
Abstract
Natural extracts used in flavour and fragrances are exposed to authentication issues. Companies working in this industrial market acquire the raw materials locally but also abroad, sourcing exotic plants with specific olfactive features or lower costs of production. The geographical origin, the botanical variety, environmental conditions, extraction processes and storage conditions represent some parameters affecting the natural extract quality. All these factors are likely to affect the sensorial properties and especially the organoleptic characteristics of the extract. In addition, fraudulent practices known as adulterations have also an impact on the quality. Sensitive and precise analytical techniques are required to identify adulterations among other sources of variability. In this context, the highly valuable rose absolute was selected as a model study for its importance in perfumery. The existence of two botanical species and several production countries are additional reasons that make this extract an interesting case study. Because the usual GC–MS metabolomic approach is not able to cover the broad range of non-volatile compounds, complementary approaches are required. An UHPLC-ToFMS fingerprinting approach was therefore developed to allow the identification of non-volatile markers of the two closely related species of the genus Rosa. Thus, 12-oxophytodienoic acid was identified as a biomarker (level 2 according to MSI guideline) enabling the distinction between R. centifolia and R. damascena. Our results finally underline the efficiency of the UHPLC-ToFMS metabolomic approach for the qualification of odorant extracts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Authentication is a major issue in the industrial sectors working with natural extracts. Geographical origins, botanical species, cultivation and harvest practices, as well as extraction processes represent some factors that may affect the composition of natural extracts (Salgueiro et al. 2010). Additionally, fraudulent practices are frequent in this lucrative industry that may change product qualities and therefore mislead customers. Among the most common practices, addition of external compounds or mixing different origins will alter the desired organoleptic properties (McHale and Sheridan 1988). The generic term used to characterize these practices is adulteration (Do et al. 2015).
Natural extracts used for flavours and fragrances are generally of high-added value and as such they are often the subject of adulteration. Despite a large panel of authentication techniques (Mazollier 2012; Fernandez et al. 2003; Jirovetz et al. 2005; Aycı et al. 2005; Li et al. 2013; Kreis and Mosandl 1992; Ravid et al. 1992; Buiarelli et al. 1991; Saraswathy et al. 2010; Schipilliti et al. 2011), none of them is exhaustive enough to determine the true level of adulteration and additional techniques are still needed. This is mainly linked to the large chemical diversity of the ingredients used in this industry. Essential oils obtained through steam distillation, hydro distillation or cold pressure are mainly composed of volatile compounds, analyzed by gas chromatography (GC). On the contrary, solvent extracts called concretes, absolutes or resinoids are largely composed of non-volatile compounds (Naves 1974) poorly detected by GC. The concrete is obtained after extraction of the raw material by apolar solvents such as hexane or petroleum ether. For the preparation of perfumes, the concrete is macerated in ethanol which is followed by a cold filtration of the insoluble apolar non-volatile compounds, usually called “waxes”. The process ends up by evaporation of the solvents leading to the absolute (Bauer 2001). The chemical analyses of concretes, absolutes and resinoids require a combination of techniques to determine their precise composition.
The recent development of metabolomics approaches using the highly sensitive UHPLC-ToFMS technology offers the ability to study in depth the chemical composition of the solvent extracts. Potential adulteration can therefore be highlighted by statistical analysis, after corrections of the biases originated from biological variability. We decided to develop a metabolomics approach aiming at the detection of adulteration for the symbolic flowers of perfumery: Rosa centifolia and Rosa damascena. Roses are cultivated in Turkey, Bulgaria, Morocco, China, India, Italia and France (Zrira 2006; Naves 1974). Mainly found in France, Italy and Morocco R. centifolia is the most precious species used for this purpose with a cost of 16.500 €/kg (absolute, France, 2013) and 2.500 €/kg for R. damascena (absolute, Bulgaria, 2013). R. damascena is used to obtain essential oils (yield: 0.03 %), concretes (yield: 0.20–0.27 %) and absolutes (yield: 50–65 % from concrete extraction) (Naves 1974; Garnero et al. 1976; Guenther 1952), whereas R. centifolia is not economically viable for essential oils.
Absolute of R. centifolia has fresh rose petals, apricot and green notes, whereas R. damascena absolute exhibits jam, honeyed, greasy notes. R. damascena and R. centifolia absolutes are known to have highly similar in their volatile chemical composition detected by GC–MS analyses. These absolutes were found to be mainly constituted of phenylethanol (63.19–71.80 %), citronellol (9.30–13.65 %) and geraniol (5.05–6.19 %) in their volatile part (data for Turkish R. damascena) (Naves 1974; Monin 2010; Aycı et al. 2005).
In this work, a LC–MS metabolomics approach was developed to distinguish botanical origins, geographical origins but also to reveal adulterations. Mixtures of extracts coming from different botanical origins can have several benefits. First, addition of R. damascena to R. centifolia samples is of interest considering the relative prices of the raw material. Addition of R. centifolia to R. damascena samples can also be used, in order to change the organoleptic properties of the extract. Moreover, the simple addition of cheap synthetic compounds like phenylethanol exhibiting the specific rose odour, or just nonadecane for dilution are other kinds of very lucrative adulterations.
Several metabolomics approaches have been used for chemotaxonomic or geographical purposes with plant non-volatile extracts (Kim et al. 2011; Mattoli et al. 2006; Hanhineva et al. 2008; Glauser et al. 2013; De Vos et al. 2007; Xiong et al. 2012; Ku et al. 2010). Non-targeted metabolomics has only been applied to citrus essential oils (Mehl et al. 2014) and fresh materials like iris rhizomes (Masson et al. 2014). Additionally, there is no report on the comparison of old raw data with new ones. This study is of importance as metabolomics involves a time consuming step of instrumental analyses. Thus, the use of such an approach in quality control services will only be robust after analyzing the stability in time of the instrument and of the composition of the extracts. This issue was tested on our samples and results are discussed in the second part of the article in order to underline the potentials and limits of our methodology.
2 Experimental
2.1 Chemical
The solvent used for the extracts dilution was ethanol (EtOH, Biosolve ULC/MS grade). The following solvents were used for the chromatographic separation: acetonitrile (CH3CN, Biosolve ULC/MS grade), formic acid (FA, Biosolve ULC/MS grade) and water from water purification system Milli-Q Barnstead Easypure II.
Standards of phenylethanol (>98 %), nonadecane (>99 %), α-linolenic acid (>99 %) and amyl cinnamaldehyde diethyl acetal (>95 %) were purchased from Sigma Aldrich.
For the synthesis of dihydrocitronellyl phenyl acetate, dihydrocitronellol (>98 %), and phenyl acetic acid (>99 %) from Sigma Aldrich were used. For the synthesis of nerolidyl propionate, nerolidol (>98 %), and propionyl chloride (>98 %) from Sigma Aldrich were used. For the synthesis of farnesyl propionate, farnesol (>95 %), and propionic acid (>99.5 %) from Sigma Aldrich were used. Toluene (Chromasolv for HPLC, 99.9 %), dichloromethane (Chromasolv for HPLC, 99.9 %), triethylamine (>99 %) and sodium carbonate from Sigma Aldrich, and sulfuric acid (0.25 mol/L), were purchased from ChemLab.
12-oxophytodienoic acid was available in our own sample library.
2.2 Plant material
Absolutes of Rosa damascena petals were collected in Morocco, Turkey and Bulgaria. In France, absolute samples of Rosa centifolia petals were provided directly by the producers (see Table 1). R. damascena and R. centifolia absolutes were collected between 2010 and 2013 and stored in a dark room at 15 °C. Different mixtures were prepared at three concentrations from pure extracts (Table 1). Pure samples and mixtures were diluted by 500 in EtOH. Then, a mixture named pool was prepared with 15 μL of each diluted samples. These diluted samples were stored at 8 °C.
In the second part of the study focused on the reuse of old raw data, samples R01, R02, R19, R25, R37 and R40 (Table 1) were analyzed after 4 months of storage at 8 °C. A second set of samples was freshly prepared at time t + 4 months. Two pools were prepared with the addition of 100 μL of each sample (R01, R02, R19, R25, R37 and R40): a first pool with the samples stored in the fridge and a second one prepared with the fresh samples.
For nonadecane and phenylethanol quantification in GC-FID, absolutes (R38, R40, R41, R01 R13 and R19) were diluted in ethanol by a factor 10 and quantified by external calibration.
Standards solutions were prepared in ethanol: from 10 to 80 mg mL−1 for phenylethanol and from 0.5 to 5 mg mL−1 for nonadecane.
2.3 GC-FID conditions
GC-FID analyses were carried out with an Agilent 7890 instrument (Agilent Technologies) equipped with a flame-ionization detector (FID), an automatic liquid sampler, and a capillary column VF-1 ms (100 % dimethylpolysiloxane, 20 m × 0.15 mm i.d., film thickness 0.15 μm; Agilent Technologies). The oven temperature was programmed rising from 60 to 250 °C at 12 °C/min and then held isothermal at 250 °C for 9 min; injector temperature 250 °C; detector temperature 250 °C; carrier gas, H2 (1.0 mL/min); split ratio 1: 160; injection volume 1 μL. The O2 and H2 flows of the FID were 350 and 35 mL/min, respectively.
2.4 UHPLC-ToFMS conditions
UHPLC analyses were performed using an Acquity UPLC system coupled to XEVO-G2-QTOF (Quadrupole-Time of flight) (Waters Corporation, Manchester, United Kingdom) equipped with an Acquity UPLC® BEH Phenyl column (130 Å, 1.7 μm, 2.1 mm × 100 mm). The ionization mode was positive electrospray. The injection volume was set at 2 μL. The column temperature was regulated at 30 °C. The method was applied using a gradient elution with water: FA (Formic Acid) 99.9:0.1 (v/v) (solvent A) CH3CN:FA 99.9:0.1 (v/v) (solvent B) at a flow rate of 0.5 mL/min: isocratic conditions (22 % B) for 1 min, linear gradient from 22 to 51 % B in 4 min, linear gradient from 51 to 88 % B for 20 min, isocratic conditions (100 % B) for 4 min and then 3 min with isocratic conditions (22 % B) to return to initial conditions. The total run time including the return to initial conditions was 32 min. The MS parameters were set as: a capillary voltage of 1 kV, sampling cone 30 V, extraction cone 3 V, source temperature 120 °C, desolvation temperature 500 °C, cone gas flow 10 L/h and desolvation gas flow 1000 L/h. Desolvation gas and cone gas was high purity N2 (>99.9995 %).The samples described above were analyzed in triplicate. 270 injections were divided up in nine batches randomly. The structure of one batch is: one blank (EtOH), 10 pools, [four samples, one pool] × 7, two samples, one pool, one blank (EtOH). The run time was 27 h. The MS source of the instrument was cleaned between each batch and the calibration procedures were applied. The pool sample is used to control the reproducibility and the stability of the mass spectrometer to supply data for potential intra and inter batches corrections during statistical treatment.
For the part on the reuse of old raw data, building of the unique batch was the same as above.
2.5 Data extraction and statistical analyses
Markerlynx XS from Waters and SIMCAP + 13.0.2 from Umetrics (Umeå, Sweden) were the extraction and statistical software used for the metabolomics data treatment.
Markerlynx XS was used for ions extraction from raw acquisition files and the building of the first numerical matrix of metabolites. The extraction parameters were the following: XIC Window: 0.02 Da, Peak Width at 5 % Height: 20 s, Peak-to-Peak Baseline Noise: 5000, no smoothing, Marker Intensity threshold: 2000, Mass Window: 0.05 Da, Retention Time Window: 0.3 min, Noise elimination level: 10, Deisotope data.
For the second part of the study, one parameter was changed: Retention Time Window: 0.8 min.
Principal Component Analysis (PCA) was conducted using Markerlynx XS before any matrix pretreatment to get a first overview of the whole data. Pareto scaling was chosen. Intra- and inter-batch effects were checked graphically. Van der Kloet pretreatment (van der Kloet et al. 2009) was applied using the pool samples to rectify the signal intensities drifts along injections. In order to correct each ion intra-batch effect, linear regression was computed on pool samples and the model obtained was used to correct intensities. Then, normalization using mean batch was used to fix inter-batch effect. After this step, the mean values of intensities were calculated for each sample in this new matrix with the signal intensities corrected. Partial Least Square Analysis–Discriminant Analysis (PLS–DA) was then carried out. R2Y, Q2 and a permutation test were used to validate the models. In this study, a value of 0.5 for the cumulated Q2 values of the whole principal components was established as a threshold. Finally, a permutation test was conducted through SIMCAP+. In the case of this study, all the groups were permutated one hundred times. It was considered valid only if the R2 curve was crossing the ordinate axis under 0.6 and the Q2 curve was crossing the ordinate axis in negative value.
2.6 Synthesis of dihydrocitronellyl phenyl acetate, farnesyl propionate and nerolidyl propionate
The chemical syntheses of dihydrocitronellyl phenyl acetate and farnesyl propionate were performed using a Dean Stark system. A solution of 0.7 g of the alcohol, 1.4 g of the corresponding acid, and 1.5 mL of H2SO4 were added to 10 mL of toluene and heated under reflux during 4 h. The mixture was then cooled at room temperature and washed three times with 15 mL of brine. The organic phase was recovered, and dried on MgSO4 before concentration under vacuum.
For nerolidyl propionate, 0.9 mL of propionyl chloride, 2.5 g of nerolidol and 1.4 mL of triethylamine were mixed and the resulting solution was heated at 30 °C during 4 h. The mixture was then cooled at room temperature. After extraction with 15 mL of dichloromethane the organic phase was washed three times with 15 mL of a Na2CO3 saturated aqueous solution, and dried on MgSO4 before concentration under vacuum.
2.7 Workflow of the study
See Fig. 1.

Workflow of studies
3 Results and discussion
3.1 Checking the raw data and normalization
The overview of the chromatograms obtained by UHPLC-ToFMS analyses showed no or very weak differences between samples, and especially between R. centifolia and R. damascena (see Figure S1) thus supporting the requirement of a metabolomics approach including statistical analyses to discriminate botanical origins.
The first key step was to observe the raw data (the whole samples from Table 1 and pools) in a PCA (see Fig. S2). The dark points called “p” for pools were spread on the whole ellipse. Van der Kloet’s (2009) normalization was then used to correct this distribution thanks to intensities adjustments inter- and intra- batches (see Fig. S3). The pools were then centered in the PCA with intensity corrections.
3.2 Studies on botanical origins, geographical origins and adulterations
With homogeneous raw data, mean values of intensities were calculated with the triplicates of each sample (randomly distributed among the batches). Models were then established to address several issues. Five of them were validated on the first PLS-DA component and permutation tests validation (100 permutations): R. centifolia versus R. damascena (R2Y: 0.92; Q2: 0.78), (R. damascena + 10/20/30 % R. centifolia) versus R. damascena (R2Y: 0.81; Q2: 0.52), (R. damascena + 10/20/30 % R. centifolia) versus R. centifolia damascena (R2Y: 0.86; Q2: 0.70), Turkey versus Morocco damascena (R2Y: 0.90; Q2: 0.85) and (R. damascena + 5/10/15 % nonadecane) versus R. damascena (R2Y: 0.72; Q2: 0.58).
3.2.1 Botanical origin: Rosa centifolia versus Rosa damascena
3.2.1.1 Rosa centifolia markers of intensity
The discrimination between R. centifolia (R36, R37, R38, R39) and R. damascena (R01, R02, R19, R20) absolutes (see Fig. 2) was very clear even if the number of samples was critical due to the high cost of these extracts. R. centifolia is an extremely rare product, only produced in France in very small amount. It is generally dedicated to the most renowned perfume companies and it is therefore difficult to obtain leading to a high price of 16,500 €/kg in 2013. On the contrary, R. damascena absolute is by far more common and easier to obtain with an average price of 2500€/kg. Even if we were able to obtain four samples of R. centifolia absolute and 32 of R. damascena absolute (from Morocco, Bulgaria and Turkey), we decided to work with only four samples of R. damascena to balance the model. Two samples of R. damascena from Morocco and two samples of R. damascena from Turkey were chosen due to the geographical diversity of this group. In addition to the permutation test used for validation, prediction models proved the correct classification of other samples of R. damascena.

a PLS-DA for the discrimination between R. centifolia and R. damascena; b permutation test (100)
Markers, with intensity ratios of R. centifolia/R. damascena higher than 2, are listed in Table 2. Three of them were also ionized in negative ESI which confirmed their elemental composition. A new PLS-DA was built only with these markers (R2Y = 0.92 and Q2 = 0.80) (see Fig. S4). Comparison with the model containing all metabolites, led to an improvement of the prediction (with 0.80 rather than 0.78) even if the explanation of the variability was the same.
M2 and M3 were linked by the loss of one molecule of H2O. Three couples of markers were in relation with high correlation coefficients (>0.99) (see Table S1).
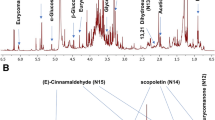
Hypotheses were proposed for these markers according to literature data. Several possibilities were found in the database of “The Good Scents Company” (available on: www.thegoodscentscompany.com). Dihydrocitronellyl phenyl acetate with its “rose” odour was an option for the molecular formula C18H28O2. Nerolidyl propionate with a floral odour was an option for C18H30O2. Farnesol being a component of rose absolute (Aycı et al. 2005), we also considered farnesyl propionate for C18H30O2. These compounds were synthesized and their structure was confirmed by GC/MS and NMR. Unfortunately none of them matched with the markers after analyses according to the method developed for metabolomics on UHPLC-ToFMS. Other hypotheses were tried: amyl cinnamaldehyde diethyl acetal for C18H28O2 because of its floral, aldehyde, green note, and α-linolenic acid for C18H30O2, with standards obtained from Sigma Aldrich. The markers at 5.57 min with m/z 293.2118 and m/z 275.2012 were identified (level 2 according to MSI guideline) comparing with a standard available in our laboratory: 12-oxo-phytodienoic acid (Figure S5). m/z 293.2118 was the [M + H]+ ion of the compound and m/z 275.2012, its main fragment after H2O loss. It is worth noticing at this stage that this marker would not have been detected using GC/MS instrument, and as such the UHPLC/MS approach gives complementary data.
3.2.1.2 Mixing of botanical origins
Two sets of data were validated through PLS-DA models: the addition of R. centifolia (10, 20, 30 %) to R. damascena samples, compared to pure R. damascena samples and then compared to pure R. centifolia samples (Figs. S6, S7). With this method, we were able to detect the adulteration, comparing to pure and known samples of R. centifolia or R. damascena.
3.2.2 Geographical origin: Turkey and Morocco
Samples from Morocco and Turkey were well separated (see Fig. S8). No important markers with minimum intensities ratio of 2 were listed as for the comparison R. centifolia/R. damascena.
Other statistical analyses between groups were considered but not validated. The mixture of Turkey with Morocco (10, 20 and 30 %) versus Morocco versus Turkey (three groups) was not validated and then differences according to origins were not large enough. This model was simplified with the comparison of the mixture of Turkey and Morocco versus Turkey (two groups) but it was not validated either. This result showed that it may be difficult to confirm an adulteration if the initial differences between pure samples are not pronounced. In this case, while Morocco and Turkey samples were well discriminated in a model where only pure samples were considered, the discrimination was not so obvious when pure samples were compared to adulterated ones. These observations proved that differences between origins are not well pronounced and confirmed that no key marker allowed the distinction between Morocco and Turkey samples. These observations support the fact that similarities or differences are not fully related to the presence of specific markers but much more to the whole set of detected ions.
3.2.3 Adulteration with nonadecane and phenylethanol
Adulterations with the rather volatile compounds nonadecane and phenylethanol were studied by UHPLC-ToFMS in order to assess the potential of this technique for volatile compounds.
Nonadecane is naturally present in rose absolute (4.35 % of the volatile fraction) (Ulusoy et al. 2009). This adulteration is particularly attractive with a cost of 1350 €/kg for nonadecane (Sigma Aldrich). Nonadecane was quantified in three samples of R. centifolia and three samples of R. damascena by GC-FID. 3.1 ± 1.0 % of nonadecane was identified in R. damascena and 1.1 ± 1.1 % in R. centifolia (in the whole extract).
The addition of nonadecane (5, 10 and 15 %) to R. damascena samples was highlighted by the PLS-DA model (see Fig. S9). The compound was not ionized in ESI, but the dilution effect of this compound in the mix allows the detection of the adulteration. Even if the use of GC/MS would be more suitable to detect and identify the component, an LC/MS approach allowed highlighting of this adulteration through a dilution factor.
Phenylethanol is an odorant compound with a “rose” note. It is also naturally present in the absolute (63.2–71.8 % of the volatile fraction) (Aycı et al. 2005) and can be purchased at a very low price of 5.50 €/kg (synthetic compound, Bernardi). Its concentration according to GC-FID analyses reached 36.7 ± 2.4 % in R. damascena absolute and 24.4 ± 10.9 % in R. centifolia absolute (in the whole extract according to GC-FID analyses). The PLS-DA model of R. damascena adulterated with phenylethanol versus pure R. damascena was not validated. Indeed, the addition of the compound at 5, 10 and 15 % was not detected. In contrast with the case of nonadecane, the dilution effect was not sufficient to discriminate. We assume that the quantities in pure absolutes vary naturally in similar proportions to that of the additions (at least for the additions of 5 and 10 %). No discrimination was then supported with these additions. In conclusion, fraudulent operations to modify the organoleptic properties by adding small quantity of phenylethanol cannot be detected by our UHPLC-MS metabolomics and in this case a GC/MS approach should be a complementary approach.
3.2.4 In summary
Metabolomics is a performing tool to highlight adulterations. In the current study on roses using UHPLC/MS, differences induced by the addition of nonadecane were detected indirectly by a dilution effect however without a clear identification of nonadecane as an adulteration source. Metabolomic analyses applied to several sample conditions have proved to be efficient when particular types of adulteration are suspicious. They can provide useful information (with the example of model A and B above about botanical origin) but in some cases they are not sufficient to conclude definitively and should be completed by GC/MS analyses for example. Metabolomics analyses have highlighted important markers of botanical origins even in the case of very similar samples like R. centifolia and R. damascena.
According to these results, an adulteration can be evidenced confirmed and sometimes interpreted, but this method alone will not give 100 % confidence on the product authenticity.
3.3 Discussion on the possibilities to compare previous raw data (time = t) to new samples (time = t + dt)
The use of old raw data is a very important concern for the success of implementation of untargeted metabolomics in control quality laboratories. Metabolomic analysis being time consuming and expensive in terms of instrumental use, the possibility to reuse data has to be assessed in the context of flavor and fragrances.
3.3.1 Study of the raw data
A new set of data was acquired independently in one single batch at “t + 4 months”. This study aimed to compare data acquired at “t” with data acquired at “t + 4 months” on the same samples stored in a fridge and data acquired at “t + 4 months” with freshly prepared samples (See Sect. 2.2. Plant material for sample references).
To work on data at “t”, and “t + 4 months” (fresh and fridge), a new extraction of ions and alignment was performed to extract the data as a single datamatrix. A quick observation of the chromatograms (Fig. S10) confirmed the similarities between “fresh t + 4 months”, “fridge t + 4 months” and t data in term of intensities and peak widths.
Pools positions for “fresh t + 4 months” data and “fridge t + 4 months” data were checked on separate PCA: they were in the center of their respective data which confirmed the successful run of this batch.
“t” data were taken as the reference on the PCA gathering “fresh t + 4 months”, “fridge t + 4 months” and t data (see Fig. 3). “Fresh t + 4 months” and “fridge t + 4 months” data were shifted by horizontal translation from “t” data. “Fresh t + 4 months” and “fridge t + 4 months” were analyzed in a same batch. The translation effect may be assigned to the storage in the fridge of the samples during 4 months. The shift between “fresh t + 4 months” and “t” is likely due to the technical variations inherent to the mass spectrometer, very sensitive to the lab environment, contaminations etc.

a Scores plot PCA of “fresh t + 4 months”, “fridge t + 4 months” and t data; b corresponding loadings plot
3.3.2 Study of models with new data
The mean values in the intensities of the three types of data (t, fresh and fridge t + 4 months) were then calculated to plot the PLS-DA corresponding to the distinction between R. damascena and R. centifolia (see Fig. S11). The model was validated with the first PLS-DA component (R2Y = 0.90 and Q2 = 0.80). The VIP (Variable Importance for the Projection) had nearly the same values for the model on initial “t” data, on t + 4 fresh data (R2Y = 0.95 and Q2 = 0.89; not shown here) and this new model mixing the data of t and the data of t + 4 months (fresh and fridge) (see Table S2). This value represents the contribution of the marker in the model.
However, the same work aimed at distinguishing Morocco and Turkey origins proved unsuccessful and the new model was therefore not validated. The possibility to introduce new analyses in old batches becomes thus problematic in this case.
Different solutions can be proposed to interpret this result. Van der Kloet’s correction used in the previous study could not be used in this case, (i) the pool sample could not be rigorously identical (new samples were not in the previous study) and (ii) the pool has to be prepared at the same time than the samples. These criteria being not respected, an additional bias would be introduced using this type of correction. Another normalization was tested: the mean values of each ion of the whole raw data were calculated for each category of samples (t, “fresh t + 4 months” and “fridge t + 4 months”); each ion intensity was then divided by the mean value corresponding to its category, and then multiplied by the mean value of the three categories.
As shown in the PCA, normalization has corrected bias between “t”, “t + 4 fresh” and “t + 4”fridge data (Fig. 4).

a PCA of t, t + 4 fresh, t + 4 fridge data after normalization; b corresponding loadings plot
The impact of this correction was evaluated again on the model R. centifolia/R. damascena. R2Y = 0.95 and Q2 = 0.93 (not shown) were improved, and the model was still validated (on the first PLS-DA component). VIP values (Table S3) were very similar to the previous models (Table S2). However no improvement was evidenced for the distinction between Turkey and Morocco.
To distinguish between these two models (R. centifolia/R. damascena: (C), Turkey/Morocco: (D)), “important” markers were highlighted for (C) (with ratio intensity levels comprised between 3 and 9 times in favor of R. centifolia), whereas no important markers were found for (D). The Turkey and Morocco profiles show some constant differences but they are not large enough to be detected as “important” markers. These samples exhibit small differences, which could be of the same range of intensity than the variations due to the analytical instrument. So, when old and new data are mixed, new sources of variations are involved that may hamper the detection of differences between samples.
As conclusion, comparison between old and new data requires the alignment of data using modified parameters for the extraction process of ions. Normalization can be applied as described above to decrease the differences in intensity and to improve results, but this process can be efficient just when important differences between groups exist. It is not possible to use Van der Kloet treatment, which points out the need to have the cleanest analyses possible in order to minimize bias. This correction is however very useful to study “simultaneously” a large set of data, and to point out slight differences between groups of samples.
Old and new data could then be treated together for issues previously demonstrated like R. damascena and R. centifolia discrimination. This conclusion led us to consider the use of such method for rapid quality control. This is unfortunately not valid for all cases like for the discrimination of the geographical origins Turkey and Morocco.
4 Conclusion
Untargeted metabolomics approach was a success for the discrimination of botanical species, geographical origins and for the detection of some cases of adulteration in complex natural extracts: rose absolutes. 12-oxo-phytodienoic acid was characterized as a marker, more abundant in R. centifolia absolute than in R. damascena absolute. These results based on LC/MS rather than GC/MS proved the performance of LC/MS metabolomics approach for the perfume industry. The other interesting question about the reuse of raw data in new models was also discussed in this work. The study on botanical species which evidenced important markers was a good candidate to assess the possibility of reusing old data and in this case our results confirmed the possibility to use old data. This result is of high importance for quality control in this industry. However, caution must be taken in the cases when markers are only present with low ratios of intensity between groups. We recommend that the use of LC/MS metabolomics should be planned for every day innovative authentication issues in the area of flavour and fragrances with some caution with very similar raw material. This method could also be combined to similar analyses with GC/MS data to give more exhaustive and precise results.
References
Aycı, F., Aydınlı, M., Bozdemir, Ö. A., & Tutaş, M. (2005). Gas chromatographic investigation of rose concrete, absolute and solid residue. Flavour Fragrance Journal, 20(5), 481–486.
Bauer, K., Garbe, D., & Surburg, H. (2001). Common fragrance and flavor materials (4th ed.). Weinheim: Wiley-VCH.
Buiarelli, F., Cartoni, G. P., Coccioli, F., & Ravazzi, E. (1991). Analysis of orange and mandarin essential oils by HPLC. Chromatographia, 31(9–10), 489–492.
De Vos, R. C., Moco, S., Lommen, A., Keurentjes, J. J., Bino, R. J., & Hall, R. D. (2007). Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nature Protocols, 2(4), 778–791.
Do, T.K.T., Hadji-Minaglou, F., Antoniotti, S., & Fernandez, X. (2015). Authenticity of essential oils. Trends in Analytical Chemistry 66, 146–157.
Fernandez, X., Lizzani-Cuvelier, L., Loiseau, A.-M., Périchet, C., & Delbecque, C. (2003). Volatile constituents of benzoin gums: Siam and Sumatra. Part 1. Flavour Fragrance Journal, 18(4), 328–333.
Garnero, J., Guichard, G., & Buil, P. (1976). L’huile essentielle et la concrète de rose de Turquie. Rivista italiana essenze profumi piante officinali aromi saponi cosmetici aerosol n° ind (1976.03).
Glauser, G., Veyrat, N., Rochat, B., Wolfender, J. L., & Turlings, T. C. (2013). Ultra-high pressure liquid chromatography-mass spectrometry for plant metabolomics: A systematic comparison of high-resolution quadrupole-time-of-flight and single stage Orbitrap mass spectrometers. Journal of Chromatography A, 1292, 151–159.
Guenther, E. (1952). The essential oils. New York: D. Van Nostrand Company.
Hanhineva, K., Rogachev, I., Kokko, H., Mintz-Oron, S., Venger, I., Kärenlampi, S., & Aharoni, A. (2008). Non-targeted analysis of spatial metabolite composition in strawberry (Fragaria x ananassa) flowers. Phytochemistry, 69(13), 2463–2481.
Jirovetz, L., Buchbauer, G., Stoyanova, A., Balinova, A., Guangjiun, Z., & Xihan, M. (2005). Solid phase microextraction/gas chromatographic and olfactory analysis of the scent and fixative properties of the essential oil of Rosa damascena L. from China. Flavour Fragrance Journal, 20(1), 7–12.
Kim, N., Kim, K., Choi, B. Y., Lee, D., Shin, Y. S., Bang, K. H., et al. (2011). Metabolomic approach for age discrimination of Panax ginseng using UPLC-Q-Tof MS. Journal of Agriculture and Food Chemistry, 59(19), 10435–10441.
Kreis, P., & Mosandl, A. (1992). Chiral compounds of essential oils. Part XII. Authenticity control of rose oils, using enantioselective multidimensional gas chromatography. Flavour Fragrance Journal, 7(4), 199–203.
Ku, K. M., Choi, J. N., Kim, J., Kim, J. K., Yoo, L. G., Lee, S. J., et al. (2010). Metabolomics analysis reveals the compositional differences of shade grown tea (Camellia sinensis L.). Journal of Agriculture and Food Chemistry, 58(1), 418–426.
Li, Y-q, Kong, D-x, & Wu, H. (2013). Analysis and evaluation of essential oil components of cinnamon barks using GC–MS and FTIR spectroscopy. Industrial Crops and Products, 41, 269–278.
Masson, J., Liberto, E., Brevard, H., Bicchi, C., & Rubiolo, P. (2014). A metabolomic approach to quality determination and authentication of raw plant material in the fragrance field. Iris rhizomes: A case study. Journal of Chromatography A, 1368, 143–154.
Mattoli, L., Cangi, F., Maidecchi, A., Ghiara, C., Ragazzi, E., Tubaro, M., et al. (2006). Metabolomic fingerprinting of plant extracts. Journal of Mass Spectrometry, 41(12), 1534–1545.
Mazollier, A. (2012). Développement de méthodologies analytiques et statistiques innovantes pour le contrôle de l’authenticité de matières premières pour les industries de la cosmétique et de la parfumerie, PhD in chemistry, Université Claude Bernard Lyon 1, Lyon.
McHale, D., & Sheridan, J. B. (1988). Detection of adulteration of cold-pressed lemon oil. Flavour Fragrance Journal, 3(3), 127–133.
Mehl, F., Marti, G., Boccard, J., Debrus, B., Merle, P., Delort, E., et al. (2014). Differentiation of lemon essential oil based on volatile and non-volatile fractions with various analytical techniques: A metabolomic approach. Food Chemistry, 143, 325–335.
Monin, C. (2010). L’Encyclopédie des Matières Premières Naturelles pour la Parfumerie. vol 1.0.0.1. Hubsoft.
Naves, Y.-R. (1974). Technologie et Chimie des Parfums Naturels. Paris: Masson et CIE.
Ravid, U., Putievsky, E., Katzir, I., Ikan, R., & Weinstein, V. (1992). Determination of the enantiomeric composition of citronellol in essential oils by chiral GC analysis on a modified γ-cyclodextrin phase. Flavour Fragrance Journal, 7(4), 235–238.
Salgueiro, L., Martins, A. P., & Correia, H. (2010). Raw materials: the importance of quality and safety. A review. Flavour Fragrance Journal, 25(5), 253–271.
Saraswathy, A., Shakila, R., & Sunilkumar, K. N. (2010). HPTLC fingerprint profile of some cinnamomum species. Pharmacognosy Journal, 2(8), 211–215.
Schipilliti, L., Dugo, G., Santi, L., Dugo, P., & Mondello, L. (2011). Authentication of bergamot essential oil by gas chromatography-combustion-isotope ratio mass spectrometer (GC-C-IRMS). Journal of Essential Oil Research, 23(2), 60–71.
Ulusoy, S., Bosgelmez-Tinaz, G., & Secilmis-Canbay, H. (2009). Tocopherol, carotene, phenolic contents and antibacterial properties of rose essential oil, hydrosol and absolute. Current Microbiology, 59(5), 554–558.
van der Kloet, F. M., Bobeldijk, I., Verheij, E. R., & Jellema, R. H. (2009). Analytical error reduction using single point calibration for accurate and precise metabolomic phenotyping. Journal of Proteome Research, 8(11), 5132–5141.
Xiong, A., Yang, L., Ji, L., Wang, Z., Yang, X., Chen, Y., et al. (2012). UPLC-MS based metabolomics study on Senecio scandens and S. vulgaris: an approach for the differentiation of two Senecio herbs with similar morphology but different toxicity. Metabolomics, 8(4), 614–623.
Zrira, S. (2006). La rose du Dadès. www.agrimaroc.net/146.pdf.
Acknowledgments
The company Payan Bertrand is acknowledged for its collaboration through the supply of samples and the knowledge on natural extracts. We are also grateful to Albert Vieille and IFF for providing additional samples. Waters Company and the ‘Association Nationale de la Recherche et de la Technologie’ (ANRT) have supported the fellowship of Laure Saint-Lary.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
L. Saint-Lary, C. Roy, J-P. Paris, J-F. Martin, O. Thomas and X. Fernandez declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Saint-Lary, L., Roy, C., Paris, JP. et al. Metabolomics as a tool for the authentication of rose extracts used in flavour and fragrance area. Metabolomics 12, 49 (2016). https://doi.org/10.1007/s11306-016-0963-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-016-0963-3