adipose tissue have been shown to enhance angiogenesis, our understanding of their function and differentiation capacity from aged tissue sources remain relatively unclear.
Aging is accompanied by the gradual decline of cellular competency and function in the body over time. One of the hallmarks of aging is thought to be stem cell exhaustion, which can manifest itself as a reduction in the number of stem cells and decreased tissue regeneration capabilities (López-Otín et al. 2013). For example, studies have reported that aged bone marrow–derived stem cells (BMSCs) showed a decline in proliferation and differentiation potential, and exhibited a higher expression of p53 and p21, both indicative of cellular senescence (Zhou et al. 2008; Yu et al. 2011). Similarly, Efimenko et al. showed aged ASCs have impaired proliferation and decreased angiogenic properties characterized by low expression levels of vascular endothelial growth factor, a major regulator of new vessel formation (Efimenko et al. 2011). These findings highlight the need to evaluate stem cells from aged tissue sources to help answer questions regarding their cellular fate and function for effective therapeutic applications. In addition, aging has been associated with microvascular alterations including changes in microvascular phenotypes, impaired microvascular growth responses, cell senescence, oxidative stress–induced microvascular injury, and altered neurovascular coupling—all of which contribute to compromised tissue function (Csiszar et al., 2017; Tarantini et al. 2017; Fulop et al. 2018; Sure et al. 2018; Ungvari et al. 2018).
Our lab has developed the rat mesentery culture model for studying endothelial cell dynamics during angiogenesis in an ex vivo microvascular environment (Stapor et al. 2013). The complexity of native microvascular networks in mesentery tissue provides our model with the advantage of matching in vivo physiology in a controlled environment. Previously, we have shown the rat mesentery culture model can be used as a tool to investigate pericyte-endothelial cell interactions during angiogenesis (Stapor et al. 2013), to evaluate VEGF-C–induced lymphangiogenesis (Sweat et al. 2014), and to evaluate anti-angiogenic drug responses using time-lapse capabilities (Azimi et al. 2015). Here we introduce the rat mesentery culture model as a platform to screen the differentiation potential of aged versus adult stem cells real-time in the presence of microvascular networks. We provide a method of seeding aged and adult stem cells from both adipose and bone marrow tissue sources onto rat mesentery tissue for time-lapse culture up to 5 days (Fig. 1). Our results validate that MSCs can be seeded onto mesentery tissue, remain viable, and incorporate into the native microvascular networks. Thus, the present manuscript provides a detailed protocol for screening stem cells from different aged donors to determine their potential for therapeutic applications.

MSC and mesentery tissue coculture. Stem cells were suspended in media at 500,000 cell/mL concentration, and 100–200 μL was placed on top of the mesentery tissue. The setup was placed in an incubator for 20 min to give the stem cells enough time to adhere to the mesentery tissue. The supernatant was then aspirated, and the coculture was transferred to a well in a nontreated 6-well tissue culture plate. The insert was covered with 4 mL of media supplemented with 1% serum and cultured for up to 5 days
Materials and methods
Culture reagents and supplies
Saline (Baxter, Deerfield, IL), Dulbecco’s phosphate-buffered saline (DPBS; Life Technologies, Carlsbad, CA), minimum essential media (MEM; Life Technologies, Carlsbad, CA), penicillin-streptomycin 10,000 U/mL (PS; Life Technologies, Carlsbad, CA), fetal bovine serum (FBS; Life Technologies, Carlsbad, CA; HyClone, Logan UT; Atlanta Biologicals; Lawrenceville, GA), formaldehyde 16% w/v (Thermo Scientific, Waltham, MA), saponin (Sigma-Aldrich, St. Louis, MO), Vybrant™ CM-DiI cell-labeling solution (DiI; Invitrogen, Carlsbad, CA), CellCrown™ inserts (Inserts; Sigma-Aldrich, St. Louis, MO), Isopore Membrane Filter (Filter membrane; Millipore, Burlington, MA), type I collagenase (Sigma-Aldrich, St. Louis, MO), powdered bovine serum albumin (powdered BSA, fraction V; Sigma-Aldrich, St. Louis, MO), Dulbecco’s modified Eagle medium: nutrient mixture F-12 (DMEM/F12; HyClone, Logan, UT), antibiotic/antimycotic (Fisher Scientific, Hampton, NH), 0.25% trypsin/1 mM EDTA (GIBCO, Grand Island, NY), α-MEM (Thermo Scientific, Waltham, MA), l-glutamine (Thermo Scientific, Waltham, MA).
Labeling reagents
Bovine serum albumin (BSA; Jackson Immunoresearch, West Grove, PA), normal goat serum (NGS; Jackson Immunoresearch, West Grove, PA), phosphate-buffered saline (PBS; Sigma-Aldrich, St. Louis, MO), glycerol (Fisher Scientific, Hampton, NH), FITC-conjugated BSI-lectin (Sigma-Aldrich, St. Louis, MO), anti-NG2 chondroitin sulfate proteoglycan antibody (NG2; Sigma-Aldrich, St. Louis, MO), and goat anti-rabbit Cy2-conjugated antibody (GAR-Cy2; Jackson Immunoresearch, West Grove, PA).
Stem cell sources
Primary human ASCs were obtained from 8 adult donors (29.3 ± 6.4 years) and 8 aged donors (62.5 ± 6.6 years) undergoing elective liposuction procedures, as previously described (Bunnell et al. 2008; Strong et al. 2015; Jones et al. 2017). All human ASC protocols were reviewed and approved by the Pennington Biomedical Research Center’s Institutional Review Board, and all human participants provided written informed consent. Primary human BMSCs were obtained from 8 adult donors (25.9 ± 5.5 years) and 8 aged donors (55.9 ± 6.1 years). Bone marrow aspirates isolated from the iliac crest, as previously described (Pachón-Peña et al. 2011; Semon et al. 2014). All human BMSC protocols were approved by Tulane University’s Institutional Review Board and the Partners Human Research Committee. The BMSCs and ASCs used for this study were characterized before coculturing. After isolation and cryopreservation, the cells were characterized by performing a series of standard assays in which the cells are characterized by their ability to form colonies in a colony-forming unit assay, as well as their ability to undergo osteogenic, adipogenic, and chondrogenic differentiation (Bunnell et al. 2008; Strong et al. 2015; Jones et al. 2017). Additionally, the cell surface antigen profile was characterized by flow cytometry. The cells are typically characterized for the expression of both positive (CD90, CD105, CD73) and negative (CD45, CD34, CD3) antigens (Pachón-Peña et al., 2011).
Mesentery tissue harvest
All animal experiments were approved by Tulane University’s Institutional Animal and Care Use Committee. Our laboratory has developed and previously published protocols for the rat mesentery tissue culture method (Stapor et al. 2013; Azimi et al. 2015), including detailed instructions to create the surgical stage for mesentery harvest and a complete list of reagents and supplies (Azimi et al. 2017). Autoclave instruments and supplies prior to surgery and perform all procedures using sterile technique.
- 1)
Anesthetize an adult male Wistar rat (350 ± 25 g) via an intramuscular injection of ketamine (80 mg/kg body weight) and xylazine (8 mg/kg body weight). Confirm that the rat is fully under anesthesia by checking for a lack of reflex response.
- 2)
Shave the abdominal region and remove remaining hair using hair removal cream. Wipe the abdominal skin three times with alternating 70% isopropyl alcohol followed by povidone-iodine.
- 3)
Make an incision along the abdominal midline starting 1 in. below the sternum. Be careful not to puncture the bowel or mesentery (1 layer of skin, 1 layer of connective tissue, and 1 layer of muscle).
- 4)
Place a drape with a precut hole over the incision and place the sterile surgical stage atop the drape, ensuring both openings align with the incision. Use sterile cotton-tipped applicators to locate and pull out the ileum through the surgical stage opening.
- 5)
Pull 6–8 mesentery tissues through the stage using the cotton-tipped applicators being careful not to touch the translucent windows. Keep the exposed tissues moist with sterile saline as needed using a sterile syringe to drip the solution.
- 6)
Euthanize the rat via intracardiac injection of Beuthanasia-D (0.2 mL per rat). Ensure the rat is euthanized by palpating the heart; there should be no pulse.
- 7)
Identify vascularized mesentery tissues and harvest using tweezers to grab the fat pad and fine scissors to cut the edges of the window, leaving a border of fat (0.2 mm).
- 8)
Wash all tissues once in warmed sterile DPBS and store in minimum MEM + 1%PS placed in an incubator until the stem cell seeding procedure.
- 9)
Return exteriorized mesentery and ileum to the abdominal cavity and dispose of animal according to institutional guidelines.
Stem cell seeding on mesentery tissues
-
1)
Suspend stem cells in media at 1 million cells/mL concentration mixed with 5 μl/mL cell tracker DiI. Incubate cell suspension at 37 °C for 30 min.
-
2)
Centrifuge the cell suspension at 1000 rpm for 5 min, followed by replacing the supernatant with fresh media. Repeat this centrifuge step two more times.
-
3)
Dilute the cell suspension using fresh media to a final concentration of 500,000 cells/mL.
-
4)
Remove harvested mesentery tissues from incubator and place in a sterile laminar flow hood. Use tweezers to transfer one tissue atop a polycarbonate filter membrane secured to an insert. Be sure to grab tissues by their fat pad to avoid damaging the translucent window.
-
5)
Quickly spread the tissue flat using the fat pad and drip 100–200 μL of cell suspension on top of the translucent window. Place the stem cell-tissue coculture in an incubator for 20 min to allow the stem cells enough time to adhere to the mesentery tissue.
-
6)
Following incubation, carefully aspirate the remaining supernatant atop the tissue. Place 1 mL of culture media (MEM + 1%PS + 1%FBS) into the well of a 6-well plate. Gently invert the stem cell seeded tissue into the well, being careful to avoid bubbles between the tissue and base of the well. Cover the tissue with an additional 3 mL of culture media.
-
7)
Continue steps 4 through 6 for the remaining tissues and culture in standard incubator conditions (5% CO2, 37 °C) for 5 days.
Immunohistochemistry
Tissue fixation
-
1)
Fix cultured tissues in the 6-well plate with 4% formaldehyde solution for 10 min at room temperature.
-
2)
Remove formaldehyde solution and rinse tissues with PBS for 10 min at room temperature three times.
-
3)
Mount tissues on glass slides using tweezers to carefully spread the window flat and remove the fat border with a scalpel blade.
Lectin labeling
-
1)
Following tissue fixation, prepare lectin antibody solution by mixing 1:40 BSI-lectin conjugated to FITC with PBS.
-
2)
Drip lectin antibody solution onto tissues and incubate for 30 min at room temperature protected from light.
-
3)
Remove lectin antibody solution and wash tissues with PBS for 10 min at room temperature three times.
-
4)
Cover tissues with 50:50 PBS and glycerol solution, place coverslip on top, and seal the glass slide edges.
NG2 labeling
-
1)
Following tissue fixation, prepare antibody buffer solution by mixing PBS with 0.1% saponin and 2% BSA.
-
2)
Prepare NG2 primary antibody solution by mixing 1:100 rabbit polyclonal NG2 with 5% NGS and antibody buffer solution. Incubate tissues for 1 h a room temperature protected from light.
-
3)
Remove NG2 primary antibody solution and wash tissues with PBS for 10 min at room temperature three times.
-
4)
Prepare secondary antibody solution by mixing 1:100 GAR-Cy2 with 5% NGS and antibody buffer solution. Incubate tissues for 1 h at room temperature protected from light.
-
5)
Remove secondary antibody solution and wash tissues with PBS for 10 min at room temperature three times.
-
6)
Cover tissues with 50:50 PBS and glycerol solution, place coverslip on top, and seal the glass slide edges.
Quantification of MSC differentiation into Pericytes
-
1)
Label live mesentery tissues with FITC-conjugated BSI-lectin as described above (labeling of live mesentery tissue).
-
2)
Image three to four randomly selected microvascular regions in mesentery tissue using a × 4 objective connected with a fluorescent microscope.
-
3)
Count the total number of DiI-positive cells and the number of DiI-positive cells in pericyte locations with typical pericyte morphology. Pericyte location is defined as cells in contact with the abluminal surface of lectin-labeled capillary blood vessels, and pericyte morphology is defined as cells with protrusions wrapping around capillary blood vessels.
-
4)
For this study, the average percentage of cells in pericyte location per experimental group was statistically compared using a one-way ANOVA. Post hoc tests were not performed as difference in the mean values among the groups were not great enough to exclude the possibility that differences were due to random sampling. Comparisons were made across groups. Statistical comparisons between donors within specific stem cell groups were not reported.
Representative results and discussion
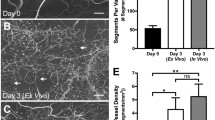
The rat mesentery culture model can be used as a novel tool to evaluate stem cell fate within intact ex vivo microvascular networks. After 5 days in culture, a subset of human derived MSCs displayed pericyte characteristics by elongating and wrapping along lectin-positive vessels within the cultured microvascular networks (Fig. 2). For the present study, paraformaldehyde fixation and immunohistochemistry of whole mounted tissues enabled the evaluation of DiI-positive stem cell location and morphology post-culture. An alternative method for tracking stem cell fate using the rat mesentery culture model includes lectin labeling of live mesentery tissues to enable real-time imaging, as demonstrated in previous studies (Stapor et al. 2013; Azimi et al. 2015). We confirmed cultured networks were angiogenic based on the observation of increased microvascular density and capillary sprouting, consistent with our previous reports (Stapor et al. 2013; Motherwell et al. 2018; Hodges et al. 2018). The pericyte identity of MSCs was defined based on cell morphology, periendothelial cell position, and DiI colocalization with NG2 labeling (Fig. 3).

MSCs differentiate into vascular pericytes. A subset of MSCs labeled with DiI cell tracker displayed pericyte location and morphology after 5 days (a). The outlined region was selected and higher magnification imaging was performed (b). The arrowheads point to examples of newly differentiated pericyte cells on lectin-positive capillaries. Images were acquired using × 4 (dry, NA = 0.1), and × 10 (dry, NA = 0.3) objectives on an inverted microscope (Olympus I× 70) coupled with a Photometrics CoolSNAP EZ camera. Scale bars = 100 μm

NG2 expression of MSCs in vascular pericyte locations. Confocal images taken from DiI-positive MSCs confirm their presence in pericyte locations (b) while expressing NG2 (b), a common pericyte marker. The arrowhead points to the cell body and the 3D projection shows DiI and NG2 co-labeling on the cell (c). Confocal microscopy images were captured with a × 40 objective (NA = 1.3) on a Nikon A1 confocal microscope equipped with 405 nm, 488 nm, and 561 nm diode laser lines, coupled with an ECLIPSE Ti Nikon inverted microscope. Optical slice thickness was less than 1 μm. Scale bars = 10 μm
The use of stem cells to manipulate angiogenesis offers a promising approach for future therapies (Ballard and Edelberg 2007; Kelly-Goss et al. 2014). Diabetic retinopathy, cardiac ischemia, and peripheral arterial disease represent age-related pathologies where the use of stem cells has already proven valuable. MSCs derived from bone marrow and adipose tissue potentially enhance angiogenesis via paracrine signaling and direct cell incorporation (Kelly-Goss et al. 2014). In order to advance our manipulation of stem cells as proangiogenic therapies, experimental models are needed to evaluate stem cell function and differentiation capacity. For aging research, critical questions remain unanswered: Do MSCs from aged populations have different capacities to become vascular cells compared with MSCs from adult populations? Does the tissue source of MSCs influence cellular fate? To answer these questions, the fate of stem cells must be tracked in a physiologically relevant microvascular network environment. The method described in this study offers an ex vivo experimental model as an alternative to current in vivo techniques.
While MSCs from aged donors are generally considered to have reduced differentiation capabilities, diminished wound repair, and decreased effectiveness at lowering inflammation (Pandey et al. 2011; Yu et al. 2011; Scruggs et al. 2013; Ungvari et al. 2018), a theme from the literature is that stem cell differentiation capacity is dependent on target cell type. For example, aging might alter adipogenic versus osteoblastic differentiation (Stenderup et al. 2001; Ding et al. 2013; Beane et al. 2014; Dufrane, 2017). The impact of aging on MSC’s ability to become specific vascular cells has not been thoroughly investigated. Our results suggest that aged MSC populations have similar cellular fate capabilities as adult MSC populations in differentiating into pericytes (Fig. 4). Pericyte differentiation is supported by the localization of the prelabeled DiI stem cells and NG2 (Fig. 3). However, it should be noted that pericytes are phenotypically and morphologically heterogeneous (Stapor et al. 2014; Kelly-Goss et al., 2014). In order to determine whether stem cells have adopted a cell type along the pericyte spectrum additional evaluation is needed. For our methods demonstration study, we defined pericytes based on NG2 labeling and morphology along blood vessels. We used NG2 as the main colabel based on our previous characterization of rat mesentery microvascular networks (Stapor et al. 2013; Stapor et al. 2014). A limitation is that other pericyte markers were not examined. We have confirmed that at least a subset of the stem cells in pericyte location can express SMA (data not shown). Similar costaining with other pericyte lineage markers, such as PDGFRβ, desmin, CD13, and/or CD146, would serve to more comprehensively identify the differentiation potential of the stem cells. Future analysis will also be needed to fully characterize whether MSCs are able to become other types of cells including endothelial cells and tissue-resident macrophages. For example, to determine endothelial fate DiI labeling can be colocalized with endothelial-specific PEACM labeling.

The average percent incorporation of stem cells into microvascular networks per experimental group. The average pericyte differentiation rate remained close across different experimental groups: (1) adult ASCs 6.72 ± 1.15%, (2) aged ASCs 5.60 ± 0.96%, (3) adult BMSCs 5.57 ± 0.82%, and (4) Aged BMSCs 6.80 ± 1.89%. Each black bar represents the average percent calculated across the 8 individual donor trials per experimental group. The data for each donor trial included analysis of approximately 10 tissues from 2 to 4 rats per donor per experimental group, and the average percent of cells in pericyte locations was calculated. Statistical analysis was carried out using one-way ANOVA. Results were considered statistically significant when p < 0.05. No significant differences were found between the experimental groups. Post hoc tests were not performed as difference in the mean values among the groups were not great enough to exclude the possibility that differences were due to random sampling. Values are averages ± SEM
Comparison of pericyte fate percentages for donors with the same age highlights donor variability (Fig. 5). In this study, the average donor age for the aged ASC and BMSC groups were 62.5 ± 6.6 years and 55.9 ± 6.1 years, respectively. Future studies will be needed to evaluate aging effects in populations older than those used in this present report. Increased donor variability with the aged MSC populations is consistent with previous reports (117). Donor variability within each experimental group is represented by the donor cell fate percentages. For example, maximum fate percentages varied for each donor group (maximums: adult ASC = 0–26%; aged ASC = 3–21%; adult BMSC = 4–23%; aged BMSC = 4–50%; minimums: adult ASC = 0–4%; aged ASC = 0–2%; adult BMSC = 0–2%; aged BMSC = 0–3%). In addition, the percentage of MSCs in pericyte location was often spatially heterogeneous within a field of view or network, where some vessels displayed a higher percentage than others (Fig. 2). The donor variability and observations of vessel- or region-specific cell fate support a phenomenon of vessel-specific recruitment and additionally raise questions regarding the importance of local microenvironments for cell recruitment and differentiation.

Representation of variability in the percent of stem cell incorporation per donor per experimental group. Each experimental group is comprised of 8 individual donors, represented by the black bars. The data for each black bar was quantified from approximately 10 tissues harvested from 2 to 4 rats, and the average percent of cells in pericyte locations was calculated. Values are averages ± SEM
An advantage of the rat mesentery culture model compared with two- and three-dimensional in vitro cell cultures or ex vivo tissue explant assays is the ability to observe single cells across the spatial hierarchy of an intact microvascular network. The mesentery’s thinness, which has been reported to be approximately 20–40 μm (Norrby 2006), enables this unique en face view. Interestingly MSCs were only observed in pericyte locations along capillaries and were not observed along larger arterioles, venules, or lymphatic vessels. Eighty-four percent of the stem cells in pericyte location were located along capillaries, and 16% were along precapillary arterioles or postcapillary venules. Zero percent of cells wrapping around vessels were located along larger arterioles/venules, and 0% were located along lymphatic vessels—an observation maybe expected because initial lymphatic vessels in the mesentery networks do not have perivascular cells. These results also suggest vessel type specific recruitment and motivate future studies to further investigate the differences in vessel type microenvironments.
The stem cell fate analysis method described here and the time-lapse capability of our model (Azimi et al. 2015; Azimi et al., 2017) motivate future studies aimed at tracking specific stem cell migration over time. For example, tracking cells over time could determine if the stem cell-derived pericytes preferentially wrap around new capillaries (formed by angiogenesis during the culture period) versus preexisting ones. Such information could be used to support the argument that stem cell-derived pericytes contribute to angiogenesis versus simply incorporating into the existing microvascular vascular network. In addition, time-lapse studies could be used to determine the specific time-course of stem cell population recruitment and differentiation.
A limitation of applying the rat mesentery culture model for aging stem cell fate studies is that it lacks vessel perfusion. However, the pericyte incorporation rate reported in our method is similar to previous reports using in vivo techniques (Mendel et al. 2013; Cronk et al. 2015). For example, in a study where human ASCs were injected into the vitreous of mice to investigate the incorporation of exogenous stem cells into the retina, they found only 3.08% of the initial cell suspension engrafted into the retina after 8 weeks (Mendel et al. 2013). Thus, we speculate our rat mesentery culture model can be used as a predictor of in vivo responses, one of the major goals for biomimetic model development. The biomimetic potential of our model is also supported by previous validation studies comparing endothelial cell phenotypes in ex vivo cultured microvascular networks and in vivo networks during angiogenesis (Motherwell et al. 2018).
Other limitations of the model include the undefined timeframe for tissue culture and physiological relevance of the mesentery compared with other tissues. While we have confirmed that non-perfused cultured tissues remain viable out to at least 7 days (Stapor et al., 2013), and have also observed viable cells along endothelial cell segments out to 14 days (data not shown), the maximum time duration for observing stem cell behaviors is unknown. For the current experiments, stem cell fate was tracked over 5 days. Regarding physiological relevance, it is again important to point out that the percentage of cells that adopt pericyte location for the current study is within the range of chronic animal studies (Kelly-Goss et al. 2014). Despite this evidence and our speculation that the rat mesentery culture model can be used as a predictor of in vivo responses, the relevance of our mesenteric model remains debatable, and it might not necessarily reflect the capacity of stem cell–derived pericytes to incorporate into other angiogenic vascular beds. Additionally, stem cell recruitment entails cell mobilization, homing to a tissue, recruitment within the tissue and differentiation. Hence, our model characterizes the recruitment of exogenously applied stem with a tissue after cells have already become tissue resident—mimicking the therapeutic scenario for which stem cells are injected into a tissue. Any effects of increased age on cell mobilization or homing to a tissue from the circulation cannot be evaluated.
In summary, here we demonstrate the rat mesentery culture model can be used to screen different stem cell populations. Both human ASCs and BMSCs can incorporate into microvascular networks when seeded onto mesenteric tissues ex vivo. The view of stem cells across an intact microvascular network highlights the value of the tissue culture model and creates new questions related to the heterogeneity of cell fate and how microenvironments within a tissue might influence behavior. The top-down tissue culture approach offers a method for comparing aged versus adult tissues harvested from healthy and pathological animal strains. The ability to exogenously introduce stem cell populations enables experiments to evaluate the influence of cell versus environment age. Fundamental questions remain. Does the aged environment matter? Does the cell age matter? The tissue culture model offers a unique view to gain spatial-temporal information about cell dynamics not possible with common single-cell or cell population analyses. In addition to the pericyte fate readout demonstrated in this study, other metrics such as cell migration, stem cell differentiation into other cell types, angiogenesis, lymphangiogenesis, immune cell activation, and stem cell pattern distribution could provide valuable insights for advancing our understanding the impact of age-related changes on stem cell-microvascular network interactions. The ability to supplement media during tissue culture also enables, for example, the evaluation of how stem cell secretomes influence network pattern changes, vessel growth, and cell proliferation or senescence. Conversely, the effects of microvascular secretome on stem cell behavior could be evaluated via immunohistochemical labeling and vessel-type specific analysis.
References
Azimi MS, Motherwell JM, Murfee WL. An ex vivo method for time-lapse imaging of cultured rat mesenteric microvascular networks. J Vis Exp. 2017. https://doi.org/10.3791/55183.
Azimi MS, Myers L, Lacey M, et al. An ex vivo model for anti-angiogenic drug testing on intact microvascular networks. PLoS One. 2015;10. https://doi.org/10.1371/journal.pone.0119227.
Beane OS, Fonseca VC, Cooper LL, et al. Impact of aging on the regenerative properties of bone marrow-, muscle-, and adipose-derived mesenchymal stem/stromal cells. PLoS One. 2014;9(12):e115963. https://doi.org/10.1371/journal.pone.0115963.
Ballard VL, Edelberg JM. Targets for regulating angiogenesis in the ageing endothelium. Expert Opin Ther Targets. 2007;11:1385–99. https://doi.org/10.1517/14728222.11.11.1385.
Bunnell BA, Flaat M, Gagliardi C, Patel B, Ripoll C. Adipose-derived stem cells: isolation, expansion and differentiation. Methods. 2008;45:115–20. https://doi.org/10.1016/j.ymeth.2008.03.006.
Cronk SM, Kelly-Goss MR, Ray HC, Mendel TA, Hoehn KL, Bruce AC, et al. Adipose-derived stem cells from diabetic mice show impaired vascular stabilization in a murine model of diabetic retinopathy: stem cells from diabetic adipose in retinopathy. Stem Cells Transl Med. 2015;4:459–67. https://doi.org/10.5966/sctm.2014-0108.
Csiszar A, Tarantini S, Fülöp GA, Kiss T, Valcarcel-Ares MN, Galvan V, et al. Hypertension impairs neurovascular coupling and promotes microvascular injury: role in exacerbation of Alzheimer’s disease. Geroscience. 2017;39(4):359–72. https://doi.org/10.1007/s11357-017-9991-9.
Ding DC, Chou HL, Hung WT, Liu HW, Chu TY. Human adipose-derived stem cells cultured in keratinocyte serum free medium: donor’s age does not affect the proliferation and differentiation capacities. J Biomed Sci. 2013;20:59. https://doi.org/10.1186/1423-0127-20-59.
Dufrane D. Impact of age on human adipose stem cells for bone tissue engineering. Cell Transplant. 2017;26(9):1496–504. https://doi.org/10.1177/0963689717721203.
Efimenko A, Starostina E, Kalinina N, Stolzing A. Angiogenic properties of aged adipose derived mesenchymal stem cells after hypoxic conditioning. J Transl Med. 2011;9:10. https://doi.org/10.1186/1479-5876-9-10.
Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol. 2009;106:326–32. https://doi.org/10.1152/japplphysiol.91353.2008.
Fuchs S, Baffour R, Zhou YF, Shou M, Pierre A, Tio FO, et al. Transendocardial delivery of autologous bone marrow enhances collateral perfusion and regional function in pigs with chronic experimental myocardial ischemia. J Am Coll Cardiol. 2001;37:1726–32. https://doi.org/10.1016/S0735-1097(01)01200-1.
Fulop GA, Kiss T, Tarantini S, Balasubramanian P, Yabluchanskiy A, Farkas E, et al. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience. 2018;40(5–6):513–21. https://doi.org/10.1007/s11357-018-0047-6.
Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314:15–23. https://doi.org/10.1007/s00441-003-0745-x.
Hao C, Shintani S, Shimizu Y, Kondo K, Ishii M, Wu H, et al. Therapeutic angiogenesis by autologous adipose-derived regenerative cells: comparison with bone marrow mononuclear cells. Am J Physiol Heart Circ Physiol. 2014;307:H869–79. https://doi.org/10.1152/ajpheart.00310.2014.
Heiss C, Keymel S, Niesler U, Ziemann J, Kelm M, Kalka C. Impaired progenitor cell activity in age-related endothelial dysfunction. J Am Coll Cardiol. 2005;45:1441–8. https://doi.org/10.1016/j.jacc.2004.12.074.
Hodges NA, Suarez-Martinez AD, Murfee WL. Understanding angiogenesis during aging: opportunities for discoveries and new models. J Appl Physiol Bethesda Md (1985). 2018;125:1843–50. https://doi.org/10.1152/japplphysiol.00112.2018.
Hoetzer GL, Van Guilder GP, Irmiger HM, et al. Aging, exercise, and endothelial progenitor cell clonogenic and migratory capacity in men. J Appl Physiol. 2007;102:847–52. https://doi.org/10.1152/japplphysiol.01183.2006.
Hou L, Kim JJ, Woo YJ, Huang NF. Stem cell-based therapies to promote angiogenesis in ischemic cardiovascular disease. Am J Physiol Heart Circ Physiol. 2016;310:H455–65. https://doi.org/10.1152/ajpheart.00726.2015.
Izadpanah R, Trygg C, Patel B, Kriedt C, Dufour J, Gimble JM, et al. Biologic properties of mesenchymal stem cells derived from bone marrow and adipose tissue. J Cell Biochem. 2006;99:1285–97. https://doi.org/10.1002/jcb.20904.
Jones R, Strong A, Gimble J, Bunnell B. Isolation and primary culture of adult human adipose-derived stromal/stem cells. BIO-Protoc. 2017;7. https://doi.org/10.21769/BioProtoc.2161.
Kachgal S, Putnam AJ. Mesenchymal stem cells from adipose and bone marrow promote angiogenesis via distinct cytokine and protease expression mechanisms. Angiogenesis. 2011;14:47–59. https://doi.org/10.1007/s10456-010-9194-9.
Kelly-Goss MR, Sweat RS, Stapor PC, Peirce SM, Murfee WL. Targeting pericytes for angiogenic therapies. Microcirculation. 2014;21:345–57. https://doi.org/10.1111/micc.12107.
Kondo K, Shintani S, Shibata R, et al. Implantation of adipose-derived regenerative cells enhances ischemia-induced angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:61–6. https://doi.org/10.1161/ATVBAHA.108.166496.
Kramerov AA, Ljubimov AV. Stem cell therapies in the treatment of diabetic retinopathy and keratopathy. Exp Biol Med. 2016;241:559–68. https://doi.org/10.1177/1535370215609692.
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039.
Mendel TA, Clabough EBD, Kao DS, et al. Pericytes derived from adipose-derived stem cells protect against retinal vasculopathy. PLoS One. 2013;8:e65691. https://doi.org/10.1371/journal.pone.0065691.
Minamino T, Miyauchi H, Yoshida T, et al. Vascular cell senescence and vascular aging. J Mol Cell Cardiol. 2004;36:175–83. https://doi.org/10.1016/j.yjmcc.2003.11.010.
Moriya J, Minamino T. Angiogenesis, cancer, and vascular aging. Front Cardiovasc Med. 2017;4. https://doi.org/10.3389/fcvm.2017.00065.
Motherwell JM, Anderson CR, Murfee WL. Endothelial cell phenotypes are maintained during angiogenesis in cultured microvascular networks. Sci Rep. 2018;8:5887. https://doi.org/10.1038/s41598-018-24081-z.
Norrby K. In vivo models of angiogenesis. J Cell Mol Med. 2006;10:588–612. https://doi.org/10.1111/j.1582-4934.2006.tb00423.x.
Ozerdem U. Targeting of pericytes diminishes neovascularization and lymphangiogenesis in prostate cancer. Prostate. 2006;66:294–304. https://doi.org/10.1002/pros.20346.
Pachón-Peña G, Yu G, Tucker A, Wu X, Vendrell J, Bunnell BA, et al. Stromal stem cells from adipose tissue and bone marrow of age-matched female donors display distinct immunophenotypic profiles. J Cell Physiol. 2011;226:843–51. https://doi.org/10.1002/jcp.22408 .
Pandey AC, Semon JA, Kaushal D, et al. MicroRNA profiling reveals age-dependent differential expression of nuclear factor κB and mitogen-activated protein kinase in adipose and bone marrow-derived human mesenchymal stem cells. Stem Cell Res Ther. 2011;2:49.
Rajantie I, Ilmonen M, Alminaite A, Ozerdem U, Alitalo K, Salven P. Adult bone marrow–derived cells recruited during angiogenesis comprise precursors for periendothelial vascular mural cells. Blood. 2004;104:2084–6. https://doi.org/10.1182/blood-2004-01-0336.
Rehman J, Traktuev D, Li J, Merfeld-Clauss S, Temm-Grove CJ, Bovenkerk JE, et al. Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation. 2004;109:1292–8. https://doi.org/10.1161/01.CIR.0000121425.42966.F1.
Scruggs BA, Semon JA, Zhang X, Zhang S, Bowles AC, Pandey AC, et al. Age of the donor reduces the ability of human adipose-derived stem cells to alleviate symptoms in the experimental autoimmune encephalomyelitis mouse model. Stem Cells Transl Med. 2013;2:797–807. https://doi.org/10.5966/sctm.2013-0026.
Semon JA, Maness C, Zhang X, Sharkey SA, Beuttler MM, Shah FS, et al. Comparison of human adult stem cells from adipose tissue and bone marrow in the treatment of experimental autoimmune encephalomyelitis. Stem Cell Res Ther. 2014;5:2. https://doi.org/10.1186/scrt391.
Stapor PC, Azimi MS, Ahsan T, Murfee WL. An angiogenesis model for investigating multicellular interactions across intact microvascular networks. Am J Physiol Heart Circ Physiol. 2013;304:H235–45. https://doi.org/10.1152/ajpheart.00552.2012.
Stapor PC, Sweat RS, Dashti DC, Betancourt AM, Murfee WL. Pericyte dynamics during angiogenesis: new insights from new identities. J Vasc Res. 2014;51(3):163–74. https://doi.org/10.1159/000362276.
Stenderup K, Justesen J, Eriksen EF, Rattan SI, Kassem M. Number and proliferative capacity of osteogenic stem cells are maintained during aging and in patients with osteoporosis. J Bone Miner Res. 2001;16(6):1120–9.
Strong AL, Bowles AC, MacCrimmon CP, Frazier TP, Lee SJ, Wu X, et al. Adipose stromal cells repair pressure ulcers in both young and elderly mice: potential role of adipogenesis in skin repair: adipose stromal/stem cells repair pressure ulcers. Stem Cells Transl Med. 2015;4:632–42. https://doi.org/10.5966/sctm.2014-0235.
Sweat RS, Sloas DC, Murfee WL. VEGF-C induces lymphangiogenesis and angiogenesis in the rat mesentery culture model. Microcirculation. 2014;21:532–40. https://doi.org/10.1111/micc.12132.
Sure VN, Sakamuri SSVP, Sperling JA, Evans WR, Merdzo I, Mostany R, et al. A novel high-throughput assay for respiration in isolated brain microvessels reveals impaired mitochondrial function in the aged mice. Geroscience. 2018;40(4):365–75. https://doi.org/10.1007/s11357-018-0037-8.
Tarantini S, Fulop GA, Kiss T, Farkas E, Zölei-Szénási D, Galvan V, et al. Demonstration of impaired neurovascular coupling responses in TG2576 mouse model of Alzheimer’s disease using functional laser speckle contrast imaging. Geroscience. 2017;39(4):465–73. https://doi.org/10.1007/s11357-017-9980-z.
Ungvari Z, Tarantini S, Kiss T, Wren JD, Giles CB, Griffin CT, et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol. 2018;15(9):555–65. https://doi.org/10.1038/s41569-018-0030-z.
Yu JM, Wu X, Gimble JM, Guan X, Freitas MA, Bunnell BA. Age-related changes in mesenchymal stem cells derived from rhesus macaque bone marrow: effect of biological aging on MSCs. Aging Cell. 2011;10:66–79. https://doi.org/10.1111/j.1474-9726.2010.00646.x.
Zhou S, Greenberger JS, Epperly MW, Goff JP, Adler C, Leboff MS, et al. Age-related intrinsic changes in human bone-marrow-derived mesenchymal stem cells and their differentiation to osteoblasts. Aging Cell. 2008;7:335–43. https://doi.org/10.1111/j.1474-9726.2008.00377.x.
Funding
The funding for this project was supported by funding from the National Institute of Aging (Grant number R01AG049821) awarded to W.M.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All experimental protocols were approved by Tulane University’s Institution of Animal Care and Use Committee (IACUC) or University of Florida’s IACUC in accordance with the National Institute of Health guidelines for animal care and use.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Azimi, M.S., Motherwell, J.M., Dutreil, M. et al. A novel tissue culture model for evaluating the effect of aging on stem cell fate in adult microvascular networks. GeroScience 42, 515–526 (2020). https://doi.org/10.1007/s11357-020-00178-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-020-00178-0