
Abstract
Particulate matters (PMs) are significant components of air pollution in the urban environment. PMs with aerodynamic diameter less than 2.5 μm (PM2.5) can penetrate to the alveolar area and introduce numerous compounds to the pneumocystis that can initiate inflammatory response. There are several questions about this exposure as follows: does PM2.5-induced inflammation lead to a specific disease? If yes, what is the form of the progressed disease? This systematic review was designed and conducted to respond to these questions. Four databases, including Web of Science, Scopus, PubMed, and Embase, were reviewed systematically to find the related articles. According to the included articles, the only available data on the inflammatory effects of PM2.5 comes from either in vitro or animal studies. Both types of studies have shown that the induced inflammation is type I and includes secretion of proinflammatory cytokines. The exposure duration of longer than 28 weeks was not observed in any of the reviewed studies. However, as there is not a specific antigenic component in the urban particulate matters and based on the available evidence, the antigen-presenting is not a common process in the inflammatory responses to PM2.5. Therefore, neither signaling to repair cells such as fibroblasts nor over-secretion of extracellular matrix (ECM) proteins can occur following PM2.5-induced inflammation. These pieces of evidence weaken the probability of the development of fibrotic diseases. On the other hand, permanent inflammation induces the destruction of ECM and alveolar walls by over-secretion of protease enzymes and therefore results in progressive obstructive effects.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
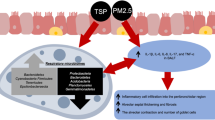
Particulate matters (PMs) with an aerodynamic diameter of less than 2.5 μm (PM2.5) are the most critical and objectionable group of pollutants in the ambient air (Wang et al. 2017). PM2.5 includes several components such as heavy metals, polycyclic aromatic hydrocarbons (PAHs), unburned or incompletely burned fuel, sulfur compounds, and nitric compounds (Prieto-Parra et al. 2017; Zhang et al. 2018). They can penetrate the alveolar region of the lung effectively and either deposit there or enter the bloodstream considering their size (Kulkarni et al. 2006). These characteristics allow PM2.5 to introduce various pollutants to respiratory cells for an extended period compared to the gaseous pollutants. Smoking is the most critical risk factor in incidence or exacerbation of respiratory diseases due to induction of permanent inflammation (Cosio et al. 2002). However, according to the epidemiological studies, various lung diseases and respiratory symptoms are attributed to the exposure to the environmental PM2.5 (Cohen et al. 2017; Dominici et al. 2006). Respiratory pathologies most commonly reported in the studies include inflammation in lung parenchyma (functional tissue of lung) or airways, oxidative stress, metabolic interruptions, and mutagenicity (Feng et al. 2016). Moreover, the relationship between exposure to particulate matters and some respiratory diseases such as exacerbation of asthma and COPD, and lung cancer has been confirmed (Raaschou-Nielsen et al. 2016). Inflammation is the most critical event in all of these diseases. Inflammation is a complicated response to exposure to pathogens or tissue injury. In normal condition, it has several steps including secretion of several cytokines as inflammatory mediators for recruitment of other immune cells, phagocytosis of damaged cells’ debris or pathogens, and finally, anti-inflammatory and tissue repairing step (Medzhitov 2008). Inflammation in the respiratory system could involve different cell types and can be chronic. Depending on the cell type involved in the inflammation in the lung, induced chronic inflammation can activate different immunological pathways and lead to individual severe diseases (Abbas et al. 2019a).
Lung diseases are generally categorized as obstructive or restrictive. Interstitial lung diseases (ILD) are the dominant group of restrictive diseases. ILD are chronic diseases with the insidious but progressive onset of dyspnea and cough. Lung fibrosis is one of the main ILD (Chilosi et al. 2012). Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and usually lethal pulmonary disease; moreover, it is more prevalent among the elders (King Jr et al. 2011). IPF involves abnormal and continuous production and deposition of extracellular matrix (ECM), including collagen/elastin fibers (Wilson and Wynn 2009). Figure 1 illustrates the histopathological appearance of IPF. The fibrosis can be diagnosed by increasing the thickness of ECM because of abnormal deposition of collagen in the tissue (King Jr et al. 2011). The regions with normal density of pneumocytes and fibrotic area are shown in Fig. 1.

Fibrosis in lung tissue (1: normal area; 2: fibrosis area)
Several pathogeneses have been proposed for the stimulation of fibrosis; however, the most accepted hypothesis is the chronic inflammation in epithelial cells in the lung parenchyma. Various inflammatory reaction pathways could cause fibrosis. They start from cell damage due to contact with irritant agents such as pathogens or particulates and finally lead to abnormal collagen secretion by fibroblasts/myofibroblasts. Several inflammatory mediators are involved in these pathways. Some of them are unique to a specific pathway, and the others are common between pathways (Kumar et al. 2014).
In a different scenario, inflammatory reaction cascade can activate different pathways and lead to obstructive lung diseases such as emphysema. The clinical definition of emphysema is the air space enlargement due to the alveolar wall distraction. The pathophysiological feature of emphysema is shown in Fig. 2. The healthy and enlarged alveoli are indicated by numbers 1 and 2 in this figure, respectively. Following chronic exposure to antigens, alveolar macrophages and airway epithelial cells can secrete various cytokines such as interleukin-8 (IL-8) and granulocyte-macrophage colony-stimulating factor (GM-CSF). These proinflammatory mediators can signal an increase in the recruitment of leukocytes in the lung (Ruwanpura et al. 2017). The leukocytes, especially neutrophils, secrete serine and cysteine proteinases. These enzymes are elastases and can destruct connective tissue components such as collagen and elastin (Taraseviciene-Stewart and Voelkel 2008). Consequently, alveolar attachment to the bronchioles would be decreased, and the alveoli area would be increased, followed by long-term secretion from them. Anatomically, it can lead to emphysema following hyperinflation, a significant reduction of forced vital capacity (FVC%), and other clinical symptoms (Cottin et al. 2017).

Emphysema in human lung (1: normal alveoli; 2: enlarged alveoli following alveolus wall destruction)
Therefore, chronic inflammation can induce both restrictive and obstructive lung diseases. Thus, numerous studies have been tried to determine the inflammatory effect of inhalable materials and further signaling pathways inducing individual lung diseases since one of the leading and frequent respiratory exposures is PM2.5 inhalation.
Exposure to urban PM2.5 is increasing worldwide. According to various studies, the number of cities having PM2.5 concentrations over international guidelines is increasing. Moreover, the incidence rate of the high level of PM2.5 is increasing in megacities. Also, PM2.5 has various components with confirmed health effects (Hsu et al. 2017; Kim et al. 2019; Lin et al. 2014; Zhang et al. 2018). Therefore, several studies have been tried to evaluate the inflammatory effect of PM2.5 inhalation, and most of them confirmed these effects after exposure to this pollutant. Nevertheless, there are some differences between their experimental approaches and their results. The examples are varied in exposure doses, exposure duration, and inflammatory response levels.
Moreover, these studies measured and reported a wide range of inflammatory mediators, which were involved in different inflammation pathways and could cause different symptoms. Consequently, the possible inflammation pathways after exposure to urban PM2.5 are unclear. However, there are many questions and controversies concerning the relation between PM2.5 exposure and inflammation. The main questions are as follows: Can inhalation and exposure to urban PM2.5 induce inflammation in lung parenchyma? Which inflammatory pathway will be activated after exposure to PM2.5? What are the available procedures to evaluate the inflammatory effect of PM2.5 inhalation? What are the possible immune response pathways that can be activated after exposure to PM2.5?
Therefore, we reviewed available studies that specifically investigated the inflammatory effects of exposure to PM2.5 based on defined search strategy to respond to these questions.
Methodology
Search strategy
This review was conducted in February 2019. Primarily, the main questions were formulated according to the available knowledge, as mentioned in the “Introduction.” The systematic search was carried out according to these questions among four databases: Web of Science, Scopus, PubMed, and Embase in the English language. The search keywords were the following: “lung + inflammation + PM2.5” and “lung + inflammation + particulate matter.” The results were filtered using the inclusion and exclusion criteria. These criteria would be mentioned in the following sections.
Inclusion criteria
Focus on urban air PM2.5 was the main inclusion criteria. Only original articles published in peer-reviewed journals were selected for this study. In vitro, animal, or human studies were all included. All of the selected articles reported an exposure dose, exposure period, and chemical analysis of used particulate matters. We also reviewed references within the returned review articles to find any further appropriate articles.
Exclusion criteria
We excluded all the studies conducted on natural particles such as dust. Moreover, all the studies about occupational situations, accidental exposures, polymorphism, genetic studies, and studies that reported inflammation by serum cytokine measurements were excluded. Besides, we removed studies that did not report the chemical analysis of PM2.5. Finally, some articles were further excluded after careful study of their abstracts or the full article contents.
Data extraction
Extracted data and information from the selected articles included the type of study (in vitro, animal, or human), subjects’ characteristics, PM2.5 source and its chemical composition, exposure procedure (dose, time, groups), type of the measured marker to determine inflammation and response level (cytokine secretion or mRNA expression), results of studies (change in marker level), and outcomes (visible histologic, anatomic, or pathologic changes and conclusion of the study). Then, extracted data/information was classified based on the similarities between studies for better visualization and semi-quantitative comparison.
Results
Included studies
The search on four databases, with inclusion and exclusion filters, returned 555 articles. After the duplication removal (N = 366), 189 articles remained. Finally, 21 articles were selected after screening by title, abstract, and main passage reviewing. Figure 3 is the PRISMA flow diagram presenting the study selection procedure.

PRISMA flow diagram
Acquired data
The acquired data from 21 included studies are summarized in Table 1. The included studies could be divided into two main groups. The first group was the animal studies, which included instillation or respiration of ambient PM2.5 by mice or rat for various periods and measurement of inflammatory mediators in serum or bronchoalveolar lavage fluid (BALF). The second group was the in vitro studies using human or animal cell lines. In these studies, PM2.5 was filtered from ambient air, and cell lines were then exposed to them in a controlled manner, and the inflammatory mediators were measured after different exposure periods.
The PM2.5 were collected from urban air in this study. Three articles reported using standard particles in their experiments, including urban and diesel particles. Numerous parameters were measured and reported in the included articles, which were various. Accordingly, we tried to classify particulates’ chemical composition into three categories: heavy metals (HMs); water-soluble ions (WSIs) including SO4−2, NO3−, and NH4+; and PAHs. Besides, we tried to convert and report all concentration values in μg/mg of particle mass. The additional extracted data from articles was exposure dose and procedures.
One of the main problems for comparison between different studies was exposure and the concentration units of inflammatory cytokines. There was no capability of converting all of the reported units to the same unit. Therefore, the exposure units were μg/ml or μg/cm2 of cell culture in in vitro studies and mg/kg or mg/mice for animal studies. Likewise, the concentration of the cytokine was reported as pg/ml or pg/gr of protein for in vitro studies. Various kinds of inflammatory cytokines were measured in the included studies. However, no justification was found for the selected kind of cytokines in each study. The only common mediator in all of the studies was IL-8. The exposure period and dose were different between in vitro and animal studies. Summary of exposure data, characteristics of particulates, and measured mediators are listed in Table 2.
Several diversities were observed in extracted information, including the analyzed components of PM2.5, the analysis of more components of PM2.5, and the response to exposure to intact PM2.5 or its components. Therefore, it was impossible to illustrate the data using the plot. Hence, the results were classified based on the observed response, PM2.5 components, and exposure period in Tables 3 and 4. According to these tables, the number of available studies reporting responses to each component is mentioned (N). Besides, the number of direct (D), reverse (R), and non-significant (NS) studies have been mentioned in these tables. The symbols, including +, −, and ±, indicate the effect of exposure period at constant dose.
Discussion
We investigated PM2.5-induced inflammation in lung epithelial cells in this systematic review. The articles included in this review were in vitro and animal studies. There are several human studies on measuring inflammatory cytokines in bronchoalveolar lavage fluid (BALF). However, they cannot be interpreted to determine the inflammatory effects of inhaled PMs because the patients who need bronchoscopy and BALF collection have serious diseases with inflammation as a common complication (El-Bayoumi and Silvestri 2008; Jin and Wang 2019; Sato et al. 2017).
As shown in Table 1, all the included articles have reported the concentration of at least one of the main groups of PM2.5 components. Table 2 shows that the exposure duration in animal studies is significantly longer than the in vitro studies. Reversely, the applied dose of PM2.5 is significantly higher in the in vitro studies.
WSIs are the most frequently measured component. WSIs can dissolve in bronchiolar or alveolar fluids, leave the particulate structure, and change airway surface liquid (ASL) osmolarity and pH (Miller et al. 2009). Dissolved WSIs can cause osmotic stress and induce inflammation. As mentioned in Table 3, the number of in vitro studies that reported a direct relationship between WSIs and cytokine concentration is significantly more than animal studies. The concentration of chemicals is constant in the in vitro studies, while in the animal body, the hemostatic effect moderates the concentration of pollutants.
PM2.5 also contains heavy metals, including Cu, Zn, and Fe, which serve as micronutrients for cells. Nevertheless, other heavy metals such as mercury, lead, cadmium, vanadium, and arsenic do not have any biological function (Gao et al. 2016). All of these toxic metals were detected in the included studies. These metals are water-soluble and can activate the oxidative stress pathway and induce inflammation (Radan et al. 2019). The PAHs are another group of measured components in urban PM2.5. They can be recognized by the aryl hydrocarbon receptor (AHR) in the cytoplasm, transferred to the nucleus, and induce DNA damage (Pieterse et al. 2013). However, their entrance from the surface of the cell into the cytoplasm is unclear. Like WSI, the number of in vitro studies that reported a direct relationship between heavy metals and PAHs and cytokine concentration is clearly more than animal studies.
In addition to chemical features, the activated pathway of inflammation is also essential. There is not robust evidence confirming the PM2.5 transfer to the parenchymal cells using endocytosis. Therefore, the induced inflammation in in vitro studies is probably related to the activation of some signaling pathways from the surface of the cell membrane. It can be true in animal studies, although the source of secreted inflammatory cytokines is not clear and can be from parenchymal cells, connective tissue cells, or immune cells.
Table 3 shows that the most prevalent secreted cytokines in in vitro studies are proinflammatory, including INF-γ, IL-1β, IL-6, IL-8, and TNF-α. Generally, the target cells of these mediators are innate immune cells such as macrophages and neutrophils. Some of them are common between in vitro and animal studies. Moreover, as indicated in Table 4, animal studies show a more complete inflammatory responses to PM2.5 exposure. Some measured mediators in animal studies such as IL-12 and IL-13 induce differentiation of naive T cells into T helper 1 cells and anti-inflammatory effects.
The other parallel mechanism, which can initiate the inflammation, is autophagy. Following the entrance to the cell, PM2.5 can induce autophagy (Deng et al. 2013). In many in vitro studies, autophagy was an uncompleted and unsuccessful mechanism against PM2.5, and it decreased the cell viability and caused cell destruction and release of the particulates into the intercellular space again. In this situation, several damage-associated molecular patterns (DAMPs) are released in the intercellular space (Zhao et al. 2019). These DAMPs are recognized using the pattern recognition receptors (PRRs) in the innate immune cells such as macrophages, dendritic cells (DCs), and monocytes and stimulate the recruitment of additional innate immune cells (Abbas et al. 2019a). After the inflammation induction, the wound healing process or the anti-inflammatory step, including repairing mechanism, would be initiated (Wolters et al. 2014), which is a critical step since the suggested pathophysiology for IPF is an abnormal wound healing process due to the uncontrolled repair. Both types of inflammation (types I and II) can happen in wound healing at normal conditions (Wilson and Wynn 2009). Type I includes secretion of IL-1β, IL-6, IL-8, INF-γ, TNF-α, and IgG2 antibodies and recruitment of innate immune cells, including macrophages and neutrophils. Afterwards, T lymphocytes, especially T helper 1 cells (Th1) and regulatory T cells (Treg), are recruited to balance inflammatory and anti-inflammatory reactions by signaling to fibroblast for collagen secretion and repairing (Abbas et al. 2019a). Type II inflammation includes secretion of IL-4, IL-5, and IL-13 to recruit the eosinophils and basophils and then to signal to Th2 cells to regulate inflammation and then to induce repair mechanisms (Gandhi et al. 2016). In IPF, continuous deposition of collagen and other ECM proteins occurs as an abnormal process, which means that anti-fibrotic mediators are not being secreted sufficiently, or the collagen deposition and fibroblast differentiation activation pathway are activated continuously. In other words, IPF is a completed but unfinished wound healing process. The initiation of this step directly depends on antigen-presenting by innate immune cells (Strieter 2008). Neutrophils do not have the antigen-presenting capability but other cells, including macrophages, natural killer (NK) cells, and DCs, which can make epitope to present antigen to lymphocytes (Abbas et al. 2019a). The PM2.5 can have various components, but, as reported in many studies, there is not any polypeptide structure in their composition (Li et al. 2016). Therefore, the innate immune cells cannot perform the antigen-presenting and lymphocytes, especially Th1, cannot be recruited. Th1 recruitment is necessary for the activation of Treg (Wing and Sakaguchi 2010). Thus, anti-inflammatory responses cannot be activated. Besides, since the lack of antigen-presenting capabilities, the recruitment of Th2 cells is unlikely too. Therefore, the inflammation will continue until full clearance of PM2.5 from the lung. Thus, recruitment and differentiation of myofibroblasts to fibroblast for the repair of the destructed tissue is rare.
Increased bronchiolar level of IL-4, IL-13, and TGF-β is reported in many studies as diagnostic indicators of IPF. However, according to the extracted data, none of the included animal studies reported a significant increase of TGF-β, IL-4, or IL-13 following exposure to PM2.5. Lack of antigen-presenting capability and absence of an increase in the levels of IL-4 and IL-13 show that IPF following the exposure to PM2.5 is rare and maybe non-existent.
The other possible disease following inflammation is emphysema. The first step of phagocytosis by innate immune cells is the secretion of several proteolytic enzymes to destruct antigens and facilitate phagocytosis (Abbas et al. 2019a). The action of proteolytic enzymes can be regulated using anti-elastase enzymes, such as α1 anti-trypsin (α1-AT). Emphysema is possible among individuals with a genetic defect in the production of α1-AT (Taraseviciene-Stewart and Voelkel 2008). In normal individuals, the most approved scenario for the development of emphysema is over-secretion of protease and elastase enzymes such as serine protease, matrix metalloproteinase-12 (MMP-12), and elastase by neutrophils and macrophages following exposure to an antigen and induction of predominant inflammation (Sharafkhaneh et al. 2008). The secretion of these proteolytic enzymes is a robust diagnostic indicator for emphysema (Taraseviciene-Stewart and Voelkel 2008). Besides these enzymes, several studies indicated that MMP-9 would be secreted following the inflammation to destruct the collagen of the basement membrane and facilitate monocyte migration from blood to the alveolar region (McMillan et al. 2004). Several studies reported that the clearance of PM2.5 from the lung takes too long. Moreover, PM2.5 have no polypeptide components. Thus, the secretion of protease enzymes can continue for a long time endlessly. Additionally, several studies reported that PM2.5 phagocytosis could damage the macrophages and destruct them and cause PM2.5 to be released again (Geng et al. 2006). Therefore, the secretion of protease enzymes will continue until the full clearance of PM2.5 from the lung. Because of continuous exposure to PM2.5, these reactions seem to be permanent. There was no data in our included animal studies regarding the measurement of these protease enzymes. However, available results confirm that inflammation in lung follows exposure to PM2.5. Therefore, the destruction of connective tissue proteins seems possible after exposure to PM2.5, while the severity of destruction is very mild and needs a very long time to make a significant tissue damage.
The secretion of INF-γ is another issue concerning the initial response of cells, which are exposed to PM2.5. It has been measured in both types of studies, and almost all of them reported significant increases in INF-γ level following exposure to PM2.5. Generally, after pathogen recognition, especially in viral infections, INF-γ is secreted by infected cells to induce cellular immune response using the stimulation of protein kinase R (PKR) production (Wingate et al. 2009). In the case of PM2.5, the produced PKR has no target antigen due to the absence of any double-stranded RNA in PM2.5 compositions. If PKR deposition continues for a long time, it can induce cell apoptosis or lead the cell to the autophagy pathway (Kim et al. 2007). Both apoptosis and autophagy can signal macrophage recruitment. In normal circumstances such as following the exposure to viruses, the apoptosis has practical function to prevent the virus from spreading, but PM2.5 induce unnecessary apoptosis in pneumocytes (Dagher et al. 2006). This event can reduce the population of parenchymal cells and cause a decrease in lung function, a condition like obstructive diseases.
Finally, the findings of this study showed that, because of the chemical structure of PM2.5, they could induce a permanent inflammation without any antigen-presenting to activate repair mechanisms. Therefore, the tissue destruction and incidence of obstructive diseases are more likely compared to fibrosis. This directly depends on the exposure period to PM2.5 and the dosage. The existence of any mechanism to induce adaptive immunity against PM2.5 is unclear and must be investigated. Also, there is no information about the other components of PM2.5 that may have receptors in pneumocytes. Moreover, it is not clear whether or not there is any pathway for the activation of anti-inflammatory response against the induced inflammation by PM2.5.
Additionally, it is not clear why some of the PM2.5 can enter the cells, and the others cannot. The response to this question could help in distinguishing between initial inflammatory responses of damaged cells and the prediction of the next steps of inflammation. Moreover, because of the absence of any human studies in this field, the pathological effects of long-term exposures to PM2.5 in human lungs are not apparent and must be further investigated to validate these findings (Deng et al. 2013; Moon et al. 2018; Roh and Sohn 2018).
Limitations
Several limitations restricted the conclusions from the included articles. The exposure procedure in several included studies was single-dose exposure, and their results were only compared with their control group. There was no capability to merge their results with other studies to find a correlation between inflammatory response and dose/duration of exposure. Pathological pieces of evidence are significant to illustrate the inflammation in tissues, but just a few articles had pathological investigations. We found several short period animal studies about the inflammatory effects of PM2.5 on lung cells, which included pathological evaluation. However, those articles lacked the chemical analysis of particulate, which is a critical data and had been excluded from this review. Another limitation of this study was related to the selected exposure doses in animal studies. The exposure doses were not selected according to the respiration rates of animals. Therefore, we could not extrapolate the animal results to the human. There was no co-culture study in in vitro studies. Thus, there were no results about innate immune cells that responded to inflammatory cytokines secreted from the lung cells.
Conclusion
According to the in vitro and animal studies, urban PM2.5 activates the inflammatory pathway that involves innate immune responses initiated from exposed/damaged lung parenchymal cells. PM2.5 cannot activate adaptive immune responses because of their chemical nature. However, this condition strengthens the development of obstructive diseases such as emphysema and weakens the probability of restrictive diseases such as IPF because the exposure to PM2.5 and following inflammation is permanent in urban areas. More detailed and robust evidence about the inflammatory effects of long-term exposure to PM2.5 would have to come from human studies such as cohort or cross-sectional studies on highly exposed persons. The adaptation mechanisms to balance inflammatory and anti-inflammatory pathways in humans are unclear and need further investigations. Moreover, the effect of seasonal changes in PM2.5 exposure dose on inflammation is unclear. Also, variations in the composition of particulates need to be investigated further.
Data availability
All of the used datasets during the current study are available online.
References
Abbas AK, Lichtman AH, Pillai S (2019a) Basic immunology e-book: functions and disorders of the immune system. Elsevier Health Sciences
Abbas I, Badran G, Verdin A, Ledoux F, Roumie M, Lo Guidice JM, Courcot D, Garçon G (2019b) In vitro evaluation of organic extractable matter from ambient PM2. 5 using human bronchial epithelial BEAS-2B cells: cytotoxicity, oxidative stress, pro-inflammatory response, genotoxicity, and cell cycle deregulation. Environ Res 171:510–522
Akhtar US, McWhinney RD, Rastogi N, Abbatt JP, Evans GJ, Scott JA (2010) Cytotoxic and proinflammatory effects of ambient and source-related particulate matter (PM) in relation to the production of reactive oxygen species (ROS) and cytokine adsorption by particles. Inhal Toxicol 22:37–47
Chilosi M, Poletti V, Rossi A (2012) The pathogenesis of COPD and IPF: distinct horns of the same devil? Respir Res 13:3
Cohen AJ, Brauer M, Burnett R, Anderson HR, Frostad J, Estep K, Balakrishnan K, Brunekreef B, Dandona L, Dandona R, Feigin V, Freedman G, Hubbell B, Jobling A, Kan H, Knibbs L, Liu Y, Martin R, Morawska L, Pope CA III, Shin H, Straif K, Shaddick G, Thomas M, van Dingenen R, van Donkelaar A, Vos T, Murray CJL, Forouzanfar MH (2017) Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet 389:1907–1918
Cosio MG, Majo J, Cosio MG (2002) Inflammation of the airways and lung parenchyma in COPD: role of T cells. Chest 121:160S–165S
Cottin V, Hansell DM, Sverzellati N, Weycker D, Antoniou KM, Atwood M, Oster G, Kirchgaessler KU, Collard HR, Wells AU (2017) Effect of emphysema extent on serial lung function in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 196:1162–1171
Dagher Z, Garçon G, Billet S, Gosset P, Ledoux F, Courcot D, Aboukais A, Shirali P (2006) Activation of different pathways of apoptosis by air pollution particulate matter (PM2. 5) in human epithelial lung cells (L132) in culture. Toxicology 225:12–24
Dagher Z, Garçon G, Gosset P, Ledoux F, Surpateanu G, Courcot D, Aboukais A, Puskaric E, Shirali P (2005) Pro-inflammatory effects of Dunkerque city air pollution particulate matter 2.5 in human epithelial lung cells (L132) in culture. J Appl Toxicol 25:166–175
Deiuliis JA et al (2012) Pulmonary T cell activation in response to chronic particulate air pollution. Am J Phys Lung Cell Mol Phys 302:L399–L409
Deng X, Zhang F, Rui W, Long F, Wang L, Feng Z, Chen D, Ding W (2013) PM2. 5-induced oxidative stress triggers autophagy in human lung epithelial A549 cells. Toxicol in Vitro 27:1762–1770
Dergham M, Lepers C, Verdin A, Cazier F, Billet S, Courcot D, Shirali P, Garçon G (2015) Temporal–spatial variations of the physicochemical characteristics of air pollution particulate matter (PM2. 5–0.3) and toxicological effects in human bronchial epithelial cells (BEAS-2B). Environ Res 137:256–267
Dominici F, Peng RD, Bell ML, Pham L, McDermott A, Zeger SL, Samet JM (2006) Fine particulate air pollution and hospital admission for cardiovascular and respiratory diseases. Jama 295:1127–1134
El-Bayoumi E, Silvestri GA (2008) Bronchoscopy for the diagnosis and staging of lung cancer. In: Seminars in respiratory and critical care medicine. vol 03. © Thieme Medical Publishers, pp 261-270
Feng S, Gao D, Liao F, Zhou F, Wang X (2016) The health effects of ambient PM2. 5 and potential mechanisms. Ecotoxicol Environ Saf 128:67–74
Gandhi NA, Bennett BL, Graham NM, Pirozzi G, Stahl N, Yancopoulos GD (2016) Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov 15:35–50
Gao J, Peng X, Chen G, Xu J, Shi G-L, Zhang Y-C, Feng Y-C (2016) Insights into the chemical characterization and sources of PM2. 5 in Beijing at a 1-h time resolution. Sci Total Environ 542:162–171
Gavett SH, Haykal-Coates N, Copeland LB, Heinrich J, Gilmour MI (2003) Metal composition of ambient PM2. 5 influences severity of allergic airways disease in mice. Environ Health Perspect 111:1471–1477
Geng H, Meng Z, Zhang Q (2006) In vitro responses of rat alveolar macrophages to particle suspensions and water-soluble components of dust storm PM2. 5. Toxicol in Vitro 20:575–584
Gualtieri M, Øvrevik J, Holme JA, Perrone MG, Bolzacchini E, Schwarze PE, Camatini M (2010) Differences in cytotoxicity versus pro-inflammatory potency of different PM fractions in human epithelial lung cells. Toxicol in Vitro 24:29–39
Happo M et al (2010) Seasonal variation in chemical composition of size-segregated urban air particles and the inflammatory activity in the mouse lung. Inhal Toxicol 22:17–32
He M, Ichinose T, Kobayashi M, Arashidani K, Yoshida S, Nishikawa M, Takano H, Sun G, Shibamoto T (2016) Differences in allergic inflammatory responses between urban PM2. 5 and fine particle derived from desert-dust in murine lungs. Toxicol Appl Pharmacol 297:41–55
He M, Ichinose T, Yoshida S, Ito T, He C, Yoshida Y, Arashidani K, Takano H, Sun G, Shibamoto T (2017) PM2. 5-induced lung inflammation in mice: differences of inflammatory response in macrophages and type II alveolar cells. J Appl Toxicol 37:1203–1218
He R-W, Shirmohammadi F, Gerlofs-Nijland ME, Sioutas C, Cassee FR (2018) Pro-inflammatory responses to PM0. 25 from airport and urban traffic emissions. Sci Total Environ 640:997–1003
Hong Z, Guo Z, Zhang R, Xu J, Dong W, Zhuang G, Deng C (2016) Airborne fine particulate matter induces oxidative stress and inflammation in human nasal epithelial cells. Tohoku J Exp Med 239:117–125
Hou T, Liao J, Zhang C, Sun C, Li X, Wang G (2018) Elevated expression of miR-146, miR-139 and miR-340 involved in regulating Th1/Th2 balance with acute exposure of fine particulate matter in mice. Int Immunopharmacol 54:68–77
Hsu C-Y et al (2017) Ambient PM2. 5 in the residential area near industrial complexes: spatiotemporal variation, source apportionment, and health impact. Sci Total Environ 590:204–214
Huang K-L, Liu S-Y, Chou CC, Lee Y-H, Cheng T-J (2017a) The effect of size-segregated ambient particulate matter on Th1/Th2-like immune responses in mice. PLoS One 12
Huang Q, Chi Y, Deng J, Liu Y, Lu Y, Chen J, Dong S (2017b) Fine particulate matter 2.5 exerted its toxicological effect by regulating a new layer, long non-coding RNA. Sci Rep 7:1–9
Jin W, Wang L (2019) Role of rapid on site evaluation in preliminary diagnosis of pulmonary infection during bronchoscopy. Eur Respiratory Soc
Kim I, Lee K, Lee S, Kim SD (2019) Characteristics and health effects of PM2. 5 emissions from various sources in Gwangju, South Korea. Sci Total Environ 696:133890
Kim KM, Pae H-O, Zheng M, Park R, Kim Y-M, Chung H-T (2007) Carbon monoxide induces heme oxygenase-1 via activation of protein kinase R–like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress. Circ Res 101:919–927
King TE Jr, Pardo A, Selman M (2011) Idiopathic pulmonary fibrosis. Lancet 378:1949–1961
Kulkarni N, Pierse N, Rushton L, Grigg J (2006) Carbon in airway macrophages and lung function in children. N Engl J Med 355:21–30
Kumar V, Abbas AK, Fausto N, Aster JC (2014) Robbins and Cotran pathologic basis of disease, professional edition e-book. elsevier health sciences
Lauer FT, Mitchell LA, Bedrick E, McDonald JD, Lee WY, Li WW, Olvera H, Amaya MA, Berwick M, Gonzales M, Currey R, Pingitore NE Jr, Burchiel SW (2009) Temporal–spatial analysis of US–Mexico border environmental fine and coarse PM air sample extract activity in human bronchial epithelial cells. Toxicol Appl Pharmacol 238:1–10
Leclercq B, Alleman LY, Perdrix E, Riffault V, Happillon M, Strecker A, Lo-Guidice JM, Garçon G, Coddeville P (2017) Particulate metal bioaccessibility in physiological fluids and cell culture media: toxicological perspectives. Environ Res 156:148–157
Lewis JB, Bodine JS, Gassman JR, Muñoz SA, Milner DC, Dunaway TM, Egbert KM, Monson TD, Broberg DS, Arroyo JA, Reynolds PR (2018) Transgenic up-regulation of Claudin-6 decreases fine diesel particulate matter (DPM)-induced pulmonary inflammation. Environ Sci Pollut Res 25:18179–18188
Li D, Zhang R, Cui L, Chu C, Zhang H, Sun H, Luo J, Zhou L, Chen L, Cui J, Chen S, Mai B, Chen S, Yu J, Cai Z, Zhang J, Jiang Y, Aschner M, Chen R, Zheng Y, Chen W (2019) Multiple organ injury in male C57BL/6J mice exposed to ambient particulate matter in a real-ambient PM exposure system in Shijiazhuang, China. Environ Pollut 248:874–887
Li H, Wang Q', Yang M, Li F, Wang J, Sun Y, Wang C, Wu H, Qian X (2016) Chemical characterization and source apportionment of PM2. 5 aerosols in a megacity of Southeast China. Atmos Res 181:288–299
Lin G, Fu J, Jiang D, Hu W, Dong D, Huang Y, Zhao M (2014) Spatio-temporal variation of PM2. 5 concentrations and their relationship with geographic and socioeconomic factors in China. Int J Environ Res Public Health 11:173–186
Liu Q, Baumgartner J, Zhang Y, Schauer JJ (2016) Source apportionment of Beijing air pollution during a severe winter haze event and associated pro-inflammatory responses in lung epithelial cells. Atmos Environ 126:28–35
Liu S, Zhang W, Zhang F, Roepstorff P, Yang F, Lu Z, Ding W (2019) TMT-based quantitative proteomics analysis reveals airborne PM2. 5-induced pulmonary fibrosis. International journal of environmental research and public health 16:98
McMillan SJ et al (2004) Matrix metalloproteinase-9 deficiency results in enhanced allergen-induced airway inflammation. J Immunol 172:2586–2594
Medzhitov R (2008) Origin and physiological roles of inflammation. Nature 454:428–435
Miller MR, Borthwick SJ, Shaw CA, McLean SG, McClure D, Mills NL, Duffin R, Donaldson K, Megson IL, Hadoke PWF, Newby DE (2009) Direct impairment of vascular function by diesel exhaust particulate through reduced bioavailability of endothelium-derived nitric oxide induced by superoxide free radicals. Environ Health Perspect 117:611–616
Moon D et al (2018) Altered proinflammatory cytokines and M1 polarization induced by PM2.5 in alveolar macrophages. Appl Ecol Environ Res 16:7699–7712. https://doi.org/10.15666/aeer/1606_76997712
Pieterse B, Felzel E, Winter R, Van Der Burg B, Brouwer A (2013) PAH-CALUX, an optimized bioassay for AhR-mediated hazard identification of polycyclic aromatic hydrocarbons (PAHs) as individual compounds and in complex mixtures. Environ Sci Technol 47:11651–11659
Prieto-Parra L, Yohannessen K, Brea C, Vidal D, Ubilla CA, Ruiz-Rudolph P (2017) Air pollution, PM2. 5 composition, source factors, and respiratory symptoms in asthmatic and nonasthmatic children in Santiago, Chile. Environ Int 101:190–200
Raaschou-Nielsen O, Beelen R, Wang M, Hoek G, Andersen ZJ, Hoffmann B, Stafoggia M, Samoli E, Weinmayr G, Dimakopoulou K, Nieuwenhuijsen M, Xun WW, Fischer P, Eriksen KT, Sørensen M, Tjønneland A, Ricceri F, de Hoogh K, Key T, Eeftens M, Peeters PH, Bueno-de-Mesquita HB, Meliefste K, Oftedal B, Schwarze PE, Nafstad P, Galassi C, Migliore E, Ranzi A, Cesaroni G, Badaloni C, Forastiere F, Penell J, de Faire U, Korek M, Pedersen N, Östenson CG, Pershagen G, Fratiglioni L, Concin H, Nagel G, Jaensch A, Ineichen A, Naccarati A, Katsoulis M, Trichpoulou A, Keuken M, Jedynska A, Kooter IM, Kukkonen J, Brunekreef B, Sokhi RS, Katsouyanni K, Vineis P (2016) Particulate matter air pollution components and risk for lung cancer. Environ Int 87:66–73
Radan M, Dianat M, Badavi M, Mard SA, Bayati V, Goudarzi G (2019) In vivo and in vitro evidence for the involvement of Nrf2-antioxidant response element signaling pathway in the inflammation and oxidative stress induced by particulate matter (PM10): the effective role of gallic acid. Free Radic Res 53:210–225
Roh JS, Sohn DH (2018) Damage-associated molecular patterns in inflammatory diseases. Immune netw 18
Ruwanpura S, McLeod L, Anderson G, Jenkins B (2017) Novel role of inflammasomes in the molecular pathogenesis of emphysema. In: D97. Impact of inflammation on acute lung injury. American Thoracic Society, pp A7328-A7328
Sato A et al (2017) Diagnosis and clinical course of patients referred with chest X-ray abnormalities and suspected of nontuberculous mycobacterial lung disease. In: B62. Non-tuberculous mycobacteria: clinical aspects and cases. American Thoracic Society, pp A3966–A3966
Sharafkhaneh A, Hanania NA, Kim V (2008) Pathogenesis of emphysema: from the bench to the bedside. Proc Am Thorac Soc 5:475–477
Strieter RM (2008) What differentiates normal lung repair and fibrosis? Inflammation, resolution of repair, and fibrosis. Proc Am Thorac Soc 5:305–310
Taraseviciene-Stewart L, Voelkel NF (2008) Molecular pathogenesis of emphysema. J Clin Invest 118:394–402
Wang S et al (2017) Exposure to high level PM2. 5 causes increased central blood pressure. Circulation 136:A19823–A19823
Wilson M, Wynn T (2009) Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol 2:103–121
Wing K, Sakaguchi S (2010) Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol 11:7–13
Wingate P, McAulay K, Anthony I, Crawford D (2009) Regulatory T cell activity in primary and persistent Epstein–Barr virus infection. J Med Virol 81:870–877
Wolters PJ, Collard HR, Jones KD (2014) Pathogenesis of idiopathic pulmonary fibrosis. Ann Rev Pathol Mech Dis 9:157–179
Zhang Y, Lang J, Cheng S, Li S, Zhou Y, Chen D, Zhang H, Wang H (2018) Chemical composition and sources of PM1 and PM2. 5 in Beijing in autumn. Sci Total Environ 630:72–82
Zhao Y, Zhang H, Yang X, Zhang Y, Feng S, Yan X (2019) Fine particulate matter (PM2. 5) enhances airway hyperresponsiveness (AHR) by inducing necroptosis in BALB/c mice. Environ Toxicol Pharmacol 68:155–163
Acknowledgements
The authors are grateful to Dr. Kaveh Sadeghi at the Virology Department, School of Public Health, Tehran University of Medical Sciences, for scientific support.
Author information
Authors and Affiliations
Contributions
HRS and SR extracted data and wrote manuscript. MY designed the study and search strategy. BJ discussed the results.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Ludek Blaha
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shamsollahi, H.R., Jahanbin, B., Rafieian, S. et al. Particulates induced lung inflammation and its consequences in the development of restrictive and obstructive lung diseases: a systematic review. Environ Sci Pollut Res 28, 25035–25050 (2021). https://doi.org/10.1007/s11356-021-13559-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-021-13559-5