
Abstract
Pyrrolizidine alkaloids show significant hepatotoxicity as they can bind to DNA or proteins after being activated in liver. Senecio vulgaris L., like many Compositae herbs containing pyrrolizidine alkaloids, was reported to have great hepatotoxicity. However, Senicio scandens Buch.-Ham., from the same genus, which was also used as a herb and documented in China Pharmacopoeia published in 2010, hardly showed any side effects or relevant toxicity. In the present study, we conducted the metabolomics study using ultra-performance liquid chromatography-mass spectrometry (UPLC-MS) to obtain the different metabolic profiles of the two Senecio herbs. In addition, principle component analysis (PCA) and orthogonal projections to latent structures-discriminant analysis (OPLS-DA) were introduced for the multivariate analysis, and MS/MS was applied to the identification of target alkaloid markers which contributed most to the established models. As a result, ten pyrrolizidine alkaloids, including adonifoline, senecionine, senecionine N-oxide, retrorsine, retrorsine N-oxide and seneciphylline, were selected and identified. Among them, adonifoline was found to be a specific marker for S. scandens while senecionine and its N-oxidative were characteristic markers for S. vulgaris. Furthermore, the hepatotoxicity studies in vivo and in vitro showed that senecionine had more potent toxicity (LD50, 57.3 mg/kg; IC50, 5.41 μM) than that of adonifonine (LD50, 163.3 mg/kg; IC50, 49.91 μM). Taken together, the present study provides not only better understanding of the different toxicity between the two Senecio herbs containing pyrrolizidine alkaloids but also a reference method, which can be applied to other genetically closed species with similar morphology but different toxicity.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Pyrrolizidine alkaloids (PAs) have shown great hepatotoxicity (Prakash et al. 1999), pheumotoxic (Thomas et al. 1997; Lame et al. 2000), genotoxic (Fu et al. 2001; Chen et al. 2010) and neurotoxic (Huxtable et al. 1996) to human beings via consumption of PAs-containing plants and their products, such as medical preparations and herbal tea (WHO 1988). PAs are found to present in more than 6000 floriferous plants, mainly belong to Familiy Compositae, Leguminosae, Orchidaceae and Boraginaceae (Smith and Culvenor 1981; Roeder 2000). Herbs from Senecio genus are one of the main sources for PAs. Senecio vulgaris L. (SV) is originated in Europe and has played a certain role in the case of functional amenorrhoea (Roeder 1995). However, several death reports were referred to the consumption of herbal tea containing SV (Ortiz et al. 1995; Vilar et al. 2000). As known, SV contains several PAs such as senecionine, seneciphylline, integerrimine and retrorsine in high concentration (Roeder 1995). Thus, preparations of this Senecio herb should neither be recommended nor administered. On the other hand, S. scandens Buch.-Ham. (SS), which is widely spread in China up to an altitude of 3200 m, has been used officially in the case of oral and pharyngeal infections (Chinese Pharmacopeia Commission 2010) due to its rich content of flavonoids (Yang et al. 2011). Although SS contains several PAs including adonifoline (Xu et al. 2006; Zhang et al. 2008), there are no any formal reports associated with its toxicity. So a key question is why SV and SS, the two genetically closed species with similar morphology, show such different toxicity.
At present time, metabolomics (Oliver 1997; Lindon et al. 2007) has been widely used in the profiling of plant metabolites (De Vos et al. 2007; Harris et al. 2007; Glauser et al. 2009), especially for the genetically closed species (Xie et al. 2008; Dai et al. 2010). A number of techniques, including nuclear magnetic resonance (NMR), liquid chromatography (LC) or gas chromatography (GC) coupled with mass spectrometry (MS), have been applied to the study (Fiehn et al. 2000; Lise et al. 2006; Wagner et al. 2006; Krishnan et al. 2005). LC-MS has higher sensitivity and resolution than NMR; while it is also much more versatile and convenient than GC–MS. Additionally, the most new technology of ultra performance LC (UPLC) has provided an even better separation, resolution and sensitivity as well as high analysis speed (Novakova et al. 2006; Yang et al. 2008) than traditional LC. Thus, UPLC has become a powerful tool for high-throughput screening and the analysis of complex biological samples, such as herbal medicines (Liu et al. 2007). In the present study, UPLC–MS was first utilized to obtain the metabolic profiles of SS and SV. Multivariate analysis techniques, including principal component analysis (PCA) and orthogonal projections to latent structures-discriminant analysis (OPLS-DA) were applied to the data mining. LC-MS and MS/MS were used for the identification of the different phytochemical compositions, which might correspond to the toxicity of the two Senecio herbs. Furthermore, to confirm the biological significance, we conducted in vivo and in vitro hepatotoxicity assays for the selected PAs markers adonifoline (ADO) and senecionine (SEN).
2 Materials and methods
2.1 Chemical and materials
Acetonitrile was of HPLC grade and purchased from Fisher Scientific Co. (Santa Clara, USA). Ammonium formate was of HPLC grade and purchased from Sigma-Aldrich Co. (Fairfield, USA). 98% Formic acid and 25% ammonia solution were of analytical grade and purchased from Shanghai Reagent Co. (Shanghai, PR China). Water was purified with a Milli-Q system (Millipore, Bedford, MA, USA). Adonifoline, senecionine, and seneciphylline were obtained from Shanghai R&D Center for Standardization of Traditional Chinese Medicines (Shanghai, P. R. China). Retrorsine was merchandised from Sigma-Aldrich Co. (Fairfield, USA). N-oxidatives of senecionine and retrorsine were prepared by oxidation of senecionine and retrorsine, respectively, according to the reference (Xiong et al. 2009). The structures of pyrrolizidine alkaloid standards are shown in Fig. 1.

Structures of PAs standards
The samples information was provided in Table 1. All the samples were collected in their flowering season and authenticated by authors. Then they were naturally dried in air. The voucher specimens were deposited in the Herbarium of Institute of Chinese Materia Medica, Shanghai University of Traditional Chinese Medicine.
2.2 Sample preparation
The herbal materials were dried at 60°C over night before use and powdered (through 50 meshes). The powder (0.5 g) was accurately weighed and extracted with 50 mL of 0.5% (v/v) formic acid solution for 40 min in an ultrasonic water bath at 30°C to get the extracts. The supernatants were filtered through a 0.22 μm syringe filter and injected to the UPLC-MS system.
2.3 UPLC-MS analysis
The analysis was performed on a Waters ACQUITY UPLC™ system (Waters Corporation, Milford, USA) with a binary solvent delivery manager, an auto sampling manager, and a TQD™ mass spectrometer (Waters Corporation, Milford, USA) equipped with an electrospray interface (ESI).
Gradient chromatographic separation was carried out on a Waters ACQUITY UPLC BEH C18 column (100 mm × 2.1 mm i.d., 1.7 μm, Waters Corporation, Milford, USA) with a supported Security Guard C18 guard column (Waters Corporation, Milford, USA). Mobile phase A was acetonitrile, while mobile phase B was 10 mM ammonium formic modified by the addition of 0.1% (v/v) 25% ammonia solution. The gradient elution started with 5% mobile phase A, increasing to 20% A at 5 min, and then to 40% A at 7 min. The mobile phase was held isocratic at 90% A for 1.5 min and another 1.5 min at 5% A before the next injection. The flow rate was 0.5 mL/min. The column was maintained at 45°C. And the injection volume was 5 μL. Each sample was injected 4 times to obtain 48 observations.
The mass spectrometer was operated in positive ion mode with the scan range from m/z 150 to m/z 650. Parameters were optimized and set as follows: capillary voltage: 3.5 kV; cone voltage: 45 V; source temperature: 150°C; desolvation temperature: 450°C; desolvation gas (nitrogen): 900 L/h; cone gas (nitrogen): 50 L/h; low mass (LM) resolution: 15; high mass (HM) resolution: 15. When executing MS/MS fragmentation, the collision energy was set at 30 V, the collision gas (helium) flow was set at 0.1 L/h while LM and HM resolution for the MS/MS function were set at 13.
2.4 Chemometrics data analysis
The UPLC-MS data of SS and SV samples were analyzed to identify potential discriminant variables. Data processing and statistical analysis was accomplished by using the MarkerLynx XS software (Waters, Manchester, UK). The parameters were as following: retention time (Rt) range 0.2–18.0 min; mass range m/z 150–650; extracted ion chromatograms (XIC) window (the mass tolerance) 0.05 Da; width of an average peak at 5% height and peak-to-peak baseline noise were automatically calculated; minimum intensity was set to 10% of base noise; mass window and retention time window for marker collection were set at 0.05 Da and 0.2 min, respectively; elimination level was set at 10.00; and isotopic peaks were excluded for analysis. After data processing, a list of the peak intensities was generated using retention time (Rt) and m/z data pairs (Rt m/z ) as an identifier for each peak. The resulting two-dimensional matrix of each peak and their intensity for each sample were further analyzed.
Multivariate analysis techniques, including unsupervised principle component analysis (PCA) and supervised orthogonal projections to latent structures-discriminant analysis (OPLS-DA), were used to discriminate between the samples and figure out the most important secondary metabolites for their difference. The data sets were mean-centered, pareto-scaled (van den Berg et al. 2006) in a column-wise manner before PCA and OPLS-DA modeling. Potential biomarkers were selected according to variable importance in the project (VIP) value, and the S-plot.
2.5 In vivo acute toxicity evaluation of adonifoline and senecionine on mice
Adonifoline and senecionine were separately dissolved in sodium chloride (containing 0.2 mol/L hydrochloric acid) and the pH value was adjusted to 5.0 with 2 mol/L sodium hydroxide solution, to prepare two stock solutions containing 300 mg/mL of adonifoline and senecionine, respectively. For the toxicity assay, the stock solutions were diluted with sodium chloride to a series of concentrations before administered to ICR mice (male, 18–22 g).
Mice were obtained from the Laboratory Animal Center of Shanghai University of Traditional Chinese Medicine (SHUTCM, Shanghai, P. R. China). All animals were fed with a commercial pellet diet and water ad libitum. Animal welfare and the animal experimental protocols were strictly consistent with the Guide for the Care and Use of Laboratory Animals (U.S. National Research Council, 1996) and related ethics regulations of SHUTCM. Before treatment, animals were fast overnight and divided randomly into ten groups with ten mice per group. Groups 1–5 were i.g. with adonifoline, and groups 6–10 with senecionine. Their median lethal doses (LD50), which were commonly used for evaluation of toxicity, were calculated by the Bliss method.
2.6 Cytotoxicity tests of adonifoline and senecionine on primary rat hepatocytes
Primary rats hepatocytes were seeded in 96-well microplates (2 × 104 cells/well), and then incubated with 1, 3, 10, 30, 100 μM of adonifoline and senecionine, respectively, for 48 h in the carbon dioxide incubator. After treatments, cells were incubated with 500 μg/mL of 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, a yellow tetrazole, which can be reduced to purple formazan in living cells.) for 4 h. At last the formazan was dissolved in 10% sodium dodecyl sulphate-5% iso-butanol-0.01 M hydrochloric acid. The optical density was measured at 570 nm with 630 nm as a reference and cell viability was normalized as a percentage of control. The half maximal inhibitory concentrations (IC50) of the two compounds were calculated.
3 Results and discussion
3.1 Liquid chromatography
Separation of the herb extracts was carried out on an UPLC system under the optimal conditions, such as the parameters of mobile phase system. Significant differences of the chromatogram separation were observed when using different aqueous mobile phase, including 0.1% formic acid (pH 2.6), 10 mM ammonium formic (pH 6.5), 0.1% concentrated ammonia solution (pH 10.0), and 10 mM ammonium formic modified by the addition of 0.1% concentrated ammonia solution (pH 8.4) (Fig. 2). Generally, the separation was better using basic mobile phase systems (Fig. 2c and d) than the other mobile phase systems (Fig. 2a and b). Finally, the modified basic mobile system (10 mM ammonium formic modified by the addition of 0.1% concentrated ammonia solution) was chosen because it yielded good separation and intensity of each composition within a shorter period of time (Fig. 2d). Figure 3 showed typical based peak intensity (BPI) chromatograms obtained from the analysis of the two Senecio herbs in positive ion mode.

Total ion chromatography of the herb extracts, separated on a Waters BEH C18 column (50 mm * 2.1 mm, 1.7 μm) with an isogradient eluent using acetonitrile and aqueous phase: a 0.1% HCOOH; b 10 mM HCOONH4; c 0.1% NH3; d 10 mM HCOONH4 + 0.1% NH3; and started at 5% acetonitrile and linear increased to 50% acetonitrile in 5 min

Comparison of UPLC chromatograms for the alkaline extracts of: a Senecio scandens (Ningguo, Anhui Province), and b S. vulgaris (Tonghua, Jilin Province) (1 senecionine N-oxide, 2 senecionine, 3 seneciphylline, 4 retrorsine N-oxide, 5 integerrimine N-oxide, 6 seneciphylline N-oxide, 7 retrorsine, 8 othonnine, 9 adonifoline, 10 platyphylline)
3.2 Multivariate analysis
Principal component analysis (PCA) was used as an unsupervised method to study the metabolome differences between SS and SV herbs for it has been universally used for achieving the natural interrelationship within the data, i.e., grouping, clustering, outliers, etc., without prior knowledge of the data set. The score plot obtained by all observations from the two species is displayed in Fig. 4a. A clear separation can be seen between SS and SV herbs. Seven principal components were calculated by cross validation, and 48.85% of the variables can be explained by the first two components (71.63% of the total variances can be explained by the seven components), indicating a significant difference between the two herbs in the profiles and levels of alkaline extracts.

a PCA scores plot of all observations (triangleSenecio scandens (SS) samples, squareS. vulgaris (SV) samples), b OPLS-DA scores plot of 39 observations in the training set (triangleSS samples in training set, squareSV samples in training set), c Predicted score plot of OPLS-DA of all observations both in training set and predicting set (triangleSS samples in training set, sqaureSV samples in training set, asterisksamples in predicting set), and d S-plot (Metabolites in the (square) were the top 10 markers selected)
However, instrumental drift, artifacts, and other experimental variations might occasionally divert the focus of a PCA model to the systematic variation unrelated to the scientific questions of interest. Thus, supervised OPLS-DA (Trygg and Wold 2002; Wiklund et al. 2008) was applied to use any priori information beneficial to the analysis and further study the differences between the two species. Compared with the partial least squares-discriminant analysis (PLS-DA) (Boulesteix and Strimmer 2006), OPLS-DA (Trygg and Wold 2002) can separate the result-non-correlated variation, so called orthogonal to the result, from the whole variation prior to data modeling; thus, it can improve the interpretational ability of resulting models and reduce model complexity. The quality of the models was described by the cross-validation parameter Q2 (the predictability of the model) and R2Y (the total explained variation for the X matrix). In present study, an OPLS-DA model (Fig. 4b) was firstly constructed using 39 observations (20 observations obtained from SS, and 19 from SV) as the training set. The R2Y and Q2 were 0.9986 and 0.9959 respectively, indicating excellent prediction ability of the model. After that, 9 observations (4 from SS, and 5 from SV) were used as testing set and applied to the established OPLS-DA model. As shown in Fig. 4c, all of the nine observations were well predicted, indicating the established OPLS-DA model had a good capability to differentiate SS from SV.
3.3 Selection, discovery, and identification of marker compounds
As is known, the S plot has become an easy way to visualize an OPLS/O2PLS discriminant analysis model of two classes thus it has mainly been used to filter out putative biomarkers from “omics” data, e.g. LC/MS metabolomics data (Wiklund et al. 2008). Both intensity and reliability is visualized in the S-plot. On one hand, the peak intensity is important as peaks with low intensity are close to the noise level and thus have a higher risk for spurious correlation. On the other hand, reliability is also very important as peaks with higher reliability mean that they are of higher effect and lower uncertainty. As shown in Fig. 4d, it combines the covariance (the contribution of variations to the model, as X axis) and the correlation (the reliability of variations to the model, as Y axis). Markers which have both significant covariance values and correlation values are most likely considered to be the potential markers discriminating between groups. The variable importance in the projection (VIP) value (Umetrics 2008), which reflects the importance of variables on the classification, was used as a guarantee for the significance of potential markers (Xie et al. 2008; Yin et al. 2009), and the variables with VIP values larger than 1 were thought to be potential markers and can be kept for further study. In present study, S-plot was used to reveal the different metabolites of the two herbs and ten markers with both significant covariance and correlation values were marked with red square (Fig. 4d). As shown in Table 1, the VIP values of all the ten compounds were larger than 10 with acceptable jack knife uncertain bars. Thus, they were finally considered as potential markers.
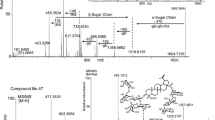
The top ten leading markers were identified by comparing their retention time and MS/MS fragmentation with those of references. Among the ten markers, which all showed diagnostic ions for PAs, nine markers were hepatotoxic pyrrolizidine alkaloids and produced diagnostic daughter ions at ions m/z 120 and 138 (Xiong et al. 2009), while the tenth (m/z 338.17, Rt 6.76 min) gave diagnostic daughter ions at ions m/z 122 and 140, indicating it was a nontoxic PA.
By comparing with PAs standards, six markers were unequivocally identified as senecionine N-oxide (marker 1), senecionine (marker 2), seneciphylline (marker 3), retrorsine N-oxide (marker 4), retrorsine (marker 7), and adonifoline (marker 9) (Table 2, Fig. 1); and the other four markers were tentatively proposed to be integerrimine N-oxide (marker 5), seneciphylline N-oxide (marker 6), othonnine (marker 8) and platyphylline (marker 10) (Table 2). Details for identification of the later four markers were as follow.
Marker 5 (m/z 352.16, Rt 4.18 min) showed the same molecular weight as senecionine N-oxide. It also showed diagnostic daughter ions for RET-PAs-N-oxides, m/z 138, 136, 120, and 118 (Xiong et al. 2009). And it was eluted at 4.18 min, a little bit earlier than that of senecionine N-oxide. Finally, marker 5 was tentatively proposed to be integerrimine N-oxide, the isomer of senecionine N-oxide.
Marker 6 (m/z 350.14, Rt 3.54 min) was 16 Da higher than seneciphylline (m/z 336.16) and showed diagnostic daughter ions for RET-PAs-N-oxides. Thus, it was identified as seneciphylline N-oxide.
Marker 8 (m/z 354.17, Rt 4.63 min) had the same molecular weight of sceleratine, othonnine and bisline, which were previously isolated from several Senecio species (Roeder 1999; Liddell 1997). MS/MS fragmentation of bisline showed ions at m/z 326 ([MH-CO]+) and 94 (100% relative intensity) (Lin et al. 1998). However, MS/MS fragmentation of marker 8 showed m/z 336 ([MH-H2O]+) and 96 (100% relative intensity) as well as m/z 293, 256, 238 and 82. Thinking about the two OH-groups at C-12 and 13 positions in othonnine, they were more likely to be othonnine or its isomer.
Marker 10 (m/z 354.17, Rt 4.63 min) showed diagnostic daughter ions at m/z 122 and 140, indicating it was a saturated retronecine type PA. It had the same molecular weight as platyphylline and its isomer neoplatyphylline, which had been found in S. nemorensis and SS (Roeder 2000; Li et al. 2008). Thus, marker 10 was tentatively proposed to be platyphylline or its isomer.
3.4 Hepatotoxicity evaluation of SS and SV
Relative content of each top ten markers (PAs) in SS and SV was compared in term of their integrated peak areas (Table 2, Fig. 5). As a result, all the PAs were accumulated much more in SV than those in SS except for adonifoline, which was a unique marker in SS. Among them, the abundant contents of senecionine N-oxide, senecionine, seneciphylline, seneciphylline N-oxide, and platyphylline in SV were 552.98, 137.63, 33.40, 146.76 and 7.19 folds of those in SS, respectively; while other four PAs including retrorsine N-oxide, integerrimine N-oxide, retrorsine and othonine accumulated a little level in SV, however, they were negative in SS. Among 10 marker alkaloids, except for platyphylline nontoxic, all the other 9 PAs possessed high toxicity. And the total content of 9 toxic PAs in SV is about 8.65 folds of that in SS (calculated as sum of peak areas of all the toxic PAs), indicating a much higher toxicity potential of SV than that of SS.

Column plot of the average intensity of the top ten leading markers in Senecio scandens (SS) and S. vulgaris (SV) by group
There are also differences between the two species when looking into samples from the same species but difference sources. SS is widely spread in China. In present study, SS samples were collected from Anhui, Guangxi, Yunnan, Fujian, and Henan province, where it is used as the main Senecio species of medical usage. On the other hand, SV is originated in Europe and spread in northeast and southwest areas of China. Thus SV samples were collected from Jilin, Heilongjiang, Yunnan province, and London. As shown in the PCA score plot in Fig. 4a, SV samples from six sources were closely gathered, indicating there were little differences among SV samples from different resources. All of them contain high contents of toxic PAs and might show similar toxicity. On the other hand, SS samples were dispersed in the PCA scores plot, indicating there were great differences among SS samples from different resources. Further study of the six SS samples revealed that two SS samples collected from Anhui Province contained relatively higher concentration of adonifoline, as well as several other toxic PAs such as senecionine, seneciphylline, and their N-oxidatives, indicating SS from this area show relative higher toxic potential and should be carefully investigated when applied for medicinal use. However, SS samples collected from Guangxi and Fujian Province contained only trace amount of adonifoline as well as other toxic PAs.
PAs with different structures showed different toxicities, as shown in the in vitro and in vivo assays of senecionie and adonifoline. By the results of our present study, senecionine and its N-oxidative are the main PAs in SV (about 38% of the detected PAs, while adonifoline is the predominant PA in SS (about 93% of the detected PAs), calculated with reference to the peak areas. The LD50 values in mice (i.g.) were 163.3 mg/kg for adonifoline versus 57.3 mg/kg for senecionine; while the IC50 values on primary hepatocytes were 49.91 μM for adonifoline versus 5.41 μM for senecionine. The in vivo and in vitro toxicities of senecionine are about 3 and 10 folds of those of adonifoline, respectively, which also contribute to the understanding of toxicity difference between SV and SS herbs.
4 Concluding remarks
Senecio scandens and S. vulgaris are two representative herbs which are genetically closed with similar morphology but different toxicity. But little investigation on the difference has been carried out. At present work, an effective strategy combining UPLC-MS with multivariate analysis has been developed for the metabolomics study of the two species to evaluate their different toxicity. Distinguished difference of the metabolic profiling was observed between the two species. Ten predominant markers were selected which were identified as the different toxic PAs such as senecionine, seneciphylline, retrorsine, and adonifoline. By a careful comparison of relative contents of toxic PAs between species as well as inner species, we proposed that high types and contents of PAs in herbs have been consistent with the potential toxicity of the Senecio species. The established method can also serve as a powerful tool for other PAs containing herbs to profile the variance of their phytochemical compositions, especially those genetically closed species with similar morphology but different toxicity. Besides, as the significant contributes of PAs to the toxicity differences of Senecio herbs, it raised the emergency of establishing a quick and high through-put method for screening of PAs in herbs as well as the finished products might containing PAs.
References
Boulesteix, A. L., & Strimmer, K. (2006). Partial least squares: A versatile tool for the analysis of high dimensional genomic data. Briefings in Bioinformatics, 8, 32–44. doi:10.1093/bib/bb1016.
Chen, T., Mei, N., & Fu, P. P. (2010). Genotoxicity of pyrrolizidine alkaloids. Journal of Applied Toxicology, 30, 183–196. doi:10.1002/jat.1504.
Chinese Pharmacopeia Commission. (2010). Pharmacopeia of the People’s Republic of China I, 32.
Dai, H., Xia, C. N., Liu, H. B., Hao, F. H., & Tang, H. R. (2010). Combined NMR and LC-DAD-MS reveals comprehensive metabonomic variations for three phenotypic cultivars of Salvia Mitiorrhiza Bunge. Journal of Proteome Research, 9, 1565–1578. doi:10.1021/pr901045c.
De Vos, R. C. H., Moco, S., Lommen, A., Keurentjes, J. J. B., Bino, R. J., & Hall, R. D. (2007). Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nature Protocols, 2, 778–791. doi:10.1038/nprot.2007.95.
Fiehn, O., Kopka, J., Dormann, P., Altmann, T., Trethewey, R. N., & Willmitzer, L. (2000). Metabolic profiling for plant functional genomics. Nature Biotechnology, 18, 1157–1161. doi:10.1038/81137.
Fu, P. P., Chou, M. W., Xia, Q. S., Yang, Y. C., Yan, J., Deorge, D. R., et al. (2001). Genotoxic pyrrolizidine alkaloids and pyrrolizidine alkaloids—mechanisms leading to DNA adduct formation and tumorigenicity. Journal of Environmental Science and Health, Part C, 19, 353–386. doi:10.1081/GNC-100107580.
Glauser, G., Guillarme, D., Glauer, G., Boccard, J., Rudaz, S., Wolfender, J. L., et al. (2009). Metabolite profiling of plant extracts by ultra-high-pressure liquid chromatography at elevated temperature coupled to time-of-flight mass spectrometry. Phytochemical Analysis, 30, 5660–5668. doi:10.1016/j.chroma.2009.05.069.
Harris, C. S., Burt, A. J., Saleem, A., Le, P. M., Martineau, L. C., Haddad, P. S., et al. (2007). A single HPLC-PDA-APCI/MS method for the quantititative comparison of phenolic compounds found in leaf, stem, root and fruit extracts of Vaccinium angustifolium. Phytochemical Analysis, 18, 161–169. doi:10.1002/pca.970.
Huxtable, R. J., Yan, C. C., Wild, S., Maxwell, S., & Cooper, R. (1996). Physicochemical and metabolic basis for the differing neurotoxicity of the pyrrolizidine alkaloids, trichodesmine and monocrotaline. Neurochemical Research, 21, 141–146. doi:10.1007/BF02529131.
Krishnan, P., Kruger, N. J., & Ratcliffe, R. G. (2005). Metabolite fingerprinting and profiling in plants using NMR. Journal of Experimental Botany, 56, 255–265. doi:10.1093/jxb/eri010.
Lame, M. W., Jones, A. D., Wilson, D. W., Dunston, S. K., & Segall, H. J. (2000). Protein targets of monocrotaline pyrrol in pulmonary artery endothelial cells. Journal of Biological Chemistry, 275, 29091–29099. doi:10.1074/jbc.M001372200.
Li, S. L., Lin, G., Fu, P. P., Chan, C. L., Li, M., Jiang, Z. H., et al. (2008). Identification of five hepatotoxic pyrrolizidine alkaloids in a commonly used traditional Chinese medicinal herb, Herba Senecionis scandentis (Qianliguang). Rapid Communications in Mass Spectrometry, 22, 591–602. doi:10.1002/rcm.3398.
Liddell, J. R. (1997). Pyrrolizidine alkaloids. Natural Products Reports, 14, 653–660. doi:10.1039/NP9971400653.
Lin, G., Zhou, K. Y., Zhao, X. G., Wang, Z. T., & But, P. P. (1998). Determination of hepatotoxic pyrrolizidine alkaloids by on-line high performance liquid. Rapid Communication in Mass Spectrometry, 12, 1445–1456. doi:10.1002/(SICI)1097-0231.
Lindon, J. C., Nicholson, J. K., & Holmes, E. (2007). The handbook of metabonomics and metabolomics (p. 35). Amsterdam: Elsevier Science BV.
Lise, J., Schauer, N., Kopka, J., Willmitzer, L., & Fernie, A. R. (2006). Gas chromatography mass spectrometry-based metabolite profiling in plants. Nature Protocols, 1, 387–396. doi:10.1038/nprot.2006.59.
Liu, M., Li, Y. G., Chou, G. X., Cheng, X. M., Zhang, M., & Wang, Z. T. (2007). Extraction and ultra-performance liquid chromatography of hydrophilic and lipophilic bioactive components in a Chinese herb Radix Salviae Miltiorrhizae. Journal of Chromatography A, 1157, 51–55. doi:10.1016/j.chroma.2007.05.018.
Novakova, L., Solichova, D., & Solich, P. (2006). Advantages of ultra performance liquid chromatography over high-performance liquid chromatography: Comparison of different analytical approaches during analysis of diclofenac gel. Journal of Separation Science, 29, 2433–2443. doi:10.1002/jssc.200600147.
Oliver, S. G. (1997). From gene to screen with yeast. Current Opinion in Genetics and Development, 7, 405–409. doi:10.1016/S0959-437X(97)80156-6.
Ortiz, C. A., Crespo, V. E., Morales, B. P., de Sáenz, S. J., Morales, C. J. M., & Ruiz, T. T. (1995). Veno-occlusive liver disease due to intake of Senecio vulgaris tea. Gastroenterology Hepatology, 18, 413–416.
Prakash, A. S., Pereira, T. N., Reilly, P. E. B., & Seawright, A. A. (1999). Pyrrolizidine alkaloids in human diet. Mutation Research/Genetic Toxicology and Environmental Mutafenesis, 443, 53–67. doi:10.1016/S1383-5742(99)00010-1.
Roeder, E. (1995). Medicinal plants in Europe containing pyrrolizidine alkaloids. Pharmazie, 50, 83–98.
Roeder, E. (1999). Analysis of pyrrolizidine alkaloids. Current Organic Chemistry, 3(6), 557–576.
Roeder, E. (2000). Medicinal plants in China containing pyrrolizidine alkaloids. Pharmazie, 55, 711–726.
Smith, L. W., & Culvenor, C. C. J. (1981). Plant sources of hepatotoxic pyrrolizidine alkaloids. Journal of Natural Products, 44, 129–152. doi:10.1021/np50014a001.
Thomas, H. C., Lame, M. W., Dunston, S. K., Segall, H. J., & Wilson, D. W. (1997). Monocrotaline pyrrole induces apoptosis in pulmonary artery endothelial cells. Toxicology and Applied Pharmacology, 151, 236–244. doi:10.1006/taap.1998.8458.
Trygg, J., & Wold, S. (2002). Orthogonal projections to latent structures (O-PLS). Journal of Chemometrics, 16, 119–128. doi:10.1002/cem.695.
Umetrics. (2008). User guide of SIMCA-P+ (p. 346). Kinnelon: Umetrics AB.
Van den Berg, R. A., Hoefsloot, H. C., Westerhuis, J. A., Smilde, A. K., & van der Werf, M. J. (2006). Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics, 7, 142–156.
Vilar, J. H., Garcia, M., & Cabrera, P. (2000). Veno-occlusive liver disease induced by Senecio vulgaris toxicity. Gastroenterology Hepatology, 23, 285–286.
Wagner, S., Scholz, K., Donegan, M., Burton, L., Wingate, J., & Volkel, W. (2006). Metabonomics and biomarker discovery: LCMS metabolic profiling and constant neutral loss scanning combined with multivariate data analysis for mercapturic acid analysis. Analytical Chemistry, 78, 1296–1305. doi:10.1021/ac051705s.
Wiklund, S., Johansson, E., Sjostrom, L., Mellerowicz, E. J., Edlund, U., Trygg, J., et al. (2008). Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Analysis Chemistry, 80, 115–122. doi:10.1021/ac0713510.
World Health Organization. (1988). Environmental Health Criteria 80: pyrrollzldine alkaloids.
Xie, G. X., Ni, Y., Su, M. M., Zhang, Y. Y., Xiao, P. G., Jia, W., et al. (2008). Application of ultra-performance LC-TOF MS metabolite profiling techniques to the analysis of medicinal Panax herbs. Metabolomics, 4, 248–260. doi:10.1007/s11306-008-0115-5.
Xiong, A. Z., Yang, L., He, Y. Q., Wang, C. H., Bligh, S. W. A., Wang, Z. T., et al. (2009). Identification of metabolites of adonifoline, a hepatotoxic pyrrolizidine alkaloid, by liquid chromatography/tandem and high-resolution mass spectrometry. Rapid Communications in Mass Spectrometry, 23, 3907–3916. doi:10.1002/rcm.4329.
Xu, X. B., Lin, H. Y., Feng, Y. S., & Chen, J. J. (2006). Preliinary phytochemical test of Scandens Buch.-Ham. and examination on chemical composition of antimicrobial part. Journal of Traditional Chinese Veterinary Medicine, 3, 10–13.
Yang, L., Xiong, A. Z., He, Y. Q., Wang, Z. Y., Wang, C. H., Wang, Z. T., et al. (2008). Bile acids metabonomic study on the CCl4- and α-Naphthylisothiocyanate-induced animal models: quantitative analysis of 22 bile acids by ultraperformance liquid chromatography-mass spectrometry. Chemical Research in Toxicology, 21, 2280–2288. doi:10.1021/tx800225q.
Yang, X. J., Yang, L., Xiong, A. Z., Li, D. X., & Wang, Z. T. (2011). Authentication of Senecio scandens and S. vulgaris based on the comprehensive secondary metabolic patterns gained by UPLC-DAD/ESI-MS. Journal of Pharmaceutical and Biomedical Analysis, 56, 165–172. doi:10.1016/j.jpba.2011.05.004.
Yin, P. Y., Wan, D. F., Zhao, C. Z., Chen, J., Xu, G. W., et al. (2009). A metabonomic study of hepatitis B-induced liver cirrhosis and hepatocellular carcinoma by using RP-LC and HILIC coupled with mass spectrometry. Molecular Biosystems, 5, 868–876. doi:10.1039/b820224a.
Zhang, F., Wang, C. H., Wang, W., Zhang, M., Bligh, S. W., Wang, Z. T., et al. (2008). Quantitative analysis by HPLC-MS2 of the pyrrolizidine alkaloid adonifoline in Senecio scandens. Phytochemical Analysis, 19(1), 25–31. doi:10.1002/pca.1009.
Acknowledgments
We thank the National Natural Science Foundation of China (30530840, 30701104 and 30572222), the Shanghai International Collaborative Project (09540704700), the Shanghai Rising-Star Program (09QA1405500) and the Scientific Innovation Project from Shanghai Municipal Education Commission (09YZ125) for their financial support of this work.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Xiong, A., Yang, L., Ji, L. et al. UPLC-MS based metabolomics study on Senecio scandens and S. vulgaris: an approach for the differentiation of two Senecio herbs with similar morphology but different toxicity. Metabolomics 8, 614–623 (2012). https://doi.org/10.1007/s11306-011-0354-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-011-0354-8