
Abstract
Introduction
Zonisamide is a new-generation anticonvulsant antiepileptic drug metabolized primarily in the liver, with subsequent elimination via the renal route.
Objectives
Our objective was to evaluate the utility of pharmacometabolomics in the detection of zonisamide metabolites that could be related to its disposition and therefore, to its efficacy and toxicity.
Methods
This study was nested to a bioequivalence clinical trial with 28 healthy volunteers. Each participant received a single dose of zonisamide on two separate occasions (period 1 and period 2), with a washout period between them. Blood samples of zonisamide were obtained from all patients at baseline for each period, before volunteers were administered any medication, for metabolomics analysis.
Results
After a Lasso regression was applied, age, height, branched-chain amino acids, steroids, triacylglycerols, diacyl glycerophosphoethanolamine, glycerophospholipids susceptible to methylation, phosphatidylcholines with 20:4 FA (arachidonic acid) and cholesterol ester and lysophosphatidylcholine were obtained in both periods.
Conclusion
To our knowledge, this is the only research study to date that has attempted to link basal metabolomic status with pharmacokinetic parameters of zonisamide.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Zonisamide, (ZNS) is a new-generation anticonvulsant with a unique chemical structure unrelated to other antiepileptic drugs (AED) (Masuda et al. 1980; Uno et al. 1979) (1,2-benzisoxazole-3-methanesulfonamide), a sulphonamide derivative. In Japan ZNS was first approved for clinical use in 1989, followed by South Korea in 1992, the USA in 2000 and Europe in 2005. Its indications includes partial seizures as monotherapy in adults with newly diagnosed epilepsy with or without secondary generalization; and adjunctive therapy for the treatment of partial seizures in adults, adolescents and children aged 6 years and above, with or without secondary generalization.
Blockage of voltage-sensitive sodium channels and T-type calcium channels are the predominant effects involved of ZNS (Kito et al. 1996; Rock et al. 1989; Schauf 1987; Suzuki et al. 1992). These mechanisms of action contribute to the stabilization of neuronal membranes and to the suppression of hypersynchronization (Romigi et al. 2015). In addition, ZNS appears to alter dopamine, 5-HT (serotonin) and acetylcholine metabolism, increasing striatal and hippocampal concentrations of dopamine and serotonin (total and extracellular) (Kaneko et al. 1993; Mizuno 1997; Okada et al. 1992, 1995, 1999). Also the gamma-aminobutyric acid and glutamate neurotransmitter systems appears to be modulated by ZNS (Mimaki et al. 1990; Okada et al. 1998; Ueda et al. 2003).
ZNS is rapidly and completely absorbed after oral administration, with a bioavailability of 100% (Kochak et al. 1998; Schulze-Bonhage 2010), and an extensively distribution in tissues (volume of distribution about 1.1–1.7 l/kg). Maximum serum concentrations achieved within 2–5 h under fasting conditions and 4–6 h with food (Sills and Brodie 2007). ZNS has a linear pharmakinetics after single doses of 100–800 mg and after multiple doses of 100–400 mg daily (Kochak et al. 1998; Sills and Brodie 2007), and it is partially bound to human serum albumin (approximately 50%) and preferentially accumulated in erythrocytes (Faught et al. 2001; Peters and Sorkin 1993). ZNS has a terminal plasma elimination half-life (t½) of ~ 60 h, independent of dose and dosing regimen, which allows it to be administered either once or twice daily (Sills and Brodie 2007). The maintenance dosage is 8 mg/kg per day in children and 100–600 mg per day in adults.
Zonisamide is mainly metabolized in the liver by the CYP3A4 isoenzyme to the open-ring metabolite 2-sulfamoylacetyl phenol, to its N-acetyl derivative by N-acetyltransferase (metabolites pharmacologically inactive) and to a lesser extent, directly conjugated to glucuronic acid. So the elimination half-life of ZNS in plasma depends on the presence of CYP3A4 enzyme inducers (e.g., phenobarbital, carbamazepine, and phenytoin), which reduce the t½ of ZNS to 25–35 h (Italiano and Perucca 2013; Levy et al. 2004; Ojemann et al. 1986). The CYP3A4 induction effects are unlikely to be clinically significant when zonisamide is added to existing therapy; however, changes in zonisamide concentrations could occur if doses of concomitant CYP3A4-inducing agents are changed or withdrawn, which might necessitate zonisamide dose adjustment (Brodie et al. 2012). Excretion in feces is a minor elimination route (Leppik 2004). Of the excreted dose, 15–30% was recovered as an unchanged drug, 20% as ZNS glucuronide and N-acetyl ZNS and 50% as the 2-sulfamoylacetyl phenol glucuronide (Frampton and Scott 2005; Sills and Brodie 2007).
Metabolomics is a multi-analytical technology that can assess the complete set of small molecules (< 1500 Da) in a specific matrix (cell, tissue or organism) under a given set of conditions (Goodacre 2007); this involves analysis of high-throughput data. Mass spectrometry, MS, couple to chromatography, gas (GC) or liquid (LC) and nuclear magnetic resonance (NMR) are the most widely used detection technologies (Fuhrer and Zamboni 2015; Lenz and Wilson 2007).
There are key aspects to take into account in a metabolomics project: the development of detection technologies, statistical methods to provide accurate and robust statistical analysis and potential patient confounders such as age, gender, comedications and diet (Gieser et al. 2011). Metabolomics has the potential to transform our understanding of mechanisms of pharmacokinetics, drug action and the molecular basis for variation in drug response. The various metabolites, their concentration and fluxes, represent the final products of cellular interactions that extend from gene sequence to gene expression, protein expression, and ultimately, to the total cellular environment (including drug exposure) (Kaddurah-Daouk and Weinshilboum 2015). Pharmacometabolomics is emerging as a discipline of metabolomics which involves determining an individual’s metabolic state as influenced by environment, genetics and gut microbiome (metabotype) to define signatures pre- and post-treatment that might explain variability in the drug pharmacokinetics (PK) or pharmacodynamics (PD) phenotype, and also predict treatment outcomes (Kaddurah-Daouk and Weinshilboum 2015; Kantae et al. 2017).
Statistical methods are used to connect the detected metabolites with the biological system. Statistical learning (Hastie et al. 2009) addresses the classical problem of control group versus treatment group or healthy patients vs. ill patients, but sparsity and robustness are needed to deal with more variables, such as high dimensional data and outliers that bias the results (Kurnaz et al. 2017). In some cases, the experimental design of the project implies a repeated measures structure and a methodological improvement is used to take into account the variability due to samples and participants. Methods such as a generalized linear mixed model with Lasso penalty is used to avoid these problems (Schelldorfer et al. 2011). Finally, once the important metabolites have been detected and their importance assessed in a robust manner, the biological interpretation and utility of the results in the daily routine are also key problems.
The aim of this work was to evaluate the utility of metabolomics in the prediction of ZNS disposition, evaluated as ZNS AUC.
2 Methods
2.1 Design and participants
The present study was performed within a randomized crossover trial with two periods to evaluate the bioequivalence of two 100-mg ZNS formulations (EUDRA-CT: 2013-004465-14). Twenty-eight healthy volunteers were included. All the participants received a test or reference formulation of ZNS in the first period (P1), and the other formulation in the second period (P2), with a washout period of at least 28 days between both periods. Blood samples for drug pharmacokinetics and metabolomics study were collected from all participants in each period, for a total of 28 × 2 observations (2 observations per participant). Bioequivalence of the two formulations was demonstrated, following the criteria accepted by current European Medicines Agency regulations (Committee for medicinal products for human use 2010). In addition, variables including age, sex, weight and height were obtained for each participant.
2.2 Pharmacokinetic study
The blood samples for drug pharmacokinetics were collected from all participants and placed in serum tubes at the following times: basal, 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 8, 10, 12, 24, 32, 48 and 72 h after drug administration. ZNS and the internal standard were measured by reversed phase high performance liquid chromatography coupled to a tandem mass spectrometry detector (LC/MS/MS). The pharmacokinetic analysis was performed using WinNonlin 6.3 software (Pharsight Corporation, Cary, USA) by means of a noncompartmental analysis.
Maximum concentration (Cmax) and the time to reach it (Tmax) were directly obtained from the plasma concentration results. AUC0-∞ (total area under the concentration–time curve; ng/ml*h) was calculated from the addition of two partial AUCs: (a) AUClast, area between the dosage time and the last time with detectable concentrations, calculated by the trapezoidal rule; and (b) AUCinf, calculated as the ratio C/k, where C is the last detectable concentration and k is the slope obtained in the lineal regression calculated from the points corresponding to the elimination phase of the drug. Pharmacokinetic data were log-transformed; Cmax and AUC were adjusted to dose/weight administered. AUCinf presents less variability than other pharmacokinetic variables; given a certain drug and a set of drug dosages and their corresponding AUC0inf, it is possible to estimate the dosage. We used〖log〗_10 (〖AUC〗_0∞) as the dependent variable.
2.3 Metabolomic profiling
The 3-ml blood samples for metabolomics analysis were collected from all the participants and placed in EDTA-K2 tubes at baseline of each period, before any medication was administered, and were stored at − 80 °C. Due to the wide concentration range of metabolites coupled with their extensive chemical diversity, there is no single platform or method to analyze the entire metabolome of a biological sample (Baker 2011; Duportet et al. 2012). In this study, multiple UPLC–MS platforms were used to optimize the coverage of the plasma metabolome (Barr et al. 2012).
Metabolite extraction was accomplished by fractionating the samples into pools of species with similar physicochemical properties, using appropriate combinations of organic solvents. Three separate UPLC–MS-based platforms were used to perform optimal profiling of fatty acids, bile acids, steroids and lysoglycerophospholipids (platform 1); glycerolipids, cholesteryl esters, sphingolipids and glycerophospholipids (platform 2) and amino acids (platform 3). Characteristics of each platform can be found in Supplementary Table S1.
Two types of quality control (QC) samples were used to assess the data quality. These were reference serum samples, which were evenly distributed over the batches and extracted and analyzed at the same time as the individual samples:
-
QC calibration sample: used to correct the various response factors between and within batches;
-
QC validation sample: used to assess how well data preprocessing procedures improved data quality.
Data preprocessing, using the TragetLynx application manager for MassLynx 4.1 software(Waters Corp., Milford, USA), generated a list of chromatographic peak areas for the metabolites detected in each sample injection. An approximate linear detection range was defined for each identified metabolite, assuming similar detector response levels for all metabolites belonging to a given chemical class represented by a single standard compound. Data points lying outside their corresponding linear detection range were replaced with missing values, and those metabolites for which more than 30% of data points were found outside their corresponding linear detection range were not used for statistical analyses.
Data normalization was performed following the procedure described by Martínez-Arranz et al. (2015), in which normalization factors were calculated for each metabolite by dividing their intensities in each sample by the recorded intensity of an appropriate internal standard in that same sample.
-
The most appropriate internal standard for each variable was defined as that which resulted in a minimum relative standard deviation after correction, as calculated from the QC calibration samples over all the analysis batches.
-
Robust linear regression (internal standard corrected response as a function of sample injection order) was used to estimate any intra-batch drift in the QC calibration samples not corrected for by internal standard correction. For all variables, internal standard corrected response in each batch was divided by its corresponding intra-batch drift trend, such that normalized abundance values of the study samples were expressed with respect to the batch-averaged QC calibration serum samples.
-
A Pareto scaling was also applied (van den Berg et al. 2006).
From this metabolite profiling data set with 521 metabolites (MET), a second data set of chemical classes was calculated as the sum of the normalized areas of all the metabolites with the same chemical characteristics, in total 80 chemical classes (CHEM), taking into account that similar looking chemicals can ionize very differently. Finally, demographic variables, such as age, weight, height, body mass index and sex were added, resulting in 527 predictors for MET and 86 for CHEM, respectively.
2.4 Statistical analysis
The chemical classes data set had no missing data, and the metabolites data set had only 25 observations missing, which were imputed using the random forest approach (Stekhoven and Bühlmann 2012).
Given that our dependent variable is a continuous variable, \({\log _{10}}\left( {AU{C_{\inf }}} \right)\), we face a regression problem, \({\mathbf{y}}={\varvec{X}}\beta +\varvec{\varepsilon}\), where \({\varvec{y}}\) is a vector of n observations, \({\varvec{X}}\) is a matrix of n observations and p predictors (527 or 86), \(\beta\) is the vector of regression coefficients and \(\varvec{\varepsilon}\) the residual error. The well-known least squares estimator of the regression coefficients is defined as follows:
In case of high dimensional data, as in our case with more predictors than observations, the previous formula is not appropriate to solve the problem. Columns of \({\varvec{X}}\) could be collinear, and on the other hand, overfitting could occur because there are many combinations of \(\beta\) which fit the data perfectly.
Modification of the sum of squared errors criterion via penalization (Witten and Tibshirani 2009) was the general approach used; we applied a Lasso penalty, with a more detailed explanation available in Hastie et al. 2009. Lasso computes some coefficients to zero; thus, these variables are not significant to explain the data.
There are also other approaches to this problem such elastic net regularization or partial least square regression. Elastic net (Zou and Hastie 2005) which select variables but allows correlation between them and methodological improvements over this procedures have been done when robust methods are applied together with elastic net (Kurnaz et al. 2017). Although Lasso is not the unique approach to this problem we decide to use it since the selection variable is done in order to built a future clinical practice ZNS dosage algorithm which should be sparse as possible in the number of variables as our previous knowledge point (Borobia et al. 2012).
Lasso regression was applied first to the CHEM, and second to the MET, by period. We fit models into two different periods to avoid the repeated measures problem.
After the variable selection via Lasso, a linear mixed model is fitted using the selected variables. To provide a measure of performance, we use the root mean of the standard error of prediction (RMSEP), building a model with variables selected in both periods.
where \({y_i}\) is the observed value, \({\hat {y}_i}\), the predicted value based on our Lasso variable selection and \(n\) the number of elements to compare.
Statistical computations were performed using the statistical environment R (R Development Core Team 2013), and the glmnet package was used to fit Lasso regression (Friedman et al. 2010) and missForest (Stekhoven and Bühlmann 2012) for imputation of missing data.
2.5 Ethics
The bioequivalence clinical trial protocol was approved by the Ethics Committee of La Paz University Hospital, Madrid (EUDRA-CT Code: 2013-004465-14). This research project nested to the bioequivalence clinical trial was approved by the same Ethics Committee (Code: ZONIP3M). All participants gave their written consent before study initiation and after reception of written and oral information related to the objectives, characteristics, procedures, risks and rights of participation in the study.
3 Results
Table 1 presents pharmacokinetic variables by period, in terms of mean, first and third quartile (Q1, Q3). This table also provides information about demographic variables.
3.1 Chemical classes
Variables selected in the chemical classes data set via Lasso (and so perceived to be important to the model) are presented in Table 2, the corresponding regression coefficients estimates are available in Table S2 in the supplementary material section. Age, height, branched-chain amino acids, steroids, triacylglycerols, diacyl glycerophosphoethanolamine, methylation-susceptible-glycerophospholipids, phosphatidylcholines with 20:4 FA (arachidonic acid) and cholesterol ester (ChoE) and lysophosphatidylcholine (LPC), were obtained in both periods. RMSEP using this model gives a value of 0.0312.
3.2 Metabolomic profile
Table 3 shows variables selected via Lasso for each period using the metabolite dataset, the corresponding regression coefficients estimates are available in Table S3 in the supplementary material section. Only height, l-cystine, diacylglycerophosphocholines and hexanoylcarnitine appear selected in both periods. Using this model, RMSEP results in 0.0295.
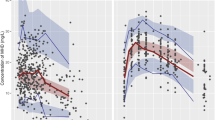
Predicted versus observed values using Lasso selected variables in a repeated measures model using both periods are shown in Fig. 1, for both the chemical class and metabolite datasets. Our data suggest that the use of metabolites instead of chemical class to predict AUCinf produces more accurate predictions even though metabolites show significant correlation between them.

Predicted versus observed values using Lasso selected variables in a repeated measures model with all data, P1 and P2. Black: chemical classes, blue: metabolite data set
4 Discussion
The results presented in this study provide an example of the potential of pharmacometabolomics in the field of pharmacokinetics and pharmacodynamics, as other authors have shown (Elbadawi-Sidhu et al. 2017; Lin et al. 2016). Related to PK, the main aim of pharmacometabolomic studies is to identify endogenous metabolite markers that allow for the stratification of patients into exposure groups, which is needed to individualize drug dosing regimens (Kantae et al. 2017). It offers an advantage over phamacogenetics, which uses genetic polymorphisms to predict individual variations in responses, but does not take into account other factors that are known to have marked impact on the PK of drugs such as tissue composition or gut microbiome. Some studies have been published associating predose metabolomics information with drug exposure: Phapale et al. (Phapale et al. 2010) correlated predose urine metabolites to the AUC of tacrolimus and inferred a metabolomics phenotype that can predict exposure to tacrolimus. Huang et al. (2015) associated pre-dose plasma metabolic profiles with atorvastatin exposure. Another report by Muhrez et al. (2017) identified 28 endogenous urine metabolites before drug administration that were predictive of the clearance of high-dose methotrexate in patients with lymphoid malignancies. The design of Phapale and Huang is similar to our bioequivalence clinical trial in sample size and predose samples.
However, most of the pharmacometabolomic studies aiming to examine PD and changes in (patho) physiology upon drug exposure by investigating differences in pre- and postdose endogenous metabolomics profiles and identifying patterns that can explain interindividual differences in treatment efficacy (Kantae et al. 2017). The applicability of this approach has been shown in several studies: Zhu et al. linked the response to sertraline for a depressed state to different metabolomics profiles pre- and post-treatment. (Zhu et al. 2013). A more recent study (Tan et al. 2017) reports differences in metabolic profiles of pretreatment serum between patients showing various responses to a standard cytarabine plus anthracycline regimen in acute myeloid leukemia.
Although a great number of metabolites related to interindividual variability in the PK–PD of drugs have been identified, its applications in clinical practice are still scarce. Descriptions of more metabolite signatures for drug response and adverse effects are required to allow the design of confirmatory personalized trials targeted to specific populations that can benefit the most from a certain drug.
In this pharmacometabolomic study we addressed the identification of endogenous metabolites predicting ZNS disposition. This approach would provide some insight in order to personalize ZNS drug dosages. To this end, we linked metabolite concentration with AUCinf, using penalized regression methods (Lin et al. 2016). These methods deal with one of the major problems in metabolomics research: more variables than observations. This study allowed us to develop a targeted metabolomic study in a new data set to validate the predictive ability of the metabolites as predictors for AUC.
Among the metabolites identified as possible predictors of ZNS bioavailability, hexanoylcarnitine pertains to the acylcarnitine group; these are vital for the transport of fatty acids into the mitochondrial matrix (Indiveri et al. 2011) and are good markers for mitochondrial function, because incomplete fatty acid oxidation results in elevated acylcarnitine concentrations (Koves et al. 2005). Cystine serves as a substrate for the cystine–glutamate antiporter. This transport system increases the concentration of cystine inside the cell; cystine is then quickly reduced to cysteine, which is the limiting precursor for glutathione synthesis. Accordingly, this antiporter system is widely reported to support antioxidant defenses in vivo (Lewerenz et al. 2013; McBean 2002). Diacylglycerophosphocholines are phospholipids, essential components of cell membranes. Regarding to the estimated coefficients via Lasso, hexanoylcarnitine has the large value in both periods with a negative sign, the larger the amount of hexanoylcarnitine, the lower the ZNS AUC. On the other hand, cystine estimated coefficient is 100 times smaller than hexanoylcarnitine with a positive sign. This means that the influence of cystine, exists but with lower impact in the final ZNS AUC. Finally, diacylglycerophosphocholines estimated coefficients have the same positive sign in both periods but with different magnitude indicating that the larger the Diacylglycerophosphocholines the higher ZNS AUC.
With the aim of reducing one of the main difficulties in a metabolomics data set—the correlation between metabolites—we adopted the chemical class approach of adding metabolites of the same chemical class in a single variable. Differences is RMSEP performance between models, CHEM versus MET, produced more error than expected when we planned these analyses. Thus we could show that a chemical classes approach would not be appropriate and correlation between metabolites should be addressed in the analysis, since similar looking chemicals can ionize very differently; however, this approach should be confirmed by further research.
The major limitations of our study are the absence of a validation set and the small sample size. Given our work is nested to a bioequivalence clinical trial in which the dependent variables are pharmacokinetics parameters, a smaller sample size results when the dependent variable is a clinical variable. Bioequivalence clinical trials are an important source of information to make advances in personalized medicine via pharmacometabolomics. Although validation data are not available for the nature of the bioequivalence clinical trial, our results report an improvement using metabolic information regarding ZNS.
Finally, this study shows the potential utility of using metabolomics within bioequivalence and other phase I trials to explore and find signatures associated with the disposition of new and old drugs. This approach could ultimately facilitate the individualization of drug therapies during and after drug development.
5 Conclusions
To our knowledge, there is no current research exploring the link between endogenous metabolomics and disposition of ZNS. More research is required before this research will allow determination of an individualized dose of ZNS according to a metabolite signature that can be applied in clinical practice. However, the first steps performed here open a path for further research on this and other drugs to accomplish this aim.
Change history
14 June 2018
The original version of this article contains a mistake.
References
Baker, M. (2011). Metabolomics: From small molecules to big ideas. Nature Methods, 8(2), 117–121.
Barr, J., Caballería, J., Martínez-Arranz, I., Domínguez-Díez, A., Alonso, C., Muntané, J., et al. (2012). Obesity-dependent metabolic signatures associated with nonalcoholic fatty liver disease progression. Journal of Proteome Research, 11(4), 2521–2532. https://doi.org/10.1021/pr201223p.
Borobia, A. M., Lubomirov, R., Ramírez, E., Lorenzo, A., Campos, A., Muñoz-Romo, R., et al. (2012). An acenocoumarol dosing algorithm using clinical and pharmacogenetic data in Spanish patients with thromboembolic disease. PLoS ONE, 7(7), e41360. https://doi.org/10.1371/journal.pone.0041360.
Brodie, M. J., Ben-Menachem, E., Chouette, I., & Giorgi, L. (2012). Zonisamide: Its pharmacology, efficacy and safety in clinical trials. Acta Neurologica Scandinavica, 126(S194), 19–28. https://doi.org/10.1111/ane.12016.
Committee for medicinal products for human use. (2010). Guideline on the investigation of bioequivalence (No. Doc. Ref.: CPMP/EWP/QWP/1401/98 Rev. 1/ Corr **). London. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf.
Duportet, X., Bastos, R., Aggio, M., Carneiro, S., & Villas-Bôas, S. G. (2012). The biological interpretation of metabolomic data can be misled by the extraction method used. Metabolomics, 8(3), 410–421.
Elbadawi-Sidhu, M., Baillie, R. A., Zhu, H., Chen, Y.-D. I., Goodarzi, M. O., Rotter, J. I., et al. (2017). Pharmacometabolomic signature links simvastatin therapy and insulin resistance. Metabolomics, 13(1), 11. https://doi.org/10.1007/s11306-016-1141-3.
Faught, E., Ayala, R., Montouris, G. G., Leppik, I. E., & Zonisamide 922 Trial Group, (2001). Randomized controlled trial of zonisamide for the treatment of refractory partial-onset seizures. Neurology, 57(10), 1774–1779.
Frampton, J. E., & Scott, L. J. (2005). Zonisamide: A review of its use in the management of partial seizures in epilepsy. CNS Drugs, 19(4), 347–367.
Friedman, J., Hastie, T., & Tibshirani, R. (2010). Regularization paths for generalized linear models via coordinate descent. Journal of Statistical Software. https://doi.org/10.18637/jss.v033.i01.
Fuhrer, T., & Zamboni, N. (2015). High-throughput discovery metabolomics. Current Opinion in Biotechnology, 31, 73–78. https://doi.org/10.1016/j.copbio.2014.08.006.
Gieser, G., Harigaya, H., Colangelo, P. M., & Burckart, G. (2011). Biomarkers in solid organ transplantation. Clinical Pharmacology and Therapeutics, 90(2), 217–220. https://doi.org/10.1038/clpt.2011.75.
Goodacre, R. (2007). Metabolomics of a superorganism. The Journal of Nutrition, 137(1 Suppl), 259S–266S.
Hastie, T., Tibshirani, R., & Friedman, J. H. (2009). The elements of statistical learning: Data mining, inference, and prediction (2nd ed.). New York: Springer.
Huang, Q., Aa, J., Jia, H., Xin, X., Tao, C., Liu, L., et al. (2015). A Pharmacometabonomic approach to predicting metabolic phenotypes and pharmacokinetic parameters of atorvastatin in healthy volunteers. Journal of Proteome Research, 14(9), 3970–3981. https://doi.org/10.1021/acs.jproteome.5b00440.
Indiveri, C., Iacobazzi, V., Tonazzi, A., Giangregorio, N., Infantino, V., Convertini, P., et al. (2011). The mitochondrial carnitine/acylcarnitine carrier: Function, structure and physiopathology. Molecular Aspects of Medicine, 32(4–6), 223–233. https://doi.org/10.1016/j.mam.2011.10.008.
Italiano, D., & Perucca, E. (2013). Clinical pharmacokinetics of new-generation antiepileptic drugs at the extremes of age: An update. Clinical Pharmacokinetics, 52(8), 627–645. https://doi.org/10.1007/s40262-013-0067-4.
Kaddurah-Daouk, R., & Weinshilboum, R. (2015). Metabolomic signatures for drug response phenotypes: Pharmacometabolomics enables precision medicine. Clinical Pharmacology & Therapeutics, 98(1), 71–75. https://doi.org/10.1002/cpt.134.
Kaneko, S., Okada, M., Hirano, T., Kondo, T., Otani, K., & Fukushima, Y. (1993). Carbamazepine and zonisamide increase extracellular dopamine and serotonin levels in vivo, and carbamazepine does not antagonize adenosine effect in vitro: Mechanisms of blockade of seizure spread. The Japanese Journal of Psychiatry and Neurology, 47(2), 371–373.
Kantae, V., Krekels, E. H. J., Esdonk, M. J., Van Lindenburg, P., Harms, A. C., Knibbe, C. A. J., et al. (2017). Integration of pharmacometabolomics with pharmacokinetics and pharmacodynamics: Towards personalized drug therapy. Metabolomics, 13(1), 9. https://doi.org/10.1007/s11306-016-1143-1.
Kito, M., Maehara, M., & Watanabe, K. (1996). Mechanisms of T-type calcium channel blockade by zonisamide. Seizure, 5(2), 115–119.
Kochak, G. M., Page, J. G., Buchanan, R. A., Peters, R., & Padgett, C. S. (1998). Steady-state pharmacokinetics of zonisamide, an antiepileptic agent for treatment of refractory complex partial seizures. Journal of Clinical Pharmacology, 38(2), 166–171.
Koves, T. R., Li, P., An, J., Akimoto, T., Slentz, D., Ilkayeva, O., et al. (2005). Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. The Journal of Biological Chemistry, 280(39), 33588–33598. https://doi.org/10.1074/jbc.M507621200.
Kurnaz, F. S., Hoffmann, I., & Filzmoser, P. (2017). Robust and sparse estimation methods for high dimensional linear and logistic regression. Retrieved from http://arxiv.org/abs/1703.04951.
Lenz, E. M., & Wilson, I. D. (2007). Analytical strategies in metabonomics. Journal of Proteome Research, 6(2), 443–458. https://doi.org/10.1021/pr0605217.
Leppik, I. E. (2004). Zonisamide: Chemistry, mechanism of action, and pharmacokinetics. Seizure, 13, S5–S9. https://doi.org/10.1016/j.seizure.2004.04.016.
Levy, R. H., Ragueneau-Majlessi, I., Garnett, W. R., Schmerler, M., Rosenfeld, W., Shah, J., & Pan, W.-J. (2004). Lack of a clinically significant effect of zonisamide on phenytoin steady-state pharmacokinetics in patients with epilepsy. Journal of Clinical Pharmacology, 44(11), 1230–1234. https://doi.org/10.1177/0091270004268045.
Lewerenz, J., Hewett, S. J., Huang, Y., Lambros, M., Gout, P. W., Kalivas, P. W., et al. (2013). The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxidants & Redox Signaling, 18(5), 522–555. https://doi.org/10.1089/ars.2011.4391.
Lin, Y. S., Kerr, S. J., Randolph, T., Shireman, L. M., Senn, T., & McCune, J. S. (2016). Prediction of intravenous busulfan clearance by endogenous plasma biomarkers using global pharmacometabolomics. Metabolomics, 12(10), 161. https://doi.org/10.1007/s11306-016-1106-6.
Martínez-Arranz, I., Mayo, R., Pérez-Cormenzana, M., Mincholé, I., Salazar, L., Alonso, C., & Mato, J. M. (2015). Enhancing metabolomics research through data mining. Journal of Proteomics, 127, 275–288. https://doi.org/10.1016/j.jprot.2015.01.019.
Masuda, Y., Karasawa, T., Shiraishi, Y., Hori, M., Yoshida, K., & Shimizu, M. (1980). 3-Sulfamoylmethyl-1,2-benzisoxazole, a new type of anticonvulsant drug. Pharmacological profile. Arzneimittel-Forschung, 30(3), 477–483.
McBean, G. J. (2002). Cerebral cystine uptake: A tale of two transporters. Trends in Pharmacological Sciences, 23(7), 299–302.
Mimaki, T., Suzuki, Y., Tagawa, T., Karasawa, T., & Yabuuchi, H. (1990). Interaction of zonisamide with benzodiazepine and GABA receptors in rat brain. Medical Journal of Osaka University, 39(1–4), 13–17.
Mizuno, K. (1997). Effects of carbamazepine and zonisamide on acetylcholine levels in rat striatum. Nihon shinkei seishin yakurigaku, 17(1), 17–23.
Muhrez, K., Benz-de Bretagne, I., Nadal-Desbarats, L., Blasco, H., Gyan, E., Choquet, S., et al. (2017). Endogenous metabolites that are substrates of organic anion transporter’s (OATs) predict methotrexate clearance. Pharmacological Research, 118, 121–132. https://doi.org/10.1016/j.phrs.2016.05.021.
Ojemann, L. M., Shastri, R. A., Wilensky, A. J., Friel, P. N., Levy, R. H., McLean, J. R., & Buchanan, R. A. (1986). Comparative pharmacokinetics of zonisamide (CI-912) in epileptic patients on carbamazepine or phenytoin monotherapy. Therapeutic Drug Monitoring, 8(3), 293–296.
Okada, M., Hirano, T., Kawata, Y., Murakami, T., Wada, K., Mizuno, K., et al. (1999). Biphasic effects of zonisamide on serotonergic system in rat hippocampus. Epilepsy Research, 34(2–3), 187–197.
Okada, M., Kaneko, S., Hirano, T., Ishida, M., Kondo, T., Otani, K., & Fukushima, Y. (1992). Effects of zonisamide on extracellular levels of monoamine and its metabolite, and on Ca2+ dependent dopamine release. Epilepsy Research, 13(2), 113–119.
Okada, M., Kaneko, S., Hirano, T., Mizuno, K., Kondo, T., Otani, K., & Fukushima, Y. (1995). Effects of zonisamide on dopaminergic system. Epilepsy Research, 22(3), 193–205.
Okada, M., Kawata, Y., Mizuno, K., Wada, K., Kondo, T., & Kaneko, S. (1998). Interaction between Ca2+, K+, carbamazepine and zonisamide on hippocampal extracellular glutamate monitored with a microdialysis electrode. British Journal of Pharmacology, 124(6), 1277–1285. https://doi.org/10.1038/sj.bjp.0701941.
Peters, D. H., & Sorkin, E. M. (1993). Zonisamide. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in epilepsy. Drugs, 45(5), 760–787.
Phapale, P. B., Kim, S.-D., Lee, H. W., Lim, M., Kale, D. D., Kim, Y.-L., et al. (2010). An Integrative approach for identifying a metabolic phenotype predictive of individualized pharmacokinetics of tacrolimus. Clinical Pharmacology & Therapeutics, 87(4), 426–436. https://doi.org/10.1038/clpt.2009.296.
R Development Core Team. (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, http://www.R-project.org/. R Foundation for Statistical Computing, Vienna, Austria.
Rock, D. M., Macdonald, R. L., & Taylor, C. P. (1989). Blockade of sustained repetitive action potentials in cultured spinal cord neurons by zonisamide (AD 810, CI 912), a novel anticonvulsant. Epilepsy Research, 3(2), 138–143.
Romigi, A., Femia, E. A., Fattore, C., Vitrani, G., Di Gennaro, G., & Franco, V. (2015). Zonisamide in the management of epilepsy in the elderly. Clinical Interventions in Aging, 10, 931–937. https://doi.org/10.2147/CIA.S50819.
Schauf, C. L. (1987). Zonisamide enhances slow sodium inactivation in Myxicola. Brain Research, 413(1), 185–188.
Schelldorfer, J., Meier, L., & Bühlmann, P. (2011). GLMMLasso: An algorithm for high-dimensional generalized linear mixed models using l1-penalization. Journal of Computational and Graphical Statistics. https://doi.org/10.1080/10618600.2013.773239.
Schulze-Bonhage, A. (2010). Zonisamide in the treatment of epilepsy. Expert Opinion on Pharmacotherapy, 11(1), 115–126. https://doi.org/10.1517/14656560903468728.
Sills, G., & Brodie, M. (2007). Pharmacokinetics and drug interactions with zonisamide. Epilepsia, 48(3), 435–441. https://doi.org/10.1111/j.1528-1167.2007.00983.x.
Stekhoven, D. J., & Bühlmann, P. (2012). Missforest-non-parametric missing value imputation for mixed-type data. Bioinformatics, 28, 112–118. https://doi.org/10.1093/bioinformatics/btr597.
Suzuki, S., Kawakami, K., Nishimura, S., Watanabe, Y., Yagi, K., Seino, M., & Miyamoto, K. (1992). Zonisamide blocks T-type calcium channel in cultured neurons of rat cerebral cortex. Epilepsy Research, 12(1), 21–27.
Tan, G., Zhao, B., Li, Y., Liu, X., Zou, Z., Wan, J., et al. (2017). Pharmacometabolomics identifies dodecanamide and leukotriene B4 dimethylamide as a predictor of chemosensitivity for patients with acute myeloid leukemia treated with cytarabine and anthracycline. Oncotarget. https://doi.org/10.18632/oncotarget.20733.
Ueda, Y., Doi, T., Tokumaru, J., & Willmore, L. J. (2003). Effect of zonisamide on molecular regulation of glutamate and GABA transporter proteins during epileptogenesis in rats with hippocampal seizures. Molecular Brain Research, 116(1–2), 1–6.
Uno, H., Kurokawa, M., Masuda, Y., & Nishimura, H. (1979). Studies on 3-substituted 1,2-benzisoxazole derivatives. 6. Syntheses of 3-(sulfamoylmethyl)-1,2-benzisoxazole derivatives and their anticonvulsant activities. Journal of Medicinal Chemistry, 22(2), 180–183.
van den Berg, R. A., Hoefsloot, H. C., Westerhuis, J. A., Smilde, A. K., & van der Werf, M. J. (2006). Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genomics, 7(1), 142. https://doi.org/10.1186/1471-2164-7-142.
Witten, D. M., & Tibshirani, R. (2009). Covariance-regularized regression and classification for high dimensional problems. Journal of the Royal Statistical Society: Series B (Statistical Methodology), 71(3), 615–636.
Zhu, H., Bogdanov, M. B., Boyle, S. H., Matson, W., Sharma, S., Matson, S., et al. (2013). Pharmacometabolomics of response to sertraline and to Placebo in major depressive disorder—Possible role for methoxyindole pathway. PLoS ONE, 8(7), e68283. https://doi.org/10.1371/journal.pone.0068283.
Zou, H., & Hastie, T. (2005). Regularization and variable selection via the elastic net. Journal of the Royal Statistical Society, Series B, 67, 301–320.
Acknowledgements
The authors would like to thank Dr. Jennifer Kirwan for her valuable comments to the manuscript. Also to the scientific committee and the participants of MOVISS 2017 Bio&Data, Vorau, Austria, for their feedback in metabolomics and interest in our data.
Funding
The ZNS bioequivalence study was funded by Laboratorios Normon, Ronda de Valdecarrizo 6, 28760 Tres Cantos, Madrid, SPAIN. The research project has been cofinanced by the Ministerio de Economia y Competitividad within the INNPACTO program (IPT-2012-0576-090000) and by the European Regional Development Fund (ERDF “A way of making Europe”).
Author information
Authors and Affiliations
Contributions
JCM-A and AJCS and AMB designed the study. JF, PG, HYT provided the pharmacokinetics study. AGB, IG, ID and LD contributed with the metabolomics results interpretation. JCM-A, AGB and AMB, wrote the manuscript. JCM-A perform the statistical analysis. All authors revised and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Martínez-Ávila, J.C., García Bartolomé, A., García, I. et al. Pharmacometabolomics applied to zonisamide pharmacokinetic parameter prediction. Metabolomics 14, 70 (2018). https://doi.org/10.1007/s11306-018-1365-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-018-1365-5