
Abstract
Taking into account difficulties in exhaustive simultaneous decarbonation and denitrogenation in biological treatment of coking wastewater (CWW), a novel full-scale CWW biological treatment sequentially combining anaerobic, aerobic, hydrolytic, and aerobic reactors (A/O1/H/O2) was designed performing excellent removal of carbon-containing pollutants in the bioreactors A and O1, while the nitrogen-containing compounds in the bioreactors H and O2. To provide an effective tool for the CWW treatment monitoring and control, the succession of microbial community in this unique toxic CWW habitat should be established and characterized in detail. The results of 16S rRNA genes revealed Acidobacteria dominating in the unique CWW habitat. The dominant groups in bioreactors A and O1 include Proteobacteria, Firmicutes, and Acidobacteria, while Proteobacteria, Acidobacteria, Nitrospirae, and Planctomycetes dominate in reactors H and O2. The genera of Rhodoplanes, Bacillus, and Leucobacter are rich in genes responsible for the xenobiotics biodegradation and metabolism pathway. The Mantel test and PCA results showed the microbial communities of A/O1/H/O2 sequence correlating strongly with SRT, and COD load and removal. The co-occurrence network analysis indicated decarbonation and denitrogenation driven by two network modules having the keystone taxa belonging to the Comamonadaceae and Hyphomicrobiaceae families. The results significantly expanded the knowledge on the diversity, structure, and function of the CWW active sludge differentiating the relationships between bacterial communities and environmental variables in CWW treatment.
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
China producing more than 60% coke in the world faces severe problems with coking wastewater (CWW) generated annually in huge amounts of 3.12 × 108 m3 (Wei et al. 2019). The wastewater is characterized by complex composition of pollutants making it one of the most toxic industrial effluents causing dramatic problems to the ecosystems and human health (Zhao et al. 2018). The toxicity of CWW inhibits bacterial activity thus reducing the removal efficiency of carbon- and nitrogen-containing pollutants in traditional CWW biological treatment processes.
Coking wastewater treatment presents a multi-billion-dollar industry in the world widely applying active sludge (AS) in various CWW treatment processes removing more than 95% of the pollutants (Zhu et al. 2019). Biological processes comprise the key step in removing carbon- and nitrogen-containing pollutants at high removal efficiency and low operational cost. The current target of engineering design in CWW treatment is decarbonation and denitrogenation achieved with minimum consumption of dissolved oxygen (DO) in aerobic treatment and with nitrification–denitrification sequence, respectively, for minimizing operational costs. Simultaneous decarbonation and denitrogenation, however, is achieved by rear combinations of anaerobic (A), aerobic (O), and hydrolytic (H) biological processes. For example, A/O, A/O/O, and A/A/O processes show good performance in nitrogen removal, whereas O/H/O and O/A/O provide excellent decarbonation results (Fukushima et al. 2013; Li et al. 2019a; Wei et al. 2021). As a rule, satisfactory denitrogenation is difficult to realize for the lack of available carbon source, making the structure and function of CWW microbial community unstable (Zheng et al. 2019). This task becomes particularly hard in respect to the nitrogen hidden in toxic substances—thiocyanates (SCN−) and cyanides (CN−)—present in CWW in high concentrations: this nitrogen has to be transferred to ammonium (NH4+-N) before nitrification and denitrification processes. In order to simultaneously remove carbon- and nitrogen-containing pollutants with high efficiency, the A/O1/H/O2 sequence of biological processes was designed showing good simultaneous decarbonation and denitrogenation at stable operation for the last 7 years at the discharge quality meeting the requirements of the national standard (GB16171-2012). The idea of the A/O1/H/O2 design was a combination of decarbonation achieved in the A/O1 bioreactors with subsequent nitrogen removal with high efficiency in the H/O2 sequence. Compared to other CWW treatment processes, the performance of A/O1/H/O2 demonstrated significant advantages in total organic carbon (TOC) and total nitrogen (TN) removal (Qin et al. 2022). The microbial community of the A/O1/H/O2 sequence, however, is not clearly known in diversity and composition of biota, the knowledge of which may provide theoretical justification of the A/O1/H/O2 combination performance.
The functionality and efficiency of CWW treatment are largely determined by the composition of microbial communities (Joshi et al. 2017). Thus, a thorough knowledge of the ecology of the CWW active sludge communities is required to improve the feasibility and stability of a biological treatment system. The advent of high-throughput sequencing technology provides new opportunities addressing the knowledge gaps by comprehensive characterization of the microbial communities in their relationships under the pressure of environmental variables. The authors failed to find surveys on microbial diversity in communities with their variations for the A/O1/H/O2 full-scale CWW treatment systems in relation to their performance in decarbonation and denitrogenation.
The structure and function of the full-scale A/O1/H/O2 microbial communities were studied using the 16S rRNA genes approach. The physicochemical characteristics of CWW measured by standard methods together with operational parameters were registered during the long-term stable operation matched with the analysis of microbial communities. Illumina MiSeq high-throughput sequencing was used to reveal the distinct species and the diversity of microbial community in the A, O1, H, and O2 bioreactors. An exploratory bioinformatics tool, phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt), was used to predict the CWW active sludge community function based on 16S rRNA genes information. The objectives pursued by this study include (1) establishing the variations in the abundance, diversity, and function of microbial communities in the A/O1/H/O2 bioreactors; (2) identification of the dominant bacterial groups and presumption of the biodegradation potential in the A/O1/H/O2 process combination in simultaneous decarbonation and denitrogenation; (3) elucidating the relationships between the bacterial communities and the impact of environmental conditions on the bioreactors. The results of the study are applicable to monitoring of the CWW active sludge’s biodegradation potential and its treatment efficiency.
Materials and methods
Full-scale A/O1/H/O2 sequence of CWW treatment: collection of samples
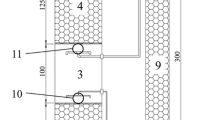
An industrial-scale CWW treatment in A/O1/H/O2 sequence exploiting the three-phase fluidized bed reactors providing simultaneous decarbonation and denitrogenation at the Baowu Group Guangdong Shaoguan Iron and Steel Co., Shaoguan city, Guangdong Province, China, was studied. The raw CWW was treated in three stages including the primary stage, the biological stage, and the advanced stage (Fig. 1). The biological treatment consists of four sequential fluidized bed bioreactors: anaerobic (A), aerobic-1 (O1), hydrolytic (H), and aerobic-2 (O2). The biological reactors sequence operates at daily capacity of approximately 2800 m3 of CWW, providing stable performance at a satisfactory result with the total hydraulic retention time (HRT) of 76 h.

Flow outline of the full-scale A/O1/H/O2 coking wastewater treatment combination: BI, biological influent; BE, biological effluent; FE, final effluent
The active sludge samples were collected in June, 2019, on daily basis from separate biological reactors three times a day with 8-h intervals. Samples taken during the day were mixed together to form an average sample of the day. Mixed liquor was also sampled in triplicate from separate bioreactors. The samples were kept in ice boxes during the sampling and transportation to the laboratory. Aliquots of 2–3 mL of each sample were centrifuged at 12,000 g for 10 min at 4 °C. The cell pellets were rinsed twice with 120 mM sodium phosphate buffer at pH 8.0 and stored at − 20 °C prior to the DNA extraction.
Physicochemical properties and operational parameters
The collected CWW samples were centrifuged at 3500 g for 3 min and the supernatants were analyzed using standard methods for the chemical oxidation demand (COD) and biochemical oxygen demand (BOD). The contents of phenolic compounds, sulfides (S2−), SCN−, and total phosphorus (TP) were analyzed by the colorimetric methods with a spectrophotometer (Genesys TM-5, Spectronic Inc. USA). Total nitrogen (TN), ammonium (NH4+-N), nitrate (NO3−), and nitrite (NO2−) were measured using their respective standard methods (Chinese SEPA 2002). After distillation, the CN− concentration was determined by the pyridine-pyrazolone method. The contents of PAHs were analyzed using the 7890A-5975C GC/MS (Agilent, USA). Total organic carbon (TOC) was measured using a TOC analyzer Shimadzu TOC-VCPH (Japan). The DO concentration was measured using the CellOX 3310i DO meter (WTW, Germany), and the pH was monitored with a pH meter (PHS-3D, China).
The design of bioreactor is known to directly influence the pollutants removal efficiency and the distribution of microbial population species. In order to deal with a high COD load rate (1.88 kg COD m−3 day−1), an internal-loop multiphase airlift–fluidized bed reactor was developed by the authors and applied in the full-scale reactors under consideration (Wei et al. 2000). The temperatures, pH, DO, hydraulic retention times (HRTs), and sludge retention times (SRTs), at which the reactors are operating, are given in Table 1. Long-SRTs are beneficial for the development of specific functional microorganism species, such as ammonium-oxidizing bacteria (AOB) and nitrite-oxidizing bacteria (NOB). The concentrations of active sludge varied across the range of 2620 to 4,490 mg L−1 over the course of operation. Sludge retention times are controlled by the removal rate of excess sludge, resulting in different mixed liquor suspended solids (MLSS) concentrations in the biological treatment processes.
DNA extraction and MiSeq sequencing of 16S rRNA genes
Microbial genomic DNA extraction was conducted using the PowerSoil™ DNA isolation kit (Mobio, USA) according to the manufacturer’s protocol. Concentration and purity of extracted DNA was determined by the ratios of 260/280 and 260/230 nm absorption measured by the ND-2000 spectrophotometer (Nanodrop Inc. Wilmington, DE, USA).
The V4 hyper variable region of 16S rRNA gene was PCR-amplified (triplicate reactions for each sample) using primers F515 (5′-GTGCCAGCMGCCGCGGTAA-3′) and R806 (5′-GGACTACVSGGGTATCTAAT-3′) (Fierer et al. 2008). A single composite DNA sample for MiSeq sequencing was prepared by combining approximately equimolar amount of purified PCR products from each sludge sample. The composition of the PCR products of V4 region of 16S rRNA genes was determined by Illumina MiSeq platform (Illumina Inc., San Diego, CA, USA). The sequences have been deposited in the NCBI Sequence Read Archive under the accession number PRJNA615645.
MiSeq sequence analysis
After MiSeq sequencing, the raw data were processed and analyzed following the pipelines of Mothur (v1.35.1) and QIIME (v. 1.9.1). Raw data were denoised using the Mothur implementation of PyroNoise algorithm. Operational taxonomic units (OTUs) defined at the 97% sequence similarity level were picked using average neighbor method after the Needleman alignment and a single-linkage pre-cluster procedure. Taxonomic assignment was obtained using RDP Classifier at the 80% confidence threshold by default. Based on the OTU information, alpha-diversity indices, including the Shannon, Simpson, Chao 1, ACE, and number of OTUs index, were calculated using R (v 3.6.2) (http://www.r-project.org/). Good’s coverage estimators were calculated using the Mothur software (v 1.35.1) and rarefaction curve was analyzed by R (v 3.6.2). The PcoA was used to show the beta-diversity, which explores differences in the microbial communities between the bioreactors A, O1, H, and O2 by R software (v 3.6.2).
To investigate the functional profiles of the microbial community dataset, a bioinformatics tool PICRUSt predicting gene family abundances was used based on 16S rRNA gene surveys, given a database of phylogenetically referenced genomes (Langille et al., 2013). For the analysis, OTUs were closed-reference picked against the May 18, 2012 Greengenes database using QIIME (v. 1.9.1) according to the online protocol. The resulting dataset was rarefied at 10,570 sequences per sample. These data were used to assess the difference between gene functions of the sequential A/O1/H/O2 active sludge.
Network analysis of microbial communities
The co-occurrence networks are used to statistically identify keystone taxa (Berry and Widder 2014). Microbial association networks were constructed to investigate the possible biotic interactions using the Molecular Ecological Network Analyses Pipeline (MENAP) (http://ieg4.rccc.ou.edu/mena) (Deng et al. 2012). The OTUs with the relative abundance exceeding 0.3% in the samples were used for network construction avoiding poorly represented OTUs and reducing network complexity. Network was visualized using Gephi (http://gephi.github.io/), and the topological property of betweenness centrality in the networks was also determined by Gephi. Local property measures degree and betweenness centrality of microbe can efficiently identify the key species in a microbial association network. In this study, positive edges correlation implied a mutualistic interaction, whereas negative edges correlation indicated competition.
Statistical analysis
The relationships between microbial community and coking wastewater characteristics/operational parameters were examined by Mantel tests analysis and principal component analysis (PCA) using R 3.6.2 (http:// www.r-project.org/) with the vegan packages. The redundancy analysis (RDA) was carried out using R (v 3.6.2) to discern possible associations among the major genera and variable wastewater characteristics. The ANOVA was performed to assess the significance of relative abundance of phyla, genera, and genes in bioreactors, having a, b, c, and d indicating significant differences at the p < 0.05 level.
Results and discussion
Performance of the A/O1/H/O2 sequence
Functional operation performance of the full-scale CWW treatment A/O1/H/O2 sequence was studied in variation of fifteen CWW indicators including COD, BOD5, concentrations of phenolic compounds, CN−, SCN−, ammonia, NO2−, NO3−, TN, total phosphorus (TP), total suspended solids (TSS), TOC, sulfides, polyaromatic hydrocarbons (PAHs), and oil. Table 2 shows the CWW quality indices inside the bioreactors comprising the A/O1/H/O2 sequence illustrating its functional stability. The COD removal in the reactors sequence comprised approximately 97%. The ratio of BOD5/COD (B/C ratio) used as a key parameter in wastewater biodegradability evaluation is also a measure of available energy and toxicity to microorganisms in active sludge. The ratio is hypothesized being responsible for the composition of the active sludge microbial community (Zhang et al. 2019a). The B/C ratios in A/O1/H/O2 sequence comprised 0.42, 0.51, 0.10, 0.27, 0.08, and 0.10 for the influent to the biological treatment, the bioreactors A to O2, and the effluent from the biological treatment, respectively, showing the majority of biodegradable organics removed in the A and O1 bioreactors (Table 2). Noteworthy, the B/C ratio increased in the bioreactor H due to the hydrolytic acidification transferring the refractory substances to the biodegradable ones. The removal of phenolic compounds reached 99.99% in the final effluent with the major eliminations of 29.9% and 69.7% in the bioreactors A and O1, respectively. The average removals of SCN− and CN− were also excellent reaching 99.96% and 99.53%, respectively. Most of the CWW quality indices were improved for more than 70% in the bioreactors A and O1. Interestingly, in the bioreactor A, the concentrations of NO3− and NH4+-N both increased from 5 to 7 mg L−1 and from 153 to 165 mg L−1, respectively, implying that nitrogen-containing heterocyclic compounds, being partly decomposed, may release nitrogen in the NH4+-N form. At the same time, the concentration of NO2− in the bioreactor O1 increased for an order of magnitude from 2.6 to 26.3 mg L−1, while NH4+-N decreased twofold from 165 to 83 mg L−1 demonstrating the performance of ammoxidation at this stage. In the bioreactor H, the concentrations of TN, NH4+-N, and NO2− dramatically dropped showing a good nitrogen removal (Table 2). Due to the hydrolytic acidification in the H-unit, the subsequent bioreactor O2 shows the removal of small-molecular pollutants: the content of NO3− increased substantially implying the complete nitrification taking place in the unit. In a word, most of the carbon-containing pollutants are removed in the bioreactors A and O1, while the denitrogentation takes place in H and O2. The results showed that the physicochemical characteristics describing the pollutants removal reflect and determine biological properties of active sludge in the reactors, determining the CWW treatment performance.
Analysis of phylogenetic diversity
High-throughput MiSeq sequencing was used to analyze the bacterial and archaeal 16S rRNA genes across the 12 active sludge samples from four bioreactors of the A/O1/H/O2 system. A total of 209,522 effective sequences and 1887 OTUs were retrieved from the samples. Based on the subset of 10,570 randomly selected sequences, the average of 688, 368, 654, and 655 OTUs were identified in the sludge samples taken from A, O1, H, and O2 bioreactors, respectively (Table 3). The OTU numbers detected in the sludge of the A/O1/H/O2 reactors are smaller than those observed at municipal WWTPs (Wu et al. 2019) as was shown earlier in the treatment of CWW and other industrial wastewaters (Zheng et al. 2019; Zhu et al. 2019). The rarefaction curves reached the plateau and the Good’s coverage ranged from 97.06 to 99.02% in all samples (Table 3), indicating that the sampling depth was sufficient to characterize the majority of microbial community.
The diversity indices, Shannon or Simpson index, showed that the biodiversity increased in the bioreactors in the sequence of O1, H, and O2. The first in the operation sequence bioreactor A, however, exhibited the highest bacterial diversity among all the treatment stages (Table 3). The Chao1 and ACE indices were used to estimate the number of OTUs present in communities; both indices are positively related to the community richness. Considering the indices of Chao 1, ACE, and the number of OTUs, one can see that the lowest and the highest community richness numbers were shown by O1 and A reactors, respectively. In general, the CWW active sludge samples demonstrate moderate biodiversity, which is more complicated than those of extreme habitats, e.g., hydrothermal and acid mine drainage (Wang et al. 2013; Zhang et al. 2019b), but simpler than soil (Nottingham et al. 2018; Sun et al. 2019). Heatmap of distance matrix and principle co-ordinates analysis (PcoA) were calculated to compare beta-diversity among the CWW bioreactor samples. The results of PcoA showed the beta-diversities of microbial communities in the bioreactors being at far distances from each other (Fig. 2), which implied the bioreactors harboring unique microbial populations. The microbial diversity in the CWW treatment reactors showed distinct community compositions highly coherent with the wastewater characteristics and environmental variables (Joshi et al. 2018).

Beta-diversity PcoA based on the OTUs abundance in active sludge samples
Taxonomic composition of microbial communities
The Ribosomal Database Project (RDP) Classifier was used to assign the effective sequences to different OTUs and phylogenetic taxa with 3% of nucleotide cutoff. Almost all the classified sequences (99.73–100%) in the A/O1/H/O2 treatment were assigned to bacteria, while only ≤ 0.27% sequences were identified as archaea in anaerobic A and hydrolytic H bioreactors (Table S1). The DO is kept at low levels in bioreactors A (0.1–0.3 mg L−1) and H (0.3–0.8 mg L−1) on the course of their long-term operation making the detection of minor anaerobic archaea in these facultative anaerobic bioreactors reasonable (Zhu et al. 2016). The relative abundance of the microbial communities was summarized at the phylum and genera levels for each sample in Tables S1, S2 and S3. A total of 40 bacterial phyla, including 6 subphyla of Proteobacteria, and 148 classified genera were identified in the A/O1/H/O2 sequence. The identified genera retrieved from the bioreactors appeared being dramatically different from each other with a long tail of low-abundance genera (Fig. 3c and S1).

Relative abundance (%) of major phyla, genera, and families in the bioreactors: the relative abundance > 1% in at least one sample defined the phyla, genera or families as major; taxa accounted for < 1% were defined as minor phyla; the average of three parallels represent the value of the corresponding sample); refer to Fig. 1 for abbreviations; a phylum (Proteobacterial subphyla); b family; c genus
In the anaerobic bioreactor A, the predominant phylum Proteobacteria was quantified including subphyla β-Proteobacteria and α-Proteobacteria in amount of 38.32% and 8.68%, respectively. Firmicutes, the subdominant phylum with its relative abundance of 19.34% is consistent with the results of previous study identifying Firmicutes as the dominant group in the first stage of anaerobic treatment of CWW (Zhu et al. 2016). Acidobacteria and Bacteroidetes follow the dominant phyla with their 7.28% and 7.11% relative abundance, respectively (Fig. 3a). Most identified OTUs of β-Proteobacteria and Acidobacteria showed positive associations with COD removal from wastewaters by means of RDA and network analysis (Jia et al. 2019). Yang et al. (2015) reported the major role of anaerobic Bacteroidetes in fermentation systems breaking down macromolecules of proteins, starch, and cellulose. At genus level, Bacillus (6.01%) was the predominant genus being able to degrade diesel oil possessing inherent potential of biosurfactant production concomitant with hydrocarbon degradation (Parthipan et al. 2017). Besides, Bacillus exhibits also high ammonification activity in the N-cycle process (Huang et al. 2019). Rhodoplanes (3.87%), Thiobacillus (3.15%), Nitrospira (1.79%), and T78 (1.75%) comprised the subdominant groups. Thiobacillus is able to degrade toxic pollutants such as CN− and SCN− in the first anaerobic reactor of CWW treatment (Joshi et al. 2017; Zhou et al. 2017). Additionally, the genus of T78, prosperous in the first anaerobic bioreactor at its relative abundance of 1.75%, is capable of anaerobic degradation of benzene compounds and oil hydrocarbons (Xu et al. 2018).
In the aerobic bioreactor O1, Acidobacteria-related sequences were the predominant phylum while over 60% of the sequences were assigned to the Ellin6075 family (Fig. 3b). Acidobacteria, particularly Gp4 and Gp6, was frequently detected in active sludge being the core genera shared by dozens of wastewater treatment systems analyzed by pyrosequencing (Ma et al. 2015). Surprisingly, Acidobacteria was found thriving in the O1 stage with its relative abundance of 62.02%. Previous study showed that the fluctuations of Acidobactera, the slowly growing species strongly associated with the biochar mineral complex and sludge, are related to the removal activity of NH4+-N (Jia et al. 2019). Proteobacteria was the subdominant group with 28.94% abundance. Especially, the relative abundance of β-Proteobacteria-related sequences also comprise more than one-fifth in O1 indicating the major function of the aerobic bioreactor as decarbonation. At the genus level, Thiobacillus was the predominant genus with relative abundance of 3.96%. Thiobacillus is known for its sulfur metabolism and denitrifying ability, making it related to the degradation of S/N-containing pollutants, including SCN−, CN−, N-heterocyclic carbenes, and PAHs (Joshi et al. 2018). At the same time, interesting finding was made in respect of Bdellovibrio genus burst out from 0.45 to 3.64% thus ranked as the second dominant group. Bdellovibrio, affiliated with Deltaproteobacteria, is a major group of predatory bacteria preying on Gram-negative ones (Niu et al. 2016). Therefore, prosperous Bdellovibrio inhibits the growth of Gram-negative bacteria, such as, e.g., Bacillus. The predatory relations thus reduce the OTU number and the diversity indices in the O1 active sludge.
In the hydrolytic bioreactor H, 47.28% of the classifiable sequences were also assigned to the predominant phylum Acidobactera. β-Proteobacteria, Nitrospirae, Chlorobi, Bacteroidetes, α-Proteobacteria, and Planctomycetes comprised the subdominant groups with the relative abundance values of 23.13%, 7.18%, 5.06%, 4.34%, 2.87%, and 2.22%, respectively. The occurrence of complex community with a large fraction of the Chlorobi, Bacteroidetes, Chloroflexi, and Proteobacteria phyla was reported in anammox bioreactors (Lawson et al. 2017). Besides, Bacteroidetes are involved in nitrification, including autotrophic metabolism with subsequent nitrite oxidation (Wu et al. 2018). Chlorobi-affiliated bacteria are highly active protein degraders, catabolizing extracellular peptides while recycling nitrate to nitrite (Lawson et al. 2017), thus making their rise in the bioreactor H reasonalbe. β-Proteobacteria play a key role in denitrification under low toxicity conditions (Zhou et al. 2019), and, being combined with the prosperous Nitrospirae, explains why the total nitrogen was removed by denitrification for more than 50% in the bioreactor H. At the genus level, Nitrospira dominates with the relative abundance of 7.18%, playing a crucial role in the nitrogen cycle enhancing the ammonia oxidation, thus contributing to the NH4+-N removal (Zhang et al. 2020). The subdominant genera groups include Thiobacillus and Rhodoplanes with their relative abundances of 6.38% and 1.17%, respectively. The previous study reported Thiobacillus and Rhodoplanes being the key microorganisms in the FeS-driven denitrification process removing both carbon- and nitrogen-containing compounds (Ma et al. 2019; Zhu et al. 2019). The Rhodoplanes genus is also diverse in metabolic capabilities playing an important role in denitrification (Sun et al. 2017).
Finally, in the aerobic bioreactor O2, Acidobacteria, Nitrospirae, Planctomyces, β-Proteobacteria, α-Proteobacteria, γ-Proteobacteria, and δ-Proteobacteria comprised the dominant phyla each containing 42.40%, 19.17%, 10.17%, 9.01%, 6.54%, 1.52%, and 1.06% of detections, respectively (Table S1). Noticeably, Nitrospirae and Planctomycetes boomed in O2 to the highest among the reactors, showing 19.17% and 10.17%, respectively. Nitrospirae species may catalyze the second step of nitrification, while the major function of phylum Planctomycetes consists of the removal of nitrogen-containing pollutants (Sorokin et al. 2012). Species of the phylum Proteobacteria, including α-, β-, and γ-Proteobacteria, are capable of autotrophic denitrification (Miao and Liu 2018). Interestingly, the relative abundance of β-Proteobacteria decreased steadily from 38.32% in the reactor A to 9.02% in the reactor O2 along the reactors’ sequence A/O1/H/O2 (Fig. 3a). The decreasing trend in β-Proteobacteria abundance is explained by its function to remove organic compounds and nutrient elements from CWW diminishing along the treatment from stage to stage in the reactors combination with decreasing decarbonation and denitrogenation rates. The genera detected in the bioreactor O2 were dominated by Nitrospira (19.16%), Thiobacillus (5.71%), Sphingobium (1.32%), and Planctomyces (1.01%). The most important finding in the bioreactor O2 consists of the prosperous NOB genus Nitrospira accounting for nearly a fifth part of the whole community and finalizing nitrification by oxidation of nitrite to nitrate (Song et al. 2020). Since more than 70% of sequences detected in the CWW active sludge were not classified to specific genera, one can consider the knowledge about the microbial resource in CWW active sludge as a limited one. Summarizing, the microbial composition analysis showed the bioreactors A and O1 responsible for the removal of carbon-containing pollutants, while the nitrogen-containing pollutants were removed mostly in the bioreactors H and O2.
Function profiles involved in removal of carbon- and nitrogen-containing pollutants
Function prediction was proposed to explore the functional pathways related to the removal of carbon- and nitrogen-containing pollutants in the A/O1/H/O2 biological CWW treatment system. The accuracy of metagenome predictions was quantified by means of the Nearest Sequenced Taxon Index (NSTI) having the lower NSTI values indicating a closer mean relationship (Langille et al. 2013). The weighted NSTI scores ranged from 0.14 to 0.25 with the average of 0.19 ± 0.06 providing a suitable data set for examination of PICRUSt predictions. The predicted results in this study provide a specific insight into the bacterial functions in the removal of carbon- and nitrogen-containing pollutants.
In the predicted metagenomes, 41 out of 43 level 2 KEGG Orthology groups (KOs) were represented. The dominating metabolism pathways providing survival of microorganisms include amino acid (10.29 ± 0.41%), carbohydrate (10.07 ± 0.22%), and energy (6.08 ± 0.75%) metabolisms (Fig. 4a). These three dominating pathways, essential for the growth of microorganisms, imply the pollutant degradation mechanisms in CWW treatment (Qiu et al. 2019). The energy metabolism pathway and the pathway of xenobiotics biodegradation and metabolism accounted for the high proportion of 5.35 − 6.74% and 2.48 − 4.21% in the A/O1/H/O2 process, respectively, contributing to the removal of carbon- and nitrogen-containing pollutants, such as NH4+, CN−, SCN− and phenols.

Microbial metabolic pathways in active sludge samples of A/O1/H/O2 coking wastewater treatment process. a. second-level pathways in the KEGG; b. sub-pathways in xenobiotics degradation pathways
Degradation pathways for carbon-containing pollutants are crucial for any wastewater treatment (Sun et al. 2014). In order to unveil those in CWW active sludge, the biodegradation and metabolism pathways of xenobiotics were considered (Fig. 4b). Generally, the xenobiotics biodegradation and metabolism pathway consists of 20 specific metabolic sub-pathways. In this case study, the relative abundance of the degradation pathways of aminobenzoate, benzoate, and caprolactam decrease along the sequential A/O1/H/O2 process (Fig. 4b), implying that these pathways are important in the removal of major carbon-containing pollutants, aromatic compounds, CN−, and phenols (Deng et al. 2020). These functional pathways are attributed to the previously detected major genera of Bacillus, Rhodoplanes, Planctomyces, and Leucobacter. So combined with the microbial community composition results, the major function of the bioreactors A and O1 consists of the removal of carbon-containing pollutants. At the same time, the pathways of drug metabolism—other enzymes, caprolactam, and nitrotoluene degradation intensify in the bioreactors H and O2, showing the pathways being important in the removal of nitrogen-containing pollutants (Fig. 4b). The predominant genus Nitrospira belonging to the Nitrospirae phylum is known for nitrification in the removal of nitrogenous pollutants and provides the gene porB for the nitrotoluene degradation pathway.
Nitrification and denitrification comprise the major TN removal processes in general, and also in the A/O1/H/O2 biological CWW treatment process combination. The predicted genes in the nitrogen metabolism sub-pathway are also related to the nitrogen recycling in nitrification and denitrification. Six nitrification-related genes, amoA, amoB, amoC, hao, nxrA, and nxrB, and 11 denitrification-related genes, narG, narH, narI, napA, napB, nasA, nasB, nirK, norB, norC, and nosZ, are involved in nitrogen removal in the considered reactors sequence. The nitrification-related genes of the amoCAB and hao show the highest abundance in O1 (Fig. 5). The ammonium monooxygenase and hydroxylamine oxidoreductase, which convert ammonia to nitrite by ammonium oxidation, are encoded by the amoCAB operon (amoC, amoA, and amoB) and hao gene, respectively (Mendez-Garcia et al. 2015; Sun et al. 2014). It is well known that NOB oxidize nitrite to nitrate by the NXR enzyme, which is membrane-associated and contains an alpha-subunit (nxrA) with the catalytic site, and a beta-subunit (nxrB) that channels electrons derived from nitrite to downstream components of the respiratory chain (Sorokin et al. 2012). The nitrifying genes nxrA and nxrB responsible for the oxidation of NO2– to NO3– demonstrate higher relative abundance in the bioreactors A and O2, as compared to O1 and H (Tang et al. 2020a). This is in a good accordance with the high abundance of Nitrospira and Planctomyces in the A- and O2-active sludge. Both denitrification and dissimilatory nitrate reduction start with the reduction of NO3− to NO2− either by means of the periplasmic nitrate reductase NapAB or the cytoplasmic nitrate reductase NarGHI (Mendez-Garcia et al. 2015; Jia et al. 2019). Subsequently, the resulting NO2− is reduced to nitrogen gas by nitrite reductase (NirK), NO reductase (norB and norC), and nitrous oxide reductase (nosZ) (Tang et al. 2020a). The denitrification nirK gene was prevalent in the bioreactors H and O2, and the relative abundance of nosZ gene was the highest in H showing the most nitrogen-containing pollutants removed in the bioreactor H by denitrification.

Relative abundance (%) of nitrogen recycling genes involved in nitrification and denitrification in the A/O1/H/O2 biological treatment system: within each group, bars with different lowercase letters are significantly different from each other (p < 0.05, ANOVA)
Relationships between microbial community and the environmental factors
The PCA- and RDA-analyses and the Mantel test were conducted to illustrate the influence of environmental factors on the microbial community composition and functions. Based on the results of Mantel test (Fig. 6), the indices of COD, BOD, TOC, phenolic compounds, CN−, SCN−, sulfides, NH4+-N, TN, and oil were strongly related with microbial community composition (p < 0.005, r > 0.7) as the main driving factors in the microbial community succession. The SRT operational parameter also appears to be the major driving factor (p < 0.001, r > 0.7). The PCA analysis clarified the main impact factors (Fig. S2), showing SRT, phenolic compounds, CN−, sulfides, and oil influencing the microbial community composition in the bioreactor A. The microbial community in the bioreactor O1 was shaped by the factors of MLSS, COD removal rate, NO2−, and TN. In the bioreactor H, the microbial community was shaped by SRT, HRT, and NO3−. In addition, NO3−, COD, and DO regulated the microbial community composition in bioreactor O2 stronger than in the others. Generally, the microbial communities of the bioreactors A and O1 were mostly affected by the factors relating to carbon-containing pollutants, while in the bioreactors H and O2 the concentration of NO3− was the principal factor.

Mantel test between operational parameters and microbial community (a), wastewater characteristics and microbial community (b), operational parameters and functional pathways (c), and wastewater characteristics and functional pathways (d) in the A/O1/H/O2 biological treatment process
The RDA analysis was conducted to quantify the impacts of the environmental factors and showed the wastewater characteristics determining the microbial community composition at phyla and genera level: the weighted average values for the first two axes comprised 93.88% and 94.22% of the total variance (Fig. 7a and b), respectively. The phylum Nitrospirae is in positive correlation with NO3−, although negatively related to NO2−, SCN−, TN, and NH4+ − N illustrating the ability of Nitrospirae species to oxidize NO2− to NO3− in the second step of nitrification, thus contributing to the metabolism of nitrogen-containing pollutants (Fig. 7a). At the genera level, Nitrospira and Sphingobium also show similar correlation (Fig. 7b) explained by enhanced ammonia oxidation helping the NH3-N removal (Tang et al. 2020b). Besides, the RDA results also showed the phylum Acidobacteria and the genus Thiobacillus being negatively related with TOC, BOD5, and concentrations of phenolic compounds, sulfides, CN−, SCN−, and oil. Additionally, the β-Proteobacteria and Firmicutes together with genera of Thiobacillus, Bdellovibrio, HA73, T78, Leucobacter, and Rhodoplanes showed negative relationships with COD, indicating an important role played in the removal of carbon-containing pollutants. All of the RDA results coincide with the structure and function of the microbial communities in the A/O1/H/O2 combination providing good simultaneous decarbonation and denitrogenation.

Redundancy analysis (RDA) of microbial species under the impact of environmental factors in the bioreactors: arrows indicate the direction and magnitude of environmental parameters associated with phylum and genus; the relationships between bacteria at phylum (a) and genus (b) level and wastewater characteristics
Network patterns reveal microbial community characteristics
Interaction of microorganisms is another deterministic factor for diversity and function of microbial community, understanding of which is achieved by identifying the keystone taxa (Zhang et al. 2019a). The association network of the microbial community consists of 51 nodes and 411 edges being separated into two modules, module 1 of positive correlations (62.75%, blue), and module 2 of negative correlations (37.25%, pink) (Fig. 8). The positive and negative edges accounted for 69.83% and 30.17% in the network analysis, respectively, suggest the microbial community being dominated by symbiotic relationships in the active sludge. Each module in the network considered a subset of species from similar ecological niches and may perform similar functions (Zhang et al. 2019a). The functions of species in the module 1 were mainly related to nitrification (Nitrosomonadaceae and Nitrospira), denitrification (Thiobacillus, Ignavibacteriacea, and Hyphomicrobiaceae), and anammox (8 OTUs belonging to Planctomycetes). Most of the OTUs in module 2 were assigned to the phylum Proteobacteria and Firmicutes responsible for the removal of carbon-containing pollutants. The modules in the network visualize different niches indicating important ecological processes (Rottjers and Faust 2018).

Network visualizing the OTU-OTU interactions in the A/O1/H/O2 reactors combination: positive correlations—blue, negative correlations—pink; nodes are colored for types of modularity classes; size of node is proportional to the degree (a) or betweenness centrality (b); labels of keystone OTUs are colored with red with amplified fonts
In this study, the threshold of nodes with degree > 23, betweenness centrality values > 100, and relative abundances > 0.3% of the microbial communities were outlined as keystone taxa (Li et al. 2019b). The keystone taxa of OTU4, OTU30, and OTU17 comprised the network hub in the co-occurrence network of the A/O1/H/O2 system. The OTU17 in the module 1 belongs to the Hyphomicrobiaceae family contributing to the denitrification process. The OTU4 and OTU30 in the module 2 belong to the Comamonadaceae family known for its contribution to the organic pollutants removal. The high-degree keystone OTUs control the microorganisms in the module 1 directly by negative reaction, while doing this by positive reaction in the module 2. The keystone taxa control the microbial community by a range of strategies being able to promote or suppress the effector groups by secreting metabolites, antibiotics, or toxins to selectively regulate community structure and functioning (Banerjee et al. 2018). Besides, the higher betweenness centrality of key OTUs indicates their higher control potential exerting over the interactions of other species in the network (Li et al. 2019b).
Conclusions
The microbial communities in the full-scale A/O1/H/O2 bioreactors combination, showing efficient simultaneous decarbonation and denitrogenation in CWW treatment, were comprehensively studied in their diversity of distinct microbial species. More than 40% of sequences in bioreactors were assigned to the phylum of Acidobacteria with its majority belonging to the taxa family Ellin6075 without specifically identified function. Nitrospira, Thiobacillus, and Planctomyces were found being enriched in genes encoding the nitrogen removal pathway. The microbial association network composed of two modules provides decarbonation and denitrogenation of CWW driven by the keystone taxa of the Comamonadaceae and Hyphomicrobiaceae families. The study demonstrated the diverse and distinct bacterial communities are determined by the composition of CWW governing the biochemical reactions. The microbial composition in bioreactors may be controlled to achieve more modes for simultaneous decarbonation and denitrogenation of CWW by regulation of operation parameters including DO, pH, SRT, organic load rate, and reflux ratio.
References
Banerjee S, Schlaeppi K, van der Heijden MGA (2018) Keystone taxa as drivers of microbiome structure and functioning. Nat Rev Microbiol 16(9):567–576
Berry D, Widder S (2014) Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front Microbiol 5:219
Deng J, Zhang B, Xie J, Wu H, Li Z, Qiu G, Wei C, Zhu S (2020) Diversity and functional prediction of microbial communities involved in the first aerobic bioreactor of coking wastewater treatment system. PLoS One 15(12):e0243748
Deng Y, Jiang YH, Yang Y, He Z, Luo F, Zhou J (2012) Molecular ecological network analyses. BMC Bioinformatics 13:113
EPA of China (2002) Water and Wastewater Monitoring Analysis Method, 4th edn. Publisher Chinese Environment Science, Beijing
Fierer N, Hamady M, Lauber CL, Knight R (2008) The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A 105(46):17994–17999
Fukushima T, Whang LM, Chen PC, Putri DW, Chang MY, Wu YJ, Lee YC (2013) Linking TFT-LCD wastewater treatment performance to microbial population abundance of Hyphomicrobium and Thiobacillus spp. Bioresour Technol 141:131–137
Huang Y, Li D, Wang L, Yong C, Sun E, Jin H, Huang H (2019) Decreased enzyme activities, ammonification rate and ammonifiers contribute to higher nitrogen retention in hyperthermophilic pretreatment composting. Bioresour Technol 272:521–528
Jia L, Jiang B, Huang F, Hu X (2019) Nitrogen removal mechanism and microbial community changes of bioaugmentation subsurface wastewater infiltration system. Bioresour Technol 294:122140
Joshi DR, Zhang Y, Gao Y, Liu Y, Yang M (2017) Biotransformation of nitrogen- and sulfur-containing pollutants during coking wastewater treatment: correspondence of performance to microbial community functional structure. Water Res 121:338–348
Joshi DR, Zhang Y, Zhang H, Gao YX, Yang M (2018) Characteristics of microbial community functional structure of a biological coking wastewater treatment system. J Environ Sci (china) 63:105–115
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31(9):814–821
Lawson CE, Wu S, Bhattacharjee AS, Hamilton JJ, McMahon KD, Goel R, Noguera DR (2017) Metabolic network analysis reveals microbial community interactions in anammox granules. Nat Commun 8:15416
Li K, Wu H, Wei J, Qiu G, Wei C, Cheng D, Zhong L (2019a) Simultaneous decarburization, nitrification and denitrification (SDCND) in coking wastewater treatment using an integrated fluidized-bed reactor. J Environ Manage 252:109661
Li Y, Wu H, Shen Y, Wang C, Wang P, Zhang W, Gao Y, Niu L (2019b) Statistical determination of crucial taxa indicative of pollution gradients in sediments of Lake Taihu, China. Environ Pollut 246:753–762
Ma JD, Wu HZ, Wang YX, Qiu GL, Fu BB, Wu CF, Wei CH (2019) Material inter-recycling for advanced nitrogen and residual COD removal from bio-treated coking wastewater through autotrophic denitrification. Bioresour Technol 289:121616
Ma Q, Qu YY, Shen WL, Zhang ZJ, Wang JW, Liu ZY, Li DX, Li HJ, Zhou JT (2015) Bacterial community compositions of coking wastewater treatment plants in steel industry revealed by Illumina high-throughput sequencing. Bioresour Technol 179:436–443
Mendez-Garcia C, Pelaez AI, Mesa V, Sanchez J, Golyshina OV, Ferrer M (2015) Microbial diversity and metabolic networks in acid mine drainage habitats. Front Microbiol 6:475
Miao L, Liu Z (2018) Microbiome analysis and -omics studies of microbial denitrification processes in wastewater treatment: recent advances. Sci China Life Sci 61(7):753–761
Niu T, Zhou Z, Shen X, Qiao W, Jiang LM, Pan W, Zhou J (2016) Effects of dissolved oxygen on performance and microbial community structure in a micro-aerobic hydrolysis sludge in situ reduction process. Water Res 90:369–377
Nottingham AT, Fierer N, Turner BL, Whitaker J, Ostle NJ, McNamara NP, Bardgett RD, Leff JW, Salinas N, Silman MR, Kruuk LEB, Meir P (2018) Microbes follow Humboldt: temperature drives plant and soil microbial diversity patterns from the Amazon to the Andes. Ecology 99(11):2455–2466
Parthipan P, Preetham E, Machuca LL, Rahman PK, Murugan K, Rajasekar A (2017) Biosurfactant and degradative enzymes mediated crude oil degradation by bacterium bacillus subtilis A1. Front Microbiol 8:193
Qin Z, Wei C, Wei T, Li ZM, Pang ZJ, Luo P, Feng CH, Qiu GL, Wei CH, Wu HZ, Peng YH, Jiang CF, Preis S (2022) Evolution of biochemical processes in coking wastewater treatment: a combined evaluation of material and energy efficiencies and secondary pollution. Sci Total Environ 807:151072
Qiu GL, Zuniga-Montanez R, Law YY, Thi SS, Nguyen TQN, Eganathan K, Liu XH, Nielsen PH, Williams RBH, Wuertz S (2019) Polyphosphate-accumulating organisms in full-scale tropical wastewater treatment plants use diverse carbon sources. Water Res 149:496–510
Rottjers L, Faust K (2018) From hairballs to hypotheses-biological insights from microbial networks. FEMS Microbiol Rev 42(6):761–780
Song Y, Jiang CY, Liang ZL, Wang BJ, Jiang Y, Yin Y, Zhu HZ, Qin YL, Cheng RX, Liu ZP, Liu Y, Jin T, Corvini PF, Rabaey K, Wang AJ, Liu SJ (2020) Casimicrobium huifangae gen. nov., sp. nov., a ubiquitous “most-wanted” core bacterial taxon from municipal wastewater treatment plants. Appl Environ Microbiol 86(4):e02209–19
Sorokin DY, Lucker S, Vejmelkova D, Kostrikina NA, Kleerebezem R, Rijpstra WI, Damste JS, Le Paslier D, Muyzer G, Wagner M, van Loosdrecht MC, Daims H (2012) Nitrification expanded: discovery, physiology and genomics of a nitrite-oxidizing bacterium from the phylum Chloroflexi. ISME J 6(12):2245–2256
Sun H, Liu F, Xu S, Wu S, Zhuang G, Deng Y, Wu J, Zhuang X (2017) Myriophyllum aquaticum constructed wetland effectively removes nitrogen in swine wastewater. Front Microbiol 8:1932
Sun S, Sun H, Zhang D, Zhang J, Cai Z, Qin G, Song Y (2019) Response of soil microbes to vegetation restoration in coal mining subsidence areas at Huaibei Coal Mine, China. Int J Environ Res Public Health 16(10):1757
Sun Y, Shen YX, Liang P, Zhou J, Yang Y, Huang X (2014) Linkages between microbial functional potential and wastewater constituents in large-scale membrane bioreactors for municipal wastewater treatment. Water Res 56:162–171
Tang J, Li X, Cui P, Lin J, Jianxiong Zeng R, Lin H, Zhou S (2020a) Nitrification plays a key role in N2O emission in electric-field assisted aerobic composting. Bioresourc Technol 297:122470
Tang VT, Rene ER, Fu DF, Singh RP, Behera SK, Pugazhendhi A (2020b) Effect of mixed microbial culture addition on enhanced river water quality: Pollutant removal and microbial community characteristics. Environ Technol Innov 18:100707
Wang S, Hou WG, Dong HL, Jiang HC, Huang LQ, Wu G, Zhang CL, Song ZQ, Zhang Y, Ren HL, Zhang J, Zhang L (2013) Control of temperature on microbial community structure in hot springs of the Tibetan Plateau. PLoS One 8(5):e62901
Wei C, Wu H, Kong Q, Wei J, Feng C, Qiu G, Wei C, Li F (2019) Residual chemical oxygen demand (COD) fractionation in bio-treated coking wastewater integrating solution property characterization. J Environ Manage 246:324–333
Wei C, Li Z, Pan J, Fu B, Wei J, Chen B, Yang X, Ye G, Wei C, Luo P, Wu C, Qiu G, Wu H (2021) An oxic–hydrolytic–oxic process at the nexus of sludge spatial segmentation, microbial functionality, and pollutants removal in the treatment of coking wastewater. ACS ES&T Water 1(5):1252–1262
Wei CH, Xie B, Xiao HL, Wang DS (2000) Volumetric mass transfer coefficient of oxygen in an internal loop airlift reactor with a convergence-divergence draft tube. Chem Eng Technol 23(7):597–603
Wu D, Zhang Z, Yu Z, Zhu L (2018) Optimization of F/M ratio for stability of aerobic granular process via quantitative sludge discharge. Bioresour Technol 252:150–156
Wu LW, Ning DL, Zhang B, Li Y, Zhang P, Shan XY, Zhang QT, Brown M, Li ZX, Van Nostrand JD, Ling FQ, Xiao NY, Zhang Y, Vierheilig J, Wells GF, Yang YF, Deng Y, Tu QC, Wang AJ, Global Water Microbiome C, Zhang T, He ZL, Keller J, Nielsen PH, Alvarez PJJ, Criddle CS, Wagner M, Tiedje JM, He Q, Curtis TP, Stahl DA, Alvarez-Cohen L, Rittmann BE, Wen XH, Zhou JZ (2019) Global diversity and biogeography of bacterial communities in wastewater treatment plants. Nat Microbiol 4(7):1183–1195
Xu WC, Zhang YX, Cao HB, Sheng YX, Li HB, Li YP, Zhao H, Gui XF (2018) Metagenomic insights into the microbiota profiles and bioaugmentation mechanism of organics removal in coal gasification wastewater in an anaerobic/anoxic/oxic system by methanol. Bioresour Technol 264:106–115
Yang Q, Xiong PP, Ding PY, Chu LB, Wang JL (2015) Treatment of petrochemical wastewater by microaerobic hydrolysis and anoxic/oxic processes and analysis of bacterial diversity. Bioresour Technol 196:169–175
Zhang B, Ning D, Van Nostrand JD, Sun C, Yang Y, Zhou J, Wen X (2020) Biogeography and assembly of microbial communities in wastewater treatment plants in China. Environ Sci Technol 54(9):5884–5892
Zhang B, Ning D, Yang Y, Van Nostrand JD, Zhou J, Wen X (2019a) Biodegradability of wastewater determines microbial assembly mechanisms in full-scale wastewater treatment plants. Water Res 169:115276
Zhang XH, Tang S, Wang M, Sun WM, Xie YW, Peng H, Zhong AM, Liu HL, Zhang XW, Yu HX, Giesy JP, Hecker M (2019b) Acid mine drainage affects the diversity and metal resistance gene profile of sediment bacterial community along a river. Chemosphere 217:790–799
Zhao WT, Sui Q, Huang X (2018) Removal and fate of polycyclic aromatic hydrocarbons in a hybrid anaerobic-anoxic-oxic process for highly toxic coke wastewater treatment. Sci Total Environ 635:716–724
Zheng MS, Zhou N, Liu SF, Dang CY, Liu YX, He SS, Zhao YJ, Liu W, Wang XK (2019) N2O and NO emission from a biological aerated filter treating coking wastewater: Main source and microbial community. J Clean Prod 213:365–374
Zhou B, Duan J, Xue L, Zhang J, Yang L (2019) Effect of plant-based carbon source supplements on denitrification of synthetic wastewater: focus on the microbiology. Environ Sci Pollut Res Int 26(24):24683–24694
Zhou J, Li HS, Chen XL, Wan DJ, Mai WN, Sun CQ (2017) Cometabolic degradation of low-strength coking wastewater and the bacterial community revealed by high-throughput sequencing. Bioresour Technol 245(Pt A):379–385
Zhu S, Wu HZ, Wei CH, Zhou L, Xie JT (2016) Contrasting microbial community composition and function perspective in sections of a full-scale coking wastewater treatment system. Appl Microbiol Biotechnol 100(2):949–960
Zhu S, Wu HZ, Wu CF, Qiu GL, Feng CH, Wei CH (2019) Structure and function of microbial community involved in a novel full-scale prefix oxic coking wastewater treatment O/H/O system. Water Res 164:114963
Funding
This work was supported by the National Natural Science Foundation of China (No. U1901218), the Program for Science and Technology of Guangdong Province, China (No. 2021A1515012256 and No. 2018A050506009), and the Program of SSIJRI (202-A017010).
Author information
Authors and Affiliations
Contributions
Shuang Zhu conceived and designed the research, participated in experimental work, and wrote the paper. Jinsi Deng performed the experiments, analyzed the data, and wrote the original draft. Xiaobao Jin and Cong Wei contributed analytical methodology, data curation, and reviewing. Haizhen Wu and Guanglei Qiu participated in the data analysis and revised the paper. Sergei Preis and Chaohai Wei revised and designed the research. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
The manuscripts reporting studies are not applicable for human participants, human data, or human tissue. The manuscript does not contain any individual person’s data in any form.
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Gerald Thouand
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhu, S., Deng, J., Jin, X. et al. Diverse and distinct bacterial community involved in a full-scale A/O1/H/O2 combination of bioreactors with simultaneous decarbonation and denitrogenation of coking wastewater. Environ Sci Pollut Res 30, 2103–2117 (2023). https://doi.org/10.1007/s11356-022-22103-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-022-22103-y